Abstract:
Pemphigus are intraepidermal autoimmune bullous dermatoses that occur with lesions on the skin and / or mucous membranes. The most frequent types are pemphigus vulgaris and pemphigus foliaceus (classic and endemic). This consensus aims to present a complete and updated review of the treatment of these two more frequent forms of pemphigus, based on the literature and the personal experience of the authors. In moderate and severe cases of pemphigus vulgaris and foliaceus, systemic corticosteroid therapy (prednisone or prednisolone) is the treatment of choice. Adjuvant drugs, usually immunosuppressive drugs (azathioprine, mycophenolate mofetil, methotrexate, cyclophosphamide) may be prescribed as corticosteroid sparers in refractory cases or with contraindications to corticosteroids to minimize side effects. In severe and nonresponsive cases, corticosteroids in the form of intravenous pulse therapy, immunoglobulin and plasmapheresis / immunoadsorption can be administered. Immunobiological drugs, particularly rituximab, appear as a promising alternative. For milder cases, smaller doses of oral corticosteroid, dapsone and topical corticosteroids are options. At the end flowcharts are presented as suggestions for a therapeutic approach for patients with pemphigus vulgaris and pemphigus foliaceus.
Keywords:
Autoimmunity; Pemphigus; Treatment
PEMPHIGUS VULGARIS
INTRODUCTION
Pemphigus is a group of rare autoimmune bullous diseases that affect the skin and mucous membranes. Its estimated incidence in central Europe is 2 new cases/million inhabitants/year. They undergo a chronic evolution, with significant morbidity and mortality, and significantly impair quality of life.11 Kridin K, Sagi SZ, Bergman R. Mortality and cause of death in Israeli patients with pemphigus. Acta Derm Venereol. 2017;97:607-611.,22 Hsu DY, Brieva J, Sinha AA, Langan SM, Silverberg JI. Comorbidities and inpatient mortality for pemphigus in the USA. Br J Dermatol. 2016;174:1290-8. They result from the production of pathogenic autoantibodies (usually IgG) that are directed against various desmosomal proteins (the desmogleins Dsg3 and Dsg1). The binding of these autoantibodies to desmosomal components compromises intraepidermal adhesion, leading to acantholysis and the formation of vesicles, blisters, and erosions on the skin and mucous membranes. 33 Di Zenzo G, Amber KT, Sayar BS, Müller EJ, Borradori L. Immune response in pemphigus and beyond: progresses and emerging concepts. Semin Immunopathol. 2016;38:57-74.
4 Mahoney MG, Wang Z, Rothenberger K, Koch PJ, Amagai M, Stanley JR. Explanations for the clinical and microscopic localization of lesions in pemphigus foliaceus and vulgaris. J Clin Invest. 1999;103:461-8.-55 Pan M, Liu X, Zheng J. The pathogenic role of autoantibodies in pemphigus vulgaris. Clin Exp Dermatol. 2011;36:703-7.
Various subtypes of pemphigus have been identified, based on their clinical and histopathological characteristics and the specific antigens against which autoantibodies are produced. The main types are pemphigus vulgaris (PV) and pemphigus foliaceus, but in the past several decades, nonclassical forms of pemphigus have been described, including paraneoplastic pemphigus, pemphigus herpetiformis, and IgA pemphigus. 66 Joly P, Litrowski N. Pemphigus group (vulgaris, vegetans, foliaceus, herpetiformis, brasiliensis). Clin Dermatol. 2011;29(4):432-6.
Regardless of subtype, the formation of autoantibodies against desmosomal components has long been considered the chief event in the pathogenesis of pemphigus. In addition to the involvement of humoral immunity, the function of cellular immunity in it has been highlighted. 77 Amber KT, Staropoli P, Shiman MI, Elgart GW, Hertl M. Autoreactive T cells in the immune pathogenesis of pemphigus vulgaris. Exp Dermatol. 2013; 22:699-704.
EPIDEMIOLOGY
PV is the main clinical form of pemphigus, accounting for 70% of cases.66 Joly P, Litrowski N. Pemphigus group (vulgaris, vegetans, foliaceus, herpetiformis, brasiliensis). Clin Dermatol. 2011;29(4):432-6. It is a rare disease, with an annual incidence that varies between 0.76 (Finland) and 16.1 (Israel) cases/million inhabitants, depending on the study population. Although it is universally occurring, PV primarily affects adults, peaking in incidence between the fourth and sixth decades of life. In Brazil, it also develops in young adults. Most studies have noted a higher incidence in women, with the female:male ratio ranging from 1.1 (Finland) to 5.0 (US). PV is considered the most severe form of pemphigus. 88 Kridin K. Pemphigus group: overview, epidemiology, mortality, and comorbidities. Immunol Res. 2018;66:255-70.,99 Bystryn JC, Rudolph JL. Pemphigus. Lancet. 2005;366:61-73.
ETIOPATHOGENESIS
The etiology of PV is unknown. Possible triggering factors include environmental agents, infections, drugs, and tumors. It is linked to specific HLAs, such as HLA-DRB1*04:02 (Jewish) and HLADQB1*05:03.1010 Pollmann R, Schmidt T, Eming R, Hertl M. Pemphigus: a Comprehensive Review on Pathogenesis, Clinical Presentation and Novel Therapeutic Approaches. Clin Rev Allergy Immunol. 2018;54:1-25
PV is characterized by acantholysis and the formation of intraepidermal blisters, resulting from the presence of IgG autoantibodies against the transmembrane desmosomal glycoprotein Dsg3 (130 kD) and, in certain cases, Dsg1 (160 kD). 1111 Hammers CM, Stanley JR. Mechanisms of disease: pemphigus and bullous pemphigoid. Annu Rev Pathol. 2016 May 23;11:175-97.
CLINICAL CONDITION
The condition generally begins with mucous lesions that, after several weeks or months, start to affect the skin. Among mucous membranes, the most frequently involved is the oral mucosa, in 70% of cases, followed by the genital in 20%. Oral lesions are painful ulcers or eroded lesions that form in any area in the oral cavity, usually accompanied by dysphagia and weight loss. Other mucous membranes can be affected, such as the conjunctiva, pharynx, larynx, esophagus, and anal mucosa.
Vesicles and flaccid blisters develop on the skin, throughout the integument, predominating on the scalp, face, and upper chest. These lesions rupture, giving rise to considerable areas of erosion that are painful. If the lesions are extensive, there might be disruptions in hydroelectrolyte balance, protein loss, and anemia. In addition, rupture of the cutaneous-mucosal barrier facilitates secondary infections. Pemphigus vegetans is a rare clinical subtype of PV that is characterized by vegetant cutaneous lesions in folds of the armpits, groin, and inframammary region. 66 Joly P, Litrowski N. Pemphigus group (vulgaris, vegetans, foliaceus, herpetiformis, brasiliensis). Clin Dermatol. 2011;29(4):432-6.,99 Bystryn JC, Rudolph JL. Pemphigus. Lancet. 2005;366:61-73.,1212 Broussard KC, Leung TG, Moradi A, Thorne JE, Fine JD. Autoimmune bullous diseases with skin and eye involvement: Cicatricial pemphigoid, pemphigus vulgaris, and pemphigus paraneoplastica. Clin Dermatol. 2016;34:205-13.,1313 Becker BA, Gaspari AA. Pemphigus vulgaris and vegetans. Dermatol Clin. 1993;11:429-52.
