Abstract
The Rendu-Osler-Weber syndrome is a rare systemic fibrovascular dysplasia, recognized by mucocutaneous telangiectasias, arteriovenous malformations, epistaxis and family history. Recurrent bleeding, hypoxemia, congestive heart failure, portosystemic encephalopathy, and symptoms related to angiodysplasia of the central nervous system may occur. Since the treatment is based on supportive measures, early recognition is of utmost importance. This article reports the case of a 53-year-old male patient who presented telangiectasias on fingers, oral cavity and nasal mucosa for 10 years, with a history of recurrent epistaxis of varying severity since childhood. Mother, sister and daughter have similar lesions.
Keywords:
Epistaxis; Genetics; Telangiectasia; hereditary hemorrhagic
INTRODUCTION
The Rendu-Osler-Weber syndrome, also known as Hereditary Hemorrhagic Telangiectasia (HHT) is a rare systemic fibrovascular dysplasia, characterized by recurrent epistaxis, mucocutaneous telangiectasias, arteriovenous malformations (AVMs) in different organs and family history.
Its incidence is 1-2/100.000 homogeneously distributed by race and sex. It has autosomal dominant transmission and high degree of penetrance, but there is no family history in around 20% of cases.11 Garcia RID, Cecatto SB, Costa KS, Veiga Jr FA, Uvo IP, Rapoport PB. Síndrome de Rendu-Osler-Weber: tratamento clínico e cirúrgico. Rev Bras Otorrinolaringol. 2003;69:577-80.
Mutations in two genes are considered as responsible for the majority of cases: endoglin (ENG) located on chromosome 9q33-q34 and activin receptor-like kinase (ALK1) located on chromosome 12q13.22 Dupuis-Girod S, Bailly S, Plauchu H. Hereditary hemorrhagic telangiectasia: from molecular biology to patient care. J Thromb Haemost. 2010;8:1447-56.
ENG mutations are observed in HHT type 1, phenotype responsible for 40% of pulmonary AVMs whereas mutations of ALK1 are observed in HHT type 2, leading to lower incidence of these AVMs (14%) associated with milder forms and late onset of the disease.33 Sadick H, Sadick M, Götte K, Naim R, Riedel F, Bran G, et al. Hereditary hemorrhagic telangiectasia: an update on clinical manifestations and diagnostic measures. Wien Klin Wochenschr. 2006;118:72-80.
The mutated genes encode proteins that modulate transforming growth factor beta (TGF-β), which stimulates vascular endothelial growth factor (VEGF) production and plays a key role in angiogenesis.44 Bose P, Holter JL, Selby GB. Bevacizumab in hereditary hemorrhagic telangiectasia. N Engl J Med. 2009;360:2143-4.
Diagnosis is based on the Curaçao criteria:55 Fuchizaki U, Miyamori H, Kitagawa S, Kaneko S, Kobayashi K. Hereditary haemorrhagic telangiectasia (Rendu-Osler-Weber disease). Lancet. 2003;362:1490-4.
1)Epistaxis- spontaneous, recurrent nosebleeds;
2)Telangiectasias- multiple, at characteristic sites (lips, oral cavity, fingers, nose);
3)Visceral lesions- such as gastrointestinal telangiectasias (with or without bleeding), pulmonary AVM, hepatic AVM, cerebral AVM, spinal AVM; and
4)Family history- a first-degree relative with HHT.
Three criteria indicate a definite diagnosis of the disorder; two a possible or suspected case.
The treatment is only palliative, with no consensus on the best treatment option. It is essential to promote control of the disease as long as possible.
