Abstract
Giant axonal neuropathy is a rare autosomal recessive neurodegenerative disease. The condition is characterized by neurons with abnormally large axons due to intracellular filament accumulation. The swollen axons affect both the peripheral and central nervous system. A 6-year old female patient had been referred to a geneticist reporting problems with walking and hypotonia. At the age of 10, she became wheelchair dependent. Scanning electron microscopy of a curly hair classified it as pili canaliculi. GAN gene sequencing demonstrated mutation c.1456G>A (p.GLU486LYS). At the age of 12, the patient died due to respiratory complications. Dermatologists should be aware of this entity since hair changes are considered suggestive of GAN.
Keywords:
Giant axonal neuropathy; Hair; Hair diseases; Microscopy, electron, scanning
INTRODUCTION
Giant Axonal Neuropathy (GAN – OMIM # 256850) is a rare hereditary autosomal recessive neurodegenerative disease with unknown prevalence. GAN was originally reported in 1972 by Berg and colleagues. The condition is characterized by neurons with abnormally large axons due to intracellular filament accumulation. The swollen axons affect both the peripheral and central nervous system.11 Kuhlenbäumer G, Timmerman V, Bomont P. Giant Axonal Neuropathy. In: Pagon RA, Adam MP, Ardinger HH, Bird TD, Dolan CR, Fong CT, Smith RJH, Stephens K, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington; 2003. [cited 2014 Jul 22]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1136/
http://www.ncbi.nlm.nih.gov/books/NBK113...
,22 Treiber-Held S, Budjarjo-Welim H, Reimann D, Richter J, Kretzschmar HA, Hanefeld F. Giant Axonal Neuropathy: A Generalized Disorder of Intermediate Filaments with Longitudinal Grooves in the Hair. Neuropediatrics. 1994;25:89-93.
GAN generally appears in infancy or early childhood, rarely at birth, and progresses to death. The onset of the disease usually presents with delay in the acquisition of abilities followed by gait disorders and progressive weakness in upper and lower limbs. It may also involve the brain nerves, resulting in facial weakness, optic atrophy and ophthalmoplegia. The disease evolves rapidly with the deterioration of the central nervous system showing epilepsy, cerebellar signs – ataxia, nystagmus and dysarthria – and signs of pyramidal tract damage, but rarely revealing mental disabilities. Another sign of the disease is dull, tightly-curled hair that is markedly different from the parents' in color and texture.22 Treiber-Held S, Budjarjo-Welim H, Reimann D, Richter J, Kretzschmar HA, Hanefeld F. Giant Axonal Neuropathy: A Generalized Disorder of Intermediate Filaments with Longitudinal Grooves in the Hair. Neuropediatrics. 1994;25:89-93.
Most individuals become wheelchair dependent in the second decade of life and eventually bedridden with severe polyneuropathy, ataxia and dementia, which may cause death in the third decade.
CASE REPORT
A 10-year-old female patient consulted with a neurologist for the first time when she started having problems with walking at the age of 5 due to progressive loss of strength. At the age of 6, she was referred to a geneticist because of difficulty in ambulation caused by hypotonia in lower and upper limbs. Clinical examination revealed genu valgus and joint hypermobility. Karyotype, muscle enzymes and radiologic studies were normal.
At 7 the patient showed growth of pubic hair and a possible menstrual episode with normal LH, FSH, estrogen and progesterone levels. Magnetic resonance imaging of the brain revealed pituitary cysts and diffuse hypomyelination of the central nervous system.
At 10 she became wheelchair dependent. Orthopedic surgery was required to correct shortening of the Achilles tendons. Dermatological examination revealed curly hair different from her parents' (Figure 1A and 1B) with eyelash involvement (Figure 1C).
After GAN was suggested, we collected DNA from peripheral blood. Sequence analysis of GAN gene demonstrated the mutation c.1456G>A (p.GLU486LYS).