The main clinical differential diagnoses in PV are bullous pemphigoid, paraneoplastic pemphigus, and Stevens-Johnson syndrome. PV should be confirmed by histopathological examination of the skin and mucosal lesions with immunofluorescence techniques.
LABORATORY DIAGNOSIS 1010 Pollmann R, Schmidt T, Eming R, Hertl M. Pemphigus: a Comprehensive Review on Pathogenesis, Clinical Presentation and Novel Therapeutic Approaches. Clin Rev Allergy Immunol. 2018;54:1-25
Histopathological examination: Whenever possible, an entire vesicle should be removed by skin biopsy. PV shows low suprabasal intraepidermal cleavage with acantholytic (rounded) cells. These cells can also be viewed, isolated or in groups, from blisters or erosion smears by Tzanck test. Acantholysis is also present in hair follicles and sebaceous gland ducts. In vegetans pemphigus, there is also papillomatosis, in addition to neutrophil and eosinophil infiltrates.
Direct immunofluorescence: IgG and C3 autoantibodies can be detected in a perilesional skin fragment (lacy pattern in the stratum spinosum).
Indirect immunofluorescence: Autoantibodies can be detected and titrated in serum in 80% to 90% of patients. Usually, the serum titer of autoantibodies correlates with the clinical activity of the PV.
TREATMENT
The treatment of autoimmune bullous dermatoses, including PV is usually based on systemic medications, because they comprise a severe group of mucosal and cutaneous diseases with significant morbidity and mortality. Treatment should be started as early as possible, and its goal is to achieve and maintain disease remission. Thus, the treatment is often prolonged and can last many years (average 5 to 10 years). Due to the rarity of PV, there are few randomized controlled clinical trials. However, several observational studies, case reports, and case series have been published and support the clinical practice of specialists in PV cases. The mortality due to PV has decreased in the past 50 years and stems primarily from the side effects of medications. 1414 Meurer M. Immunosuppressive therapy for autoimmune bullous diseases. Clin Dermatol. 2012;30:78-83.,1515 Bystryn JC, Steinman NM. The adjuvant therapy of pemphigus. An update. Arch Dermatol. 1996;132:203-12.
Evaluation before starting treatment
Clinical evaluation: Weight, height, and blood pressure
Laboratory tests: Blood count; electrolytes; hepatic and renal function; blood glucose and glycated hemoglobin; vitamin D; lipids; serologies for hepatitis B and C, syphilis, and HIV; urine I; pregnancy test if applicable; chest x-ray; and bone densitometry (should be repeated after 6 months and then annually).
Ophthalmological evaluation: Initial and then annually.
Systemic treatment
Corticosteroids
Systemic corticosteroids are the basis of the treatment for PV. They have potent anti-inflammatory and immunosuppressive activities. The introduction of these drugs in the 1950s was followed by a reduction in mortality from 75% to 30%. 1515 Bystryn JC, Steinman NM. The adjuvant therapy of pemphigus. An update. Arch Dermatol. 1996;132:203-12.
16 Hertl M, Jedlickova H, Karpati S, Marinovic B, Uzun S, Yayli S, et al. Pemphigus. S2 guideline for diagnosis and treatment-guided by the European Dermatology Forum (EDF) in cooperation with the European Academy of Dermatology and Venereology (EADV). J Eur Acad Dermatol Venereol. 2015;29:405-14.-1717 Tavakolpour S. Current and future treatment options for pemphigus: Is it time to move towards more effective treatments? Int Immunopharmacol. 2017;53:133-42.
- Oral administration: Prednisone is the most commonly used oral corticosteroid, followed by prednisolone and deflazacort. Although several groups prefer to administer a dosage of 40 to 60mg/day (prednisone) for patients with mild PV and 60 to 100mg/day for more severe conditions, most supply a full dose (1 to 2mg/kg/day po) for all patients from the outset, avoiding a progressive increase in dose. However, extremely high dosages, as used previously (3 to 4mg/kg/day), have been shown to be disadvantageous due to their frequent and severe side effects.
Corticosteroids act rapidly in PV, effecting improvement in several days and impeding the emergence of new lesions after 2 to 3 weeks. Complete re-epithelization can take up to 2 months. After the condition is controlled, defined as the disruption of the emergence of new lesions and total re-epithelialization of existing lesions, the corticosteroid dose is slowly reduced. The rate of this decrease should decline toward the end, which can sometimes take years, due to the lack of uniform protocols for this practice. Certain groups recommend that starting from a specific daily dose (usually 40mg/day prednisone), the drug should be administered every other day, which would minimize the side effects. Similarly, there is no consensus on how to increase the dose in cases of recurrence. Generally, relapse is milder than the initial presentation of the disease and requires doses of prednisone that are equal to or lower than the initial dose for control.1818 Ruocco E, Wolf R, Ruocco V, Brunetti G, Romano F, Lo Schiavo A. Pemphigus: Associations and management guidelines: Facts and controversies. Clin Dermatol. 2013;31:382-90.,1919 Harman KE, Brown D, Exton LS, Groves RW, Hampton PJ, Mohd Mustapa MF, et al. British Association of Dermatologists' guidelines for the management of pemphigus vulgaris 2017. Br J Dermatol. 2017;177:1170-201
- Pulse therapy: Corticosteroids can also be administered as pulse therapy for cases in which control with prednisone at dosages of over 1mg/kg/day is not achieved. To this end, methylprednisolone 1g/day IV and dexamethasone 300mg/day IV are used, both for 3 consecutive days. The advantage of pulse therapy is that it allows for a faster reduction in the prednisone dose, minimizing its side effects.2020 Rao PN, Lakshmi TS. Pulse therapy and its modifications in pemphigus: a six year study. Indian J Dermatol Venereol Leprol. 2003;69:329-33.,2121 Chams-Davatchi C, Esmaili N, Daneshpazhooh M, Valikhani M, Balighi K, Hallaji Z, et al. Randomized controlled open-label trial of four treatment regimens for pemphigus vulgaris. J Am Acad Dermatol. 2007;57:622-8.
Although corticosteroids are effective in controlling PV in most patients, they have frequent and potentially severe side effects, the most significant of which are hypertension, diabetes mellitus, cutaneous and systemic infections, gastric ulcer, osteoporosis, femoral head necrosis, glaucoma, and cortisone cataract. These side effects are partly responsible for the morbidity and lethality of the disease, often due to the increase in the frequency of consultations, laboratory tests, and hospital admissions. All patients should receive gastric mucosal protectors and vitamin D supplementation.2222 Ahmed AR, Moy R. Death in pemphigus. J Am Acad Dermatol. 1982;7:221-8.
To minimize the side effects, morbidity, and mortality of PV, contrary to what was advocated several decades ago, it is recommended that the daily dosage of prednisone does not exceed 1.5mg/kg/day-above this value, the likelihood of skin infection and evolution to septicemia (the main death cause in these patients) increases progressively. Thus, other drugs are recommended, in association with corticosteroids-termed adjuvant drugs (corticosteroid-sparing agents).2323 Frew JW, Martin LK, Murrell DF. Evidence-based treatments in pemphigus vulgaris and pemphigus foliaceus. Dermatol Clin. 2011;29:599-606.
Adjuvant drugs
When the condition is not controlled solely with corticosteroids or when the patient has clinical contraindications to high-dose corticosteroids (e.g., hypertension, diabetes mellitus, glaucoma, osteoporosis-all of which are frequent in the age group in which the prevalence of PV peaks), other drugs, called adjuvants or corticosteroid-sparing agents, should be incorporated. Adjuvant drugs also prevent relapses in previously controlled patients.2424 Atzmony L, Hodak E, Leshem YA, Rosenbaum O, Gdalevich M, Anhalt GJ, et al. The role of adjuvant therapy in pemphigus: A systematic review and meta-analysis. J Am Acad Dermatol. 2015;73:264-71.