CASE REPORT
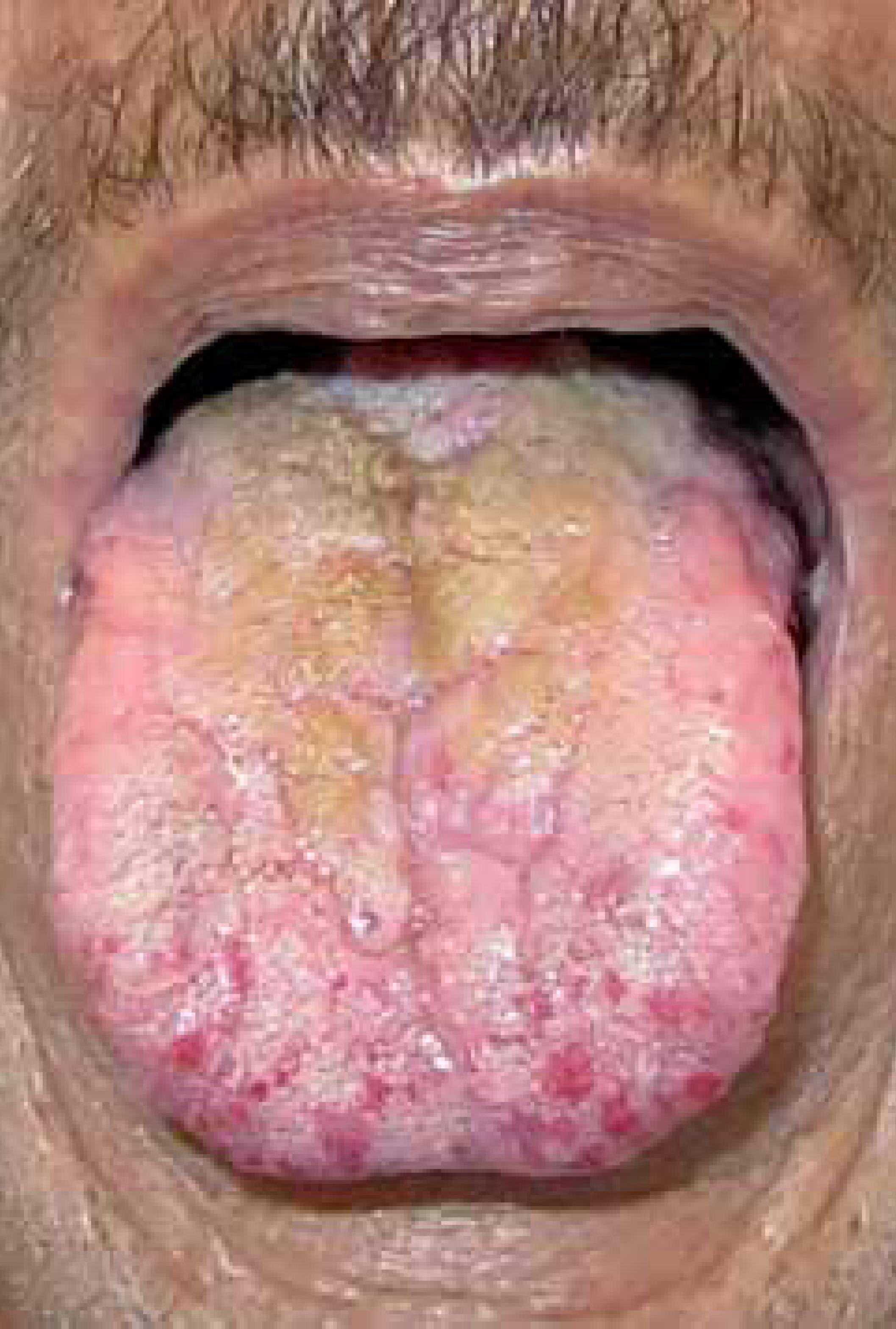
Male patient, 53 years old, presented vegetating plaque on the back of the tongue and telangiectasias on the lips, gums, palate, oral and nasal mucosa as well as on fingers for 10 years (Figure 1). He complains also of recurrent epistaxis of varying intensity ever since his childhood. Routine laboratory tests showed mild anemia and iron deficiency (hemoglobin 11.9 g/dL; serum iron levels 56 mcg/dL), since there was no major bleeding in the past months.
Mother (who died at age 45 due to stroke), sister and daughter have similar lesions. Histology of nasal injury point to vascular dilation and deposition of hemosiderin (Figure 2).
A. Histology shows vascular dilation and pigment leakage (40 X magnified); B. Erythrocytes in the center of the field (100 X magnified)
Bronchoscopy, esophagogastroduodenoscopy and colonoscopy show diffuse telangiectasias without signs of recent bleeding (Figures 3and4). As the most serious complaint of the patient was nose bleeding, he was referred to otolaryngology and is being monitored to exclude other possible foci of AVMs.
Esophagus: telangiectasias covering the entire mucosa without evidence of bleeding in upper gastrointestinal endoscopy
DISCUSSION
Hereditary Hemorrhagic Telangiectasia presents mucocutaneous macular telangiectasia with 1 to 3 mm of diameter that, generally, appears 10 to 30 years after epistaxis episodes (60% of patients). Palms and fingers are the only sites affected in 71% of patients, lips and tongue in 66%. Twenty to 40% have complete cutaneous manifestations, compatible with the present report, which met the four Curaçao criteria.