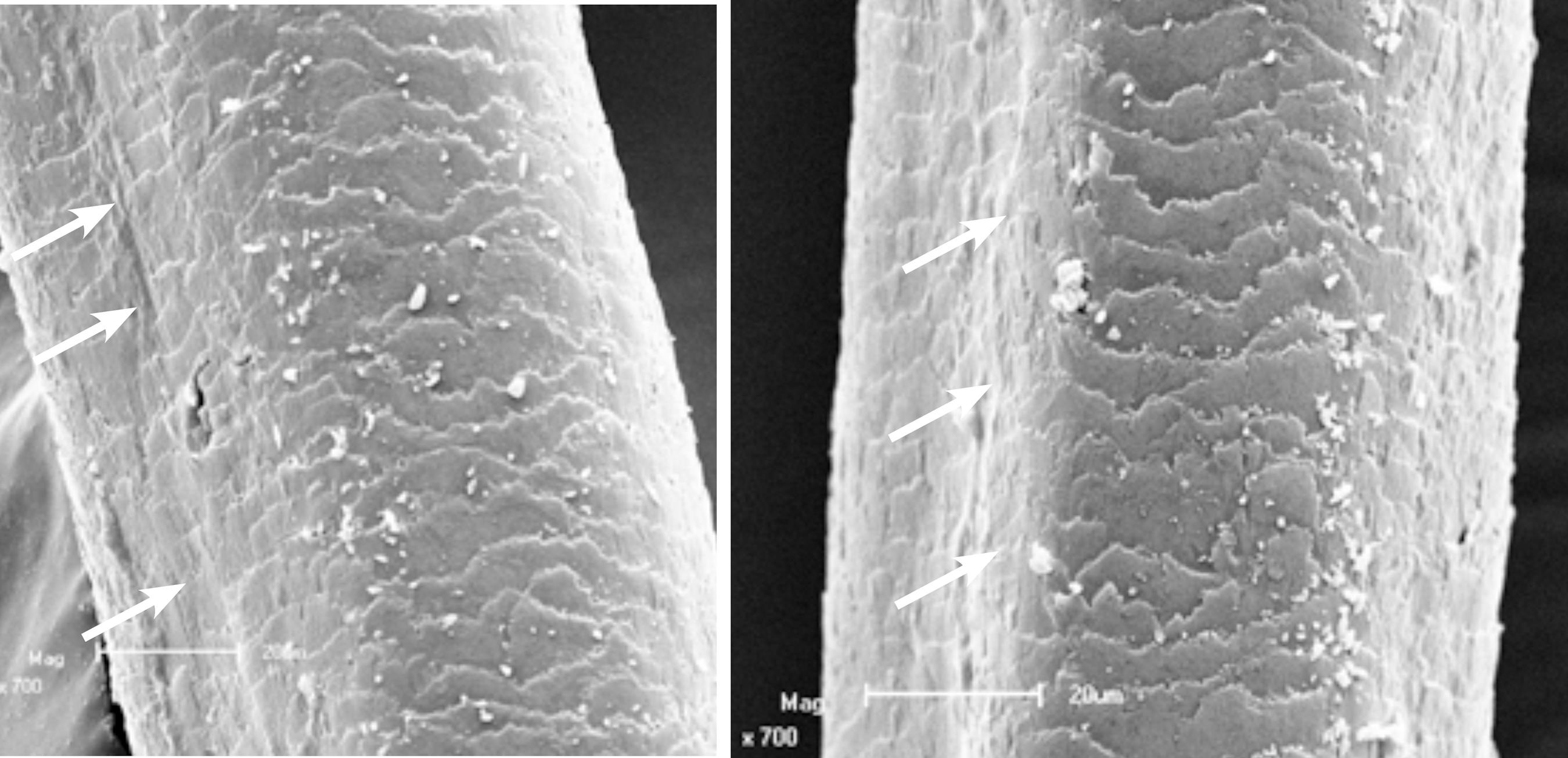
Scanning electron microscopy revealed longitudinal grooves on the surface of the hair at low magnification and a polygonal shape at higher magnification (Figures 2 and 3), typical findings of pili canaliculi.
Scanning electron microscopy – low magnification showing longitudinal grooving (arrows) (x 200 and x 350).
Scanning electron microscopy – high magnification showing grooving (arrows) and polygonal hair shape (x 700)
The patient died at the age of 12 due to respiratory complications.
DISCUSSION
Giant axonal neuropathy is caused by mutations in the GAN gene, which affect of the protein gigaxonin, leading to disorganization of neurofilaments (intermediate filaments of neuronal cells). The mutation may also lead to the axonal accumulation of proteins, hence the denomination giant axons.