Azathioprine (AZA)
Azathioprine is a cytotoxic drug that is used in most autoimmune diseases. It is an imidazole derivative of mercaptopurine, which antagonizes purine metabolism and inhibits the synthesis of DNA, RNA, and proteins. It can also interfere with cellular metabolism and impede mitosis. AZA affects several aspects of the immune system. It reversibly reduces the number of monocytes and Langerhans cells and inhibits gamma globulin synthesis, T lymphocyte function, T helper-dependent B cell responses, and B cell suppressor function. 2525 Maltzman JS, Koretzky GA. Azathioprine: old drug, new actions. J Clin Invest. 2003;111:1122-4. The efficacy of AZA as a corticosteroid-sparing agent in autoimmune bullous diseases, particularly in PV, is well documented and is the oldest and most prescribed immunosuppressive medication in this context. 1919 Harman KE, Brown D, Exton LS, Groves RW, Hampton PJ, Mohd Mustapa MF, et al. British Association of Dermatologists' guidelines for the management of pemphigus vulgaris 2017. Br J Dermatol. 2017;177:1170-201,2626 Chams-Davatchi C, Mortazavizadeh A, Daneshpazhooh M, Davatchi F, Balighi K, Esmaili N, et al. Randomized double blind trial of prednisolone and azathioprine, vs. prednisolone and placebo, in the treatment of pemphigus vulgaris. J Eur Acad Dermatol Venereol. 2013;27:1285-92.,2727 Chaidemenos G, Apalla Z, Koussidou T, Papagarifallou I, Ioannides D. High dose oral prednisone vs. prednisone plus azathioprine for the treatment of oral pemphigus: a retrospective, bi-centre, comparative study. J Eur Acad Dermatol Venereol. 2011;25:206-10.
The recommended dosage of AZA in PV is 100 to 200mg/ day (1 to 3mg/kg/day), orally, divided into 2 doses. Its therapeutic effect begins after 4 to 6 weeks, which restricts its use as monotherapy. Three months of use should elapse before replacing it with another adjuvant when there is no satisfactory clinical response.1818 Ruocco E, Wolf R, Ruocco V, Brunetti G, Romano F, Lo Schiavo A. Pemphigus: Associations and management guidelines: Facts and controversies. Clin Dermatol. 2013;31:382-90.,2828 Committee for Guidelines for the Management of Pemphigus Disease, Amagai M, Tanikawa A, Shimizu T, Hashimoto T, Ikeda S, et al. Japanese guidelines for the management of pemphigus. J Dermatol. 2014;41:471-86.
Its main side effects are leukopenia, thrombocytopenia, anemia, pancytopenia, and hepatotoxicity. Long-term immunosuppression can increase the risk of infections and neoplasms. Individuals with a genetic deficiency in thiopurine methyltransferase (TPMT) present with greater sensitivity to AZA-induced myelotoxicity. This medication is contraindicated in pregnant women and nursing mothers.1717 Tavakolpour S. Current and future treatment options for pemphigus: Is it time to move towards more effective treatments? Int Immunopharmacol. 2017;53:133-42.
Mycophenolate mofetil (MMF)
After oral administration, MMF is absorbed and converted into its active metabolite, mycophenolic acid. This, in turn, selectively inhibits inosine monophosphate dehydrogenase, impeding purine synthesis in B and T cells and thus slowing their proliferation.2929 Olejarz W, Bryk D, Zapolska-Downar D. Mycophenolate mofetil-a new atheropreventive drug? Acta Pol Pharm. 2014;71:353-61.
MMF has been used as an adjuvant to corticosteroids in patients with PV as first-line treatment and in non-responders to AZA. Several groups prefer MMF to AZA as the first-line adjuvant therapy in PV, due to its lower hepatotoxicity and comparable efficacy. Compared with AZA, MMF is a poorer corticosteroid-sparing agent but is more effective in controlling PV.1818 Ruocco E, Wolf R, Ruocco V, Brunetti G, Romano F, Lo Schiavo A. Pemphigus: Associations and management guidelines: Facts and controversies. Clin Dermatol. 2013;31:382-90.,3030 Cholera M, Chainani-Wu N. Management of Pemphigus Vulgaris. Adv Ther. 2016;33:910-58.
31 Vyas N, Patel NS, Cohen GF. Mycophenolate mofetil as a first-line steroid sparing agent in the treatment of pemphigus vulgaris. J Drugs Dermatol. 2013;12:210-6.-3232 Martin LK, Werth VP, Villaneuva EV, Murrell DF. A systematic review of randomized controlled trials for pemphigus vulgaris and pemphigus foliaceus. J Am Acad Dermatol. 2011;64:903-8.
The recommended dosage of MMF in PV is 2-3g/day, divided into 2 doses. Its main side effects are altered bowel habits, neutropenia, lymphopenia, and myalgia. Therapeutic failure should be considered only after 3 months of use at a dosage of 3g/day.3030 Cholera M, Chainani-Wu N. Management of Pemphigus Vulgaris. Adv Ther. 2016;33:910-58.,3333 Beissert S, Mimouni D, Kanwar AJ, Solomons N, Kalia V, Anhalt GJ. Treating pemphigus vulgaris with prednisone and mycophenolate mofetil: a multicenter, randomized, placebo controlled trial. J Invest Dermatol. 2010;130:2041-8.
Rituximab
Chimeric anti-CD20 monoclonal antibody (which depletes normal and pathogenic B lymphocytes) has been used for cases of severe and refractory PV since 2006.3434 Ahmed AR, Spigelman Z, Cavacini LA, Posner MR. Treatment of pemphigus vulgaris with rituximab and intravenous immuneglobulin. N Engl J Med. 2006;355:1772-9. Following the administration of rituximab, there is a rapid and sustained depletion of circulating and tissue B lymphocytes that persists for at least 6 to 12 months. Recent evidence demonstrates that it also affects T lymphocytes.3535 Eming R, Nagel A, Wolff-Franke S, Podstawa E, Debus D, Hertl M. Rituximab exerts a dual effect in pemphigus vulgaris. J Invest Dermatol. 2008;128:2850-8. In June 2018, the US FDA approved rituximab for PV.
There are many prospective and retrospective studies that have proven its efficacy, leading to complete and sustained remission in most patients in 3 to 4 months.3434 Ahmed AR, Spigelman Z, Cavacini LA, Posner MR. Treatment of pemphigus vulgaris with rituximab and intravenous immuneglobulin. N Engl J Med. 2006;355:1772-9.,3636 Huang A, Madan RK, Levitt J. Future therapies for pemphigus vulgaris:Rituximab and beyond. J Am Acad Dermatol. 2016;74:746-53.
37 Wang HH, Liu CW, Li YC, Huang YC. Efficacy of rituximab for pemphigus: a systematic review and meta-analysis of different regimens. Acta Derm Venereol. 2015;95:928-32.
38 Joly P, Maho-Vaillant M, Prost-Squarcioni C, Hebert V, Houivet E, Calbo S, et al., First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): a prospective, multicentre, parallel-group, open-label randomised trial. Lancet. 2017;389:2031-40-3939 Ingen-Housz-Oro S, Valeyrie-Allanore L, Cosnes A, Ortonne N, Hüe S, Paul M, et al. First-line treatment of pemphigus vulgaris with a combination of rituximab and high-potency topical corticosteroids. JAMA Dermatol. 2015;151:200-3. A recent systematic review that included 114 studies and 1085 patients concluded that rituximab is an excellent treatment for refractory cases.4040 Tavakolpour S, Mahmoudi H, Balighi K, Abedini R, Daneshpazhooh M. Sixteen-year history of rituximab therapy for 1085 pemphigus vulgaris patients: A systematic review. Int Immunopharmacol. 2018;54:131-8
Rituximab should be administered IV as a slow infusion (4 to 6 hours). There are no standardized protocols for the use of rituximab in autoimmune bullous diseases, but studies have been published using the lymphoma protocol (375mg/m2, 1x/week for 4 weeks) and that for rheumatoid arthritis (1000mg with an interval of 2 weeks; can be repeated after 6 months).3636 Huang A, Madan RK, Levitt J. Future therapies for pemphigus vulgaris:Rituximab and beyond. J Am Acad Dermatol. 2016;74:746-53.,4141 Cianchini G, Lupi F, Masini C, Corona R, Puddu P, De Pità O. Therapy with rituximab for autoimmune pemphigus: results from a single-center observational study on 42 cases with long-term follow-up. J Am Acad Dermatol. 2012;67:617-22.
42 Heelan K, Al-Mohammedi F, Smith MJ, Knowles S, Lansang P, Walsh S, et al. Durable remission of pemphigus with a fixed-dose rituximab protocol. JAMA Dermatol. 2014;150:703-8.-4343 Kanwar AJ, Vinay K, Sawatkar GU, Dogra S, Minz RW, Shear NH, et al. Clinical and immunological outcomes of high and low dose rituximab treatments in pemphigus patients:a randomized comparative observer blinded study. Br J Dermatol. 2014;170:1341-9. There seems to be no difference in percentage in remission or disease-free interval between these protocols. Rituximab can be used alone or in combination with IVIG, plasmapheresis, or immunoadsorption (the latter appears to prolong the response time with respect to rituximab alone). It can also be administered to patients who are already taking prednisone and immunosuppressants, and the dose reduction and suspension of the latter should be accelerated due to the increased risk of infection.4040 Tavakolpour S, Mahmoudi H, Balighi K, Abedini R, Daneshpazhooh M. Sixteen-year history of rituximab therapy for 1085 pemphigus vulgaris patients: A systematic review. Int Immunopharmacol. 2018;54:131-8,4444 Amber KT, Hertl M. An assessment of treatment history and its association with clinical outcomes and relapse in 155 pemphigus patients with response to a single cycle of rituximab. J Eur Acad Dermatol Venereol. 2015;29:777-82.
45 Joly P, Mouquet H, Roujeau JC, D'Incan M, Gilbert D, Jacquot S, et al. A single cycle of rituximab for the treatment of severe pemphigus. N Engl J Med. 2007;357:545-52.
46 Londhe PJ, Kalyanpad Y, Khopkar US. Intermediate doses of rituximab used as adjuvant therapy in refractory pemphigus. Indian J Dermatol Venereol Leprol. 2014;80:300-5.
47 Ahmed AR, Nguyen T, Kaveri S, Spigelman ZS. First line treatment of pemphigus vulgaris with a novel protocol in patients with contraindications to systemic corticosteroids and immunosuppressive agents: preliminary retrospective study with a seven year follow-up. Int Immunopharmacol. 2016;34:25-31.-4848 Behzad M, Möbs C, Kneisel A, Möller M, Hoyer J, Hertl M, et al. Combined treatment with immunoadsorption and rituximab leads to fast and prolonged clinical remission in difficult-to-treat pemphigus vulgaris. Br J Dermatol. 2012;166:844-52.,3434 Ahmed AR, Spigelman Z, Cavacini LA, Posner MR. Treatment of pemphigus vulgaris with rituximab and intravenous immuneglobulin. N Engl J Med. 2006;355:1772-9.
Rituximab is generally well tolerated, and serious adverse effects are rare. Infusion reactions (which can be reduced with prior administration of analgesics, antihistamines, and corticosteroids) include anaphylaxis, fever, hypotension, chills, headache, nausea, pruritus, and skin rash. In addition, neutropenia, hypogammaglobulinemia, and infections, including sepsis, are rarely reported. Certain authors and expert groups recomend rituximab as a first-line treatment option for PV.1818 Ruocco E, Wolf R, Ruocco V, Brunetti G, Romano F, Lo Schiavo A. Pemphigus: Associations and management guidelines: Facts and controversies. Clin Dermatol. 2013;31:382-90.,3636 Huang A, Madan RK, Levitt J. Future therapies for pemphigus vulgaris:Rituximab and beyond. J Am Acad Dermatol. 2016;74:746-53.,3838 Joly P, Maho-Vaillant M, Prost-Squarcioni C, Hebert V, Houivet E, Calbo S, et al., First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): a prospective, multicentre, parallel-group, open-label randomised trial. Lancet. 2017;389:2031-40
39 Ingen-Housz-Oro S, Valeyrie-Allanore L, Cosnes A, Ortonne N, Hüe S, Paul M, et al. First-line treatment of pemphigus vulgaris with a combination of rituximab and high-potency topical corticosteroids. JAMA Dermatol. 2015;151:200-3.-4040 Tavakolpour S, Mahmoudi H, Balighi K, Abedini R, Daneshpazhooh M. Sixteen-year history of rituximab therapy for 1085 pemphigus vulgaris patients: A systematic review. Int Immunopharmacol. 2018;54:131-8,4949 Graves JE, Nunley K, Heffernan MP. Off-label uses of biologics in dermatology: rituximab, omalizumab, infliximab, etanercept, adalimumab, efalizumab, and alefacept (part 2 of 2). J Am Acad Dermatol. 2007;56:e55-79.
50 Lunardon L, Tsai KJ, Propert KJ, Fett N, Stanley JR, Werth VP, et al. Adjuvant rituximab therapy of pemphigus: a single-center experience with 31 patients. Arch Dermatol. 2012;148:1031-6.
51 Murrell DF, Peña S, Joly P, et al.Diagnosis and Management of Pemphigus: recommendations by an International Panel of Experts. J Am Acad Dermatol. 2018. pii: S0190-9622(18)30207-X [Epub ahead of print]-5252 Leshem YA, Hodak E, David M, Anhalt GJ, Mimouni D. Successful treatment of pemphigus with biweekly 1-g infusions of rituximab: a retrospective study of 47 patients. J Am Acad Dermatol. 2013;68:404-11.
Cyclophosphamide
Cyclophosphamide is an alkylating agent that selectively affects B lymphocytes and antibody production. It can be administered orally in PV (1 to 3mg/kg/day) or intravenously, with or without dexamethasone IV, in the form of pulse therapy.1616 Hertl M, Jedlickova H, Karpati S, Marinovic B, Uzun S, Yayli S, et al. Pemphigus. S2 guideline for diagnosis and treatment-guided by the European Dermatology Forum (EDF) in cooperation with the European Academy of Dermatology and Venereology (EADV). J Eur Acad Dermatol Venereol. 2015;29:405-14. In such cases, dexamethasone is administered at 100mg/day IV for 3 days, with cyclophosphamide 500mg/day IV being administered on the first day. This pulse therapy is repeated every 2 to 4 weeks, between which an oral dose of cyclophosphamide 50mg/day and prednisone 1mg/kg/day is maintained. Treatment failure should be considered after 3 months of use at 2mg/kg/day. 1919 Harman KE, Brown D, Exton LS, Groves RW, Hampton PJ, Mohd Mustapa MF, et al. British Association of Dermatologists' guidelines for the management of pemphigus vulgaris 2017. Br J Dermatol. 2017;177:1170-201,3636 Huang A, Madan RK, Levitt J. Future therapies for pemphigus vulgaris:Rituximab and beyond. J Am Acad Dermatol. 2016;74:746-53.,5353 Shaik F, Botha J, Aboobaker J, Mosam A. Corticosteroid /cyclophosphamide pulse treatment in south African patients with pemphigus. Clin Exp Dermatol. 2010;35:245-50.
Its main toxic effects are infertility, predisposition to neoplasia, lymphopenia, and sepsis. Due to its greater toxicity, it can be considered as an adjuvant only in cases that are refractory to AZA and MMF. 1919 Harman KE, Brown D, Exton LS, Groves RW, Hampton PJ, Mohd Mustapa MF, et al. British Association of Dermatologists' guidelines for the management of pemphigus vulgaris 2017. Br J Dermatol. 2017;177:1170-201,2626 Chams-Davatchi C, Mortazavizadeh A, Daneshpazhooh M, Davatchi F, Balighi K, Esmaili N, et al. Randomized double blind trial of prednisolone and azathioprine, vs. prednisolone and placebo, in the treatment of pemphigus vulgaris. J Eur Acad Dermatol Venereol. 2013;27:1285-92.,545 Pan M, Liu X, Zheng J. The pathogenic role of autoantibodies in pemphigus vulgaris. Clin Exp Dermatol. 2011;36:703-7.
6 Joly P, Litrowski N. Pemphigus group (vulgaris, vegetans, foliaceus, herpetiformis, brasiliensis). Clin Dermatol. 2011;29(4):432-6.
7 Amber KT, Staropoli P, Shiman MI, Elgart GW, Hertl M. Autoreactive T cells in the immune pathogenesis of pemphigus vulgaris. Exp Dermatol. 2013; 22:699-704.
8 Kridin K. Pemphigus group: overview, epidemiology, mortality, and comorbidities. Immunol Res. 2018;66:255-70.
9 Bystryn JC, Rudolph JL. Pemphigus. Lancet. 2005;366:61-73.
10 Pollmann R, Schmidt T, Eming R, Hertl M. Pemphigus: a Comprehensive Review on Pathogenesis, Clinical Presentation and Novel Therapeutic Approaches. Clin Rev Allergy Immunol. 2018;54:1-25
11 Hammers CM, Stanley JR. Mechanisms of disease: pemphigus and bullous pemphigoid. Annu Rev Pathol. 2016 May 23;11:175-97.
12 Broussard KC, Leung TG, Moradi A, Thorne JE, Fine JD. Autoimmune bullous diseases with skin and eye involvement: Cicatricial pemphigoid, pemphigus vulgaris, and pemphigus paraneoplastica. Clin Dermatol. 2016;34:205-13.
13 Becker BA, Gaspari AA. Pemphigus vulgaris and vegetans. Dermatol Clin. 1993;11:429-52.
14 Meurer M. Immunosuppressive therapy for autoimmune bullous diseases. Clin Dermatol. 2012;30:78-83.
15 Bystryn JC, Steinman NM. The adjuvant therapy of pemphigus. An update. Arch Dermatol. 1996;132:203-12.
16 Hertl M, Jedlickova H, Karpati S, Marinovic B, Uzun S, Yayli S, et al. Pemphigus. S2 guideline for diagnosis and treatment-guided by the European Dermatology Forum (EDF) in cooperation with the European Academy of Dermatology and Venereology (EADV). J Eur Acad Dermatol Venereol. 2015;29:405-14.
17 Tavakolpour S. Current and future treatment options for pemphigus: Is it time to move towards more effective treatments? Int Immunopharmacol. 2017;53:133-42.
18 Ruocco E, Wolf R, Ruocco V, Brunetti G, Romano F, Lo Schiavo A. Pemphigus: Associations and management guidelines: Facts and controversies. Clin Dermatol. 2013;31:382-90.
19 Harman KE, Brown D, Exton LS, Groves RW, Hampton PJ, Mohd Mustapa MF, et al. British Association of Dermatologists' guidelines for the management of pemphigus vulgaris 2017. Br J Dermatol. 2017;177:1170-201
20 Rao PN, Lakshmi TS. Pulse therapy and its modifications in pemphigus: a six year study. Indian J Dermatol Venereol Leprol. 2003;69:329-33.
21 Chams-Davatchi C, Esmaili N, Daneshpazhooh M, Valikhani M, Balighi K, Hallaji Z, et al. Randomized controlled open-label trial of four treatment regimens for pemphigus vulgaris. J Am Acad Dermatol. 2007;57:622-8.
22 Ahmed AR, Moy R. Death in pemphigus. J Am Acad Dermatol. 1982;7:221-8.
23 Frew JW, Martin LK, Murrell DF. Evidence-based treatments in pemphigus vulgaris and pemphigus foliaceus. Dermatol Clin. 2011;29:599-606.
24 Atzmony L, Hodak E, Leshem YA, Rosenbaum O, Gdalevich M, Anhalt GJ, et al. The role of adjuvant therapy in pemphigus: A systematic review and meta-analysis. J Am Acad Dermatol. 2015;73:264-71.
25 Maltzman JS, Koretzky GA. Azathioprine: old drug, new actions. J Clin Invest. 2003;111:1122-4.
26 Chams-Davatchi C, Mortazavizadeh A, Daneshpazhooh M, Davatchi F, Balighi K, Esmaili N, et al. Randomized double blind trial of prednisolone and azathioprine, vs. prednisolone and placebo, in the treatment of pemphigus vulgaris. J Eur Acad Dermatol Venereol. 2013;27:1285-92.
27 Chaidemenos G, Apalla Z, Koussidou T, Papagarifallou I, Ioannides D. High dose oral prednisone vs. prednisone plus azathioprine for the treatment of oral pemphigus: a retrospective, bi-centre, comparative study. J Eur Acad Dermatol Venereol. 2011;25:206-10.
28 Committee for Guidelines for the Management of Pemphigus Disease, Amagai M, Tanikawa A, Shimizu T, Hashimoto T, Ikeda S, et al. Japanese guidelines for the management of pemphigus. J Dermatol. 2014;41:471-86.
29 Olejarz W, Bryk D, Zapolska-Downar D. Mycophenolate mofetil-a new atheropreventive drug? Acta Pol Pharm. 2014;71:353-61.
30 Cholera M, Chainani-Wu N. Management of Pemphigus Vulgaris. Adv Ther. 2016;33:910-58.
31 Vyas N, Patel NS, Cohen GF. Mycophenolate mofetil as a first-line steroid sparing agent in the treatment of pemphigus vulgaris. J Drugs Dermatol. 2013;12:210-6.
32 Martin LK, Werth VP, Villaneuva EV, Murrell DF. A systematic review of randomized controlled trials for pemphigus vulgaris and pemphigus foliaceus. J Am Acad Dermatol. 2011;64:903-8.
33 Beissert S, Mimouni D, Kanwar AJ, Solomons N, Kalia V, Anhalt GJ. Treating pemphigus vulgaris with prednisone and mycophenolate mofetil: a multicenter, randomized, placebo controlled trial. J Invest Dermatol. 2010;130:2041-8.
34 Ahmed AR, Spigelman Z, Cavacini LA, Posner MR. Treatment of pemphigus vulgaris with rituximab and intravenous immuneglobulin. N Engl J Med. 2006;355:1772-9.
35 Eming R, Nagel A, Wolff-Franke S, Podstawa E, Debus D, Hertl M. Rituximab exerts a dual effect in pemphigus vulgaris. J Invest Dermatol. 2008;128:2850-8.
36 Huang A, Madan RK, Levitt J. Future therapies for pemphigus vulgaris:Rituximab and beyond. J Am Acad Dermatol. 2016;74:746-53.
37 Wang HH, Liu CW, Li YC, Huang YC. Efficacy of rituximab for pemphigus: a systematic review and meta-analysis of different regimens. Acta Derm Venereol. 2015;95:928-32.
38 Joly P, Maho-Vaillant M, Prost-Squarcioni C, Hebert V, Houivet E, Calbo S, et al., First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): a prospective, multicentre, parallel-group, open-label randomised trial. Lancet. 2017;389:2031-40
39 Ingen-Housz-Oro S, Valeyrie-Allanore L, Cosnes A, Ortonne N, Hüe S, Paul M, et al. First-line treatment of pemphigus vulgaris with a combination of rituximab and high-potency topical corticosteroids. JAMA Dermatol. 2015;151:200-3.
40 Tavakolpour S, Mahmoudi H, Balighi K, Abedini R, Daneshpazhooh M. Sixteen-year history of rituximab therapy for 1085 pemphigus vulgaris patients: A systematic review. Int Immunopharmacol. 2018;54:131-8
41 Cianchini G, Lupi F, Masini C, Corona R, Puddu P, De Pità O. Therapy with rituximab for autoimmune pemphigus: results from a single-center observational study on 42 cases with long-term follow-up. J Am Acad Dermatol. 2012;67:617-22.
42 Heelan K, Al-Mohammedi F, Smith MJ, Knowles S, Lansang P, Walsh S, et al. Durable remission of pemphigus with a fixed-dose rituximab protocol. JAMA Dermatol. 2014;150:703-8.
43 Kanwar AJ, Vinay K, Sawatkar GU, Dogra S, Minz RW, Shear NH, et al. Clinical and immunological outcomes of high and low dose rituximab treatments in pemphigus patients:a randomized comparative observer blinded study. Br J Dermatol. 2014;170:1341-9.
44 Amber KT, Hertl M. An assessment of treatment history and its association with clinical outcomes and relapse in 155 pemphigus patients with response to a single cycle of rituximab. J Eur Acad Dermatol Venereol. 2015;29:777-82.
45 Joly P, Mouquet H, Roujeau JC, D'Incan M, Gilbert D, Jacquot S, et al. A single cycle of rituximab for the treatment of severe pemphigus. N Engl J Med. 2007;357:545-52.
46 Londhe PJ, Kalyanpad Y, Khopkar US. Intermediate doses of rituximab used as adjuvant therapy in refractory pemphigus. Indian J Dermatol Venereol Leprol. 2014;80:300-5.
47 Ahmed AR, Nguyen T, Kaveri S, Spigelman ZS. First line treatment of pemphigus vulgaris with a novel protocol in patients with contraindications to systemic corticosteroids and immunosuppressive agents: preliminary retrospective study with a seven year follow-up. Int Immunopharmacol. 2016;34:25-31.
48 Behzad M, Möbs C, Kneisel A, Möller M, Hoyer J, Hertl M, et al. Combined treatment with immunoadsorption and rituximab leads to fast and prolonged clinical remission in difficult-to-treat pemphigus vulgaris. Br J Dermatol. 2012;166:844-52.
49 Graves JE, Nunley K, Heffernan MP. Off-label uses of biologics in dermatology: rituximab, omalizumab, infliximab, etanercept, adalimumab, efalizumab, and alefacept (part 2 of 2). J Am Acad Dermatol. 2007;56:e55-79.
50 Lunardon L, Tsai KJ, Propert KJ, Fett N, Stanley JR, Werth VP, et al. Adjuvant rituximab therapy of pemphigus: a single-center experience with 31 patients. Arch Dermatol. 2012;148:1031-6.
51 Murrell DF, Peña S, Joly P, et al.Diagnosis and Management of Pemphigus: recommendations by an International Panel of Experts. J Am Acad Dermatol. 2018. pii: S0190-9622(18)30207-X [Epub ahead of print]
52 Leshem YA, Hodak E, David M, Anhalt GJ, Mimouni D. Successful treatment of pemphigus with biweekly 1-g infusions of rituximab: a retrospective study of 47 patients. J Am Acad Dermatol. 2013;68:404-11.
53 Shaik F, Botha J, Aboobaker J, Mosam A. Corticosteroid /cyclophosphamide pulse treatment in south African patients with pemphigus. Clin Exp Dermatol. 2010;35:245-50.
54 Zhao CY, Murrell DF. Pemphigus Vulgaris: An Evidence-Based Treatment Update. Drugs. 2015;75:271-84.
55 Sharma VK, Khandpur S. Evaluation of cyclophosphamide pulse therapy as an adjuvant to oral corticosteroid in the management of pemphigus vulgaris. Clin Exp Dermatol. 2013;38:659-64.
56 Rose E, Wever S, Zilliken D, Linse R, Haustein UF, Bröcker EB, et al. Intravenous dexamethasone-cyclophosphamide pulse therapy in comparison with oral methylprednisolone-azathioprine therapy in patients with pemphigus: results of a multicenter prospectively randomized study. J Dtsch Dermatol Ges. 2005;3:200-6.
57 Sethy PK, Khandpur S, Sharma VK. Randomized open comparative trial of dexamethasone-cyclophosphamide pulse and daily oral cyclophosphamide versus cyclophosphamide pulse and daily oral prednisolone in pemphigus vulgaris. Indian J Dermatol Venereol Leprol. 2009;75:476-82.-5858 Parmar NV, Kanwar AJ, Minz RW, Parsad D, Vinay K, Tsuruta D, et al. Assessment of the therapeutic benefit of dexamethasone cyclophosphamide pulse versus only oral cyclophosphamide in phase II of the dexamethasone cyclophosphamide pulse therapy: a preliminary prospective randomized controlled study. Indian J Dermatol Venereol Leprol. 2013;79:70-6.
Methotrexate
Based on its anti-inflammatory activity and inhibition of cell proliferation through the suppression of dihydrofolate reductase, methotrexate can be added as an adjuvant in PV at 10 to 20mg/ week in cases of therapeutic failure to other adjuvants. The most frequent side effects are gastrointestinal intolerance, hematological toxicity, and infection.5959 Gürcan HM, Ahmed AR. Analysis of current data on the use of methotrexate in the treatment of pemphigus and pemphigoid. Br J Dermatol. 2009;161:723-31.
60 Baum S, Greenberger S, Samuelov L, Solomon M, Lyakhovitsky A, Trau H, et al. Methotrexate is an effective and safe adjuvant therapy for pemphigus vulgaris. Eur J Dermatol. 2012;22:83-7.-6161 Tran KD, Wolverton JE, Soter NA. Methotrexate in the treatment of pemphigus vulgaris: experience in 23 patients. Br J Dermatol. 2013;169:916-21.
Dapsone
Drug with anti-inflammatory and anti-TNF activity that can be attempted as adjuvant medication in PV at 50 to 200mg/day orally, but there are conflicting reports in the literature. Its side effects are usually dose-dependent and reversible. 1919 Harman KE, Brown D, Exton LS, Groves RW, Hampton PJ, Mohd Mustapa MF, et al. British Association of Dermatologists' guidelines for the management of pemphigus vulgaris 2017. Br J Dermatol. 2017;177:1170-201,6262 Gürcan HM, Ahmed AR. Efficacy of dapsone in the treatment of pemphigus and pemphigoid: analysis of current data. Am J Clin Dermatol. 2009;10:383-96.,6363 Baum S, Debby A, Gilboa S, Trau H, Barzilai A. Efficacy of Dapsone in the treatment of pemphigus vulgaris: a single-center case study. Dermatology. 2016;232:578-85.
Cyclosporine
Cyclosporine is a calcineurin inhibitor with potent immunosuppressive activity against B and T lymphocytes. It is effective as an adjuvant in the treatment of PV in rare cases at dosages of 3 to 5mg/kg/day, po or IV. 6464 Olszewska M, Kolacinska-Strasz Z, Sulej J, Labecka H, Cwikla J, Natorska U, et al. Efficacy and safety of cyclophosphamide, azathioprine, and cyclosporine (ciclosporin) as adjuvant drugs in pemphigus vulgaris. Am J Clin Dermatol. 2007;8:85-92.
Intravenous immunoglobulin (IVIG)
Derived from a donor pool, the mode of action of IVIG in PV is complex, with several mechanisms acting synergistically (selectively removing pathogenic antibodies; altering the expression and function of Fc receptors; affecting the activation, differentiation, and effector functions of T and B cells; and interfering with the activation of cytokines and complement). Its advantage is its safety profile, with few side effects (headache, dyspnea, tachycardia, abdominal discomfort). IVIG is used in cases of PV that do not respond to other treatments or those that present with severe side effects, and it is effective in certain cases at a dosage of 0.4g/kg/day for 5 days, always as an adjunct to corticosteroid therapy once per month. It is expensive and takes 3 to 6 cycles on average. It can be used in pregnant women.3232 Martin LK, Werth VP, Villaneuva EV, Murrell DF. A systematic review of randomized controlled trials for pemphigus vulgaris and pemphigus foliaceus. J Am Acad Dermatol. 2011;64:903-8.,6565 Chaigne B, Mouthon L. Mechanisms of action of intravenous immunoglobulin. Transfus Apher Sci. 2017;56:45-9.
66 Svecova D. IVIG therapy in pemphigus vulgaris has corticosteroid-sparing and immunomodulatory effects. Australas J Dermatol. 2016;57:141-4.-6767 Amagai M, Ikeda S, Shimizu H, Iizuka H, Hanada K, Aiba S, et al. A randomized double-blind trial of intravenous immunoglobulin for pemphigus. J Am Acad Dermatol. 2009;60:595-603.
Anti-TNF drugs
TNF-α is one of the cytokines that are involved in acantholysis. Case reports with the use of infliximab and etanercept have suggested its efficacy in PV, but other studies contradict these findings.1717 Tavakolpour S. Current and future treatment options for pemphigus: Is it time to move towards more effective treatments? Int Immunopharmacol. 2017;53:133-42.,6868 Sinha AA, Hoffman MB, Janicke EC. Pemphigus vulgaris: approach to treatment. Eur J Dermatol. 2015;25:103-13.
Plasmapheresis/Immunoadsorption
Plasmapheresis was first used in 1978 for PV to remove pathogenic autoantibodies from circulation. However, it triggered a rebound effect, causing greater production of these autoantibodies after their withdrawal from circulation. For this reason, it is recommended that corticosteroids and immunosuppressants (e.g., pulsotherapy with methylprednisolone and cyclophosphamide) be used in monthly cycles for up to 1 year.1818 Ruocco E, Wolf R, Ruocco V, Brunetti G, Romano F, Lo Schiavo A. Pemphigus: Associations and management guidelines: Facts and controversies. Clin Dermatol. 2013;31:382-90.,6868 Sinha AA, Hoffman MB, Janicke EC. Pemphigus vulgaris: approach to treatment. Eur J Dermatol. 2015;25:103-13. IVIG can be used in place of cyclophosphamide to prevent the rebound effect in autoantibody production. Plasmapheresis is an exceptional alternative for severe cases of PV that are unresponsive to other therapeutic modalities.17 It is available in few hospitals and is expensive. Its main side effect is septicemia.
Immunoadsorption, introduced in 1984, is a more selective method that does not remove other antibodies or plasma components from circulation, unlike plasmapheresis. Performed in cycles of 4 consecutive days every 4 weeks, it has fewer side effects than plasmapheresis. 6969 Eming R, Rech J, Barth S, Kalden JR, Schuler G, Harrer T, et al. Prolonged clinical remission of patients with severe pemphigus upon rapid removal of desmoglein- reactive autoantibodies by immunoadsorption. Dermatology. 2006;212:177-87.,7070 Meyersburg D, Schmidt E, Kasperkiewicz M, Zillikens D. Immunoadsorption in dermatology. Ther Apher Dial. 2012;16:311-20.
Topical treatment
Always used as an adjuvant to systemic therapies, topical treatment of PV lesions aims to reduce pain and prevent secondary infections. It is usually applied as corticosteroid creams and antibiotics. There are reports on the use of tacrolimus, particularly in facial lesions.7171 Gach JE, Ilchyshyn A. Beneficial effects of topical tacrolimus on recalcitrant erosions of pemphigus vulgaris. Clin Exp Dermatol. 2004;29:271-2. For extensive cases, antiseptic solutions, such as potassium permanganate (1:10,000 or 1:20,000) and chlorhexidine, can be used. More potent corticoid gels (clobetasol dipropionate) can be applied to the oral mucosa. Triamcinolone acetonide (10 mg/ml) can be administered as an intralesional injection for refractory skin lesions (e.g., pemphigus vegetans).1717 Tavakolpour S. Current and future treatment options for pemphigus: Is it time to move towards more effective treatments? Int Immunopharmacol. 2017;53:133-42.,1818 Ruocco E, Wolf R, Ruocco V, Brunetti G, Romano F, Lo Schiavo A. Pemphigus: Associations and management guidelines: Facts and controversies. Clin Dermatol. 2013;31:382-90.
Future therapies
New anti-B cell immunobiologicals are being examined in clinical research regarding their efficacy, safety, and cost in patients with PV, including veltuzumab (anti-CD 20 antibody, SC administration), obinutuzumab, ofatumumab, ocaratuzumab, PRO 121921, anti-BAFF, and anti-BAFF-R. 3636 Huang A, Madan RK, Levitt J. Future therapies for pemphigus vulgaris:Rituximab and beyond. J Am Acad Dermatol. 2016;74:746-53.
Systemic antibiotic therapy
Systemic antibiotic therapy is indicated only in cases with clinical or laboratory evidence of a secondary infection-never prophylactically. Preferably, its choice should be guided by a blood or skin fragment culture and an antibiogram.
Treatment plan
PV treatment should comprise 2 phases: the induction of remission and the maintenance of remission. 1919 Harman KE, Brown D, Exton LS, Groves RW, Hampton PJ, Mohd Mustapa MF, et al. British Association of Dermatologists' guidelines for the management of pemphigus vulgaris 2017. Br J Dermatol. 2017;177:1170-201,7272 Murrell DF, Dick S, Ahmed AR, Amagai M, Barnadas MA, Borradori L, et al. Consensus statement on definitions of disease, end points, and therapeutic response for pemphigus. J Am Acad Dermatol. 2008;58:1043-6.
73 Czernik A, Bystryn JC. Kinetics of response to conventional treatment in patients with pemphigus vulgaris. Arch Dermatol. 2008;144:682-3.
74 Martin LK, Werth V, Villanueva E, Segall J, Murrell DF. Interventions for pemphigus vulgaris and pemphigus foliaceus. Cochrane Database Syst Rev. 2009;CD006263.
75 Herbst A, Bystryn JC. Patterns of remission in pemphigus vulgaris. J Am Acad Dermatol. 2000;42:422-7.-7676 Almugairen N, Hospital V, Bedane C, Duvert-Lehembre S, Picard D, Tronquoy AF, et al. Assessment of the rate of long-term complete remission off therapy in patients with pemphigus treated with different regimens including medium- and high-dose corticosteroids. J Am Acad Dermatol. 2013;69:583-8.
Induction of remission
The objective is to control the disease, interrupting the emergence of new bullous lesions and the re-epithelialization of lesions that are already present. Corticosteroids are the most effective and fastest-acting therapeutic option for controlling PV, rendering them important at this stage. Disease control may take several weeks (on average 3 weeks), and dose escalation might be required for it to occur.
Adjuvant medications can be initiated at this stage, but their benefit is limited, because their onset of activity is slow. For this reason, its isolated use for the initial control of PV is not recommended.
Medication doses should be maintained until the condition is controlled, defined as re-epithelization of approximately 80% of skin and mucosal lesions and no emergence of new lesions for at least 2 weeks. Oral mucosal lesions usually resolve more slowly than skin lesions. At this stage, the corticosteroid dose can be reduced slowly.
Maintenance of remission
The dose of the medication should be decreased gradually to minimize side effects. The ultimate goal is to keep the disease controlled with a dose of prednisone of up to 10mg/day. PV is a chronic disease, and in one study, 36% of patients received treatment for over 10 years.
At this stage, the role of adjuvant medications becomes more significant, although there are no prospective controlled studies that have clearly demonstrated the benefits of these drugs. For this reason, many groups do not use them routinely in PV, unless there are contraindications or notable side effects with corticosteroids or if the disease recurs on dose reduction. One exception is rituximab, for which, in 2017, the first randomized controlled trial to demonstrate the superiority of its combination with prednisolone over prednisolone alone for controlling PV after 2 years (89% versus 28% in complete remission) was published.
Treatment suspension
Complete remission can occur, having been observed in 38%, 50%, and 75% of patients after 3, 5, and 10 years after diagnosis, respectively. Another study found that 59% of patients were without treatment after 3 years. However, premature withdrawal should be avoided, being rarely possible before 1 year.
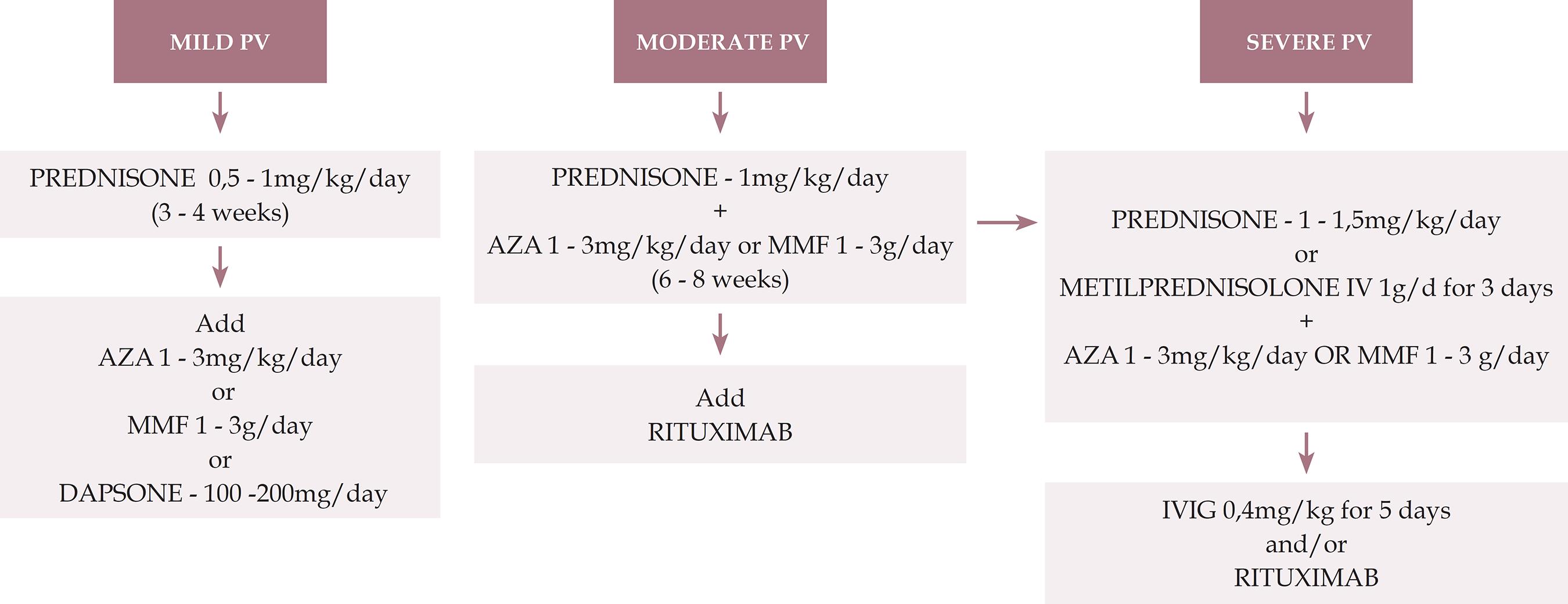
We present an algorithm for the treatment of pemphigus vulgaris (Figure 1).
Treatment algorithm for pemphigus vulgaris
PV: pemphigus vulgaris; AZA: azathioprine; MMF: mycophenolate mofetil; IVIG: intravenous immunoglobulin
PV only in mucous membrane:
Mild: only in oral mucosa / Moderate: extensive lesions in oral mucosa/ Severe: lesions in oral mucosa and others (for example, esophagus, larynx)
PV mucocutaneous:
Mild: < 1% de BSA / Moderate: 1 - 10% of BSA / Severe: > 10% of BSA:
BSA: body surface area (1% means the sum of injured areas corresponding to the palmar surface of the hand with the five digits)
* This severity rating is not definitive and aims to provide initial guidance. Each case should be evaluated individually, including considering the speed of onset and progression of the lesions, comorbidities, contraindications to the use of corticosteroids and response to the treatment applied.
** Prednisone can be replaced by oral prednisolone
EVOLUTION AND PROGNOSIS
Before the advent of corticosteroids and immunosuppressants, PV had a 2-year mortality rate of 50%. Since then, the mortality rate has dropped to approximately 10%. The main cause of death in patients with PV is septicemia. Patients often present as people with large burns, experiencing loss of the skin-mucosal barrier, which promotes infections and hydroelectrolytic and metabolic disorders. Oral lesions are usually more resistant to treatment and can persist for years, significant impairing patients’ quality of life. Generally, it is possible to obtain total disease control, which allows the medication to be suspended, but the patient should be kept under observation, because relapses are frequent. 88 Kridin K. Pemphigus group: overview, epidemiology, mortality