Pulmonary manifestations include hemoptysis, hemothorax, right-to-left-shunting and paradoxical emboli (venous circulation embolic event that reaches the systemic circulation). These symptoms arise from AVMs and fistulas, present in 5-23% of patients. The paradoxical embolism, found in 40-50% of patients, can cause transitory ischemic events and strokes. In 8% of patients, massive pulmonary hemorrhage can occur. Other symptoms include hypoxia, dyspnea, cyanosis, polycythemia and clubbing of the distal phalange.
Complications in the central nervous system (CNS) vary from transitory ischemic events to an abscess or intracranial bleeding. It is estimated that 8-41% of patients with HHT will present some neurological complication during their lifetime. Clinical manifestations may include convulsions, headache, coma and different degrees of neurological deficits.
In the gastrointestinal tract, manifestations include telangiectasias, AVMs and varicosities, which can lead to upper gastrointestinal bleeding, liver dysfunction and encephalopathy. Bleeding tends to be recurrent and progressive, usually occurring after 40 years of age.11 Garcia RID, Cecatto SB, Costa KS, Veiga Jr FA, Uvo IP, Rapoport PB. Síndrome de Rendu-Osler-Weber: tratamento clínico e cirúrgico. Rev Bras Otorrinolaringol. 2003;69:577-80.
The treatment traditionally relies on surgical procedures, preferably endoscopic, to control iron deficiency anemia, bleeding and potentially lethal aneurysms, in addition to the use of hormonal therapies (estrogen and progesterone).66 Juares AJC, Dell'Aringa AR, Nardi JC, Kobari K, Rodrigues VLMGM, Perches-Filho RM. Síndrome de Rendu-Osler-Weber: relato de caso e revisão de literatura. Rev Bras Otorrinolaringol. 2008;74:452-7.,77 Gontijo B, Pereira LB, Silva CMR. Vascular malformations. An Bras Dermatol. 2004;79:7-25.In recent research, bevacizumab (anti-VEGF monoclonal antibody) has been evaluated with promising results, but it is still not considered definitive due primarily to its various long-term side effects.44 Bose P, Holter JL, Selby GB. Bevacizumab in hereditary hemorrhagic telangiectasia. N Engl J Med. 2009;360:2143-4.
Prognosis is favorable in cases where the bleeding can be controlled, and the reported mortality due to disease complications is less than 10% of cases.
Knowledge of dermatological aspects of this rare and potentially severe condition may be essential for early diagnosis, enabling the investigation of pulmonary and CNS malformations and preventing possible catastrophic effects of the disease.
-
Financial Support: None.
-
How to cite this article: Barbosa AB, Hans-Filho G, Vicari CFS, Medeiros MZ, Couto DV, Takita LC. Rendu-Osler-Weber syndrome: dermatological approach. An Bras Dermatol. 2015;90(3 Suppl 1):S226-8.
-
*
Work performed at the Serviço de Dermatologia Dr. Günter Hans Núcleo de Hospital Universitário da Universidade Federal do Mato Grosso do Sul (NHUUFMS) - Campo Grande (MS), Brazil.
References
-
1Garcia RID, Cecatto SB, Costa KS, Veiga Jr FA, Uvo IP, Rapoport PB. Síndrome de Rendu-Osler-Weber: tratamento clínico e cirúrgico. Rev Bras Otorrinolaringol. 2003;69:577-80.
-
2Dupuis-Girod S, Bailly S, Plauchu H. Hereditary hemorrhagic telangiectasia: from molecular biology to patient care. J Thromb Haemost. 2010;8:1447-56.
-
3Sadick H, Sadick M, Götte K, Naim R, Riedel F, Bran G, et al. Hereditary hemorrhagic telangiectasia: an update on clinical manifestations and diagnostic measures. Wien Klin Wochenschr. 2006;118:72-80.
-
4Bose P, Holter JL, Selby GB. Bevacizumab in hereditary hemorrhagic telangiectasia. N Engl J Med. 2009;360:2143-4.
-
5Fuchizaki U, Miyamori H, Kitagawa S, Kaneko S, Kobayashi K. Hereditary haemorrhagic telangiectasia (Rendu-Osler-Weber disease). Lancet. 2003;362:1490-4.
-
6Juares AJC, Dell'Aringa AR, Nardi JC, Kobari K, Rodrigues VLMGM, Perches-Filho RM. Síndrome de Rendu-Osler-Weber: relato de caso e revisão de literatura. Rev Bras Otorrinolaringol. 2008;74:452-7.
-
7Gontijo B, Pereira LB, Silva CMR. Vascular malformations. An Bras Dermatol. 2004;79:7-25.
Publication Dates
-
Publication in this collection
June 2015
History
-
Received
21 Feb 2014 -
Accepted
01 July 2014