One of the functions of the gigaxonin protein may be to maintain the architecture of other intermediate filaments such as keratins, which could explain one characteristic sign of the disease: the hair involvement.55 Opal P, Goldman RD. Explaining intermediate filament accumulation in giant axonal neuropathy. Rare Dis. 2013;1:e25378.,66 Cleveland DW, Yamanaka K, Bomont P. Gigaxonin controls vimentin organization through a tubulin chaperone-independent pathway. Hum Mol Genet. 2009;18:1384-94. Some studies analyzed families affected by pili canaliculi – a cutaneous genetic manifestation without neurological involvement – in which patients presented uncombable hair or gradual hypotrichosis33 Filho Rheingantz da Cunha R, Larangeira de Almeida H Jr, Suita de Castro LA, Moreira Rocha N, Abrantes V. Pili Canaliculi: clinical and microscopic investigation of the first Brazilian family. Int J Dermatol. 2007;46:190-3.,44 Cunha Filho RR, Almeida Jr HL, Rocha NM, Castro LAS. Síndrome dos cabelos impenteáveis (pili canaliculi): variabilidade clínica em 12 membros de uma família. An Bras Dermatol. 2008;83:53-5.. Ultrastructural hair examination in those patients revealed grooves on hair surface giving polygonal shapes (triangular, square, reniform) to the hair shaft.44 Cunha Filho RR, Almeida Jr HL, Rocha NM, Castro LAS. Síndrome dos cabelos impenteáveis (pili canaliculi): variabilidade clínica em 12 membros de uma família. An Bras Dermatol. 2008;83:53-5. The results are similar to those presented by GAN patients' hair. Treiber-Held et al. reported cases of grooved hair in GAN patients, confirming our results.22 Treiber-Held S, Budjarjo-Welim H, Reimann D, Richter J, Kretzschmar HA, Hanefeld F. Giant Axonal Neuropathy: A Generalized Disorder of Intermediate Filaments with Longitudinal Grooves in the Hair. Neuropediatrics. 1994;25:89-93.
Since the results of three-dimensional ultrastructural hair analysis in our patient are suggestive of pili canaliculi, dermatologists should be aware of this entity because hair changes are considered suggestive of GAN.11 Kuhlenbäumer G, Timmerman V, Bomont P. Giant Axonal Neuropathy. In: Pagon RA, Adam MP, Ardinger HH, Bird TD, Dolan CR, Fong CT, Smith RJH, Stephens K, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington; 2003. [cited 2014 Jul 22]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1136/
http://www.ncbi.nlm.nih.gov/books/NBK113...
,77 Demir E, Bomont P, Erdem S, Cavalier L, Demirci M, Kose G, et al. Giant axonal neuropathy: clinical and genetic study in six cases. J Neurol Neurosurg Psychiatry. 2005;76:825-32.
-
*
Work performed at Faculdade de Medicina e Laboratório de Microscopia Eletrônica. Universidade Federal de Pelotas (UFPel) – Pelotas (RS), Brazil.
-
Financial Support: None
References
-
1Kuhlenbäumer G, Timmerman V, Bomont P. Giant Axonal Neuropathy. In: Pagon RA, Adam MP, Ardinger HH, Bird TD, Dolan CR, Fong CT, Smith RJH, Stephens K, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington; 2003. [cited 2014 Jul 22]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1136/
» http://www.ncbi.nlm.nih.gov/books/NBK1136/ -
2Treiber-Held S, Budjarjo-Welim H, Reimann D, Richter J, Kretzschmar HA, Hanefeld F. Giant Axonal Neuropathy: A Generalized Disorder of Intermediate Filaments with Longitudinal Grooves in the Hair. Neuropediatrics. 1994;25:89-93.
-
3Filho Rheingantz da Cunha R, Larangeira de Almeida H Jr, Suita de Castro LA, Moreira Rocha N, Abrantes V. Pili Canaliculi: clinical and microscopic investigation of the first Brazilian family. Int J Dermatol. 2007;46:190-3.
-
4Cunha Filho RR, Almeida Jr HL, Rocha NM, Castro LAS. Síndrome dos cabelos impenteáveis (pili canaliculi): variabilidade clínica em 12 membros de uma família. An Bras Dermatol. 2008;83:53-5.
-
5Opal P, Goldman RD. Explaining intermediate filament accumulation in giant axonal neuropathy. Rare Dis. 2013;1:e25378.
-
6Cleveland DW, Yamanaka K, Bomont P. Gigaxonin controls vimentin organization through a tubulin chaperone-independent pathway. Hum Mol Genet. 2009;18:1384-94.
-
7Demir E, Bomont P, Erdem S, Cavalier L, Demirci M, Kose G, et al. Giant axonal neuropathy: clinical and genetic study in six cases. J Neurol Neurosurg Psychiatry. 2005;76:825-32.
Publication Dates
-
Publication in this collection
Sep-Oct 2016
History
-
Received
29 Apr 2015 -
Accepted
22 July 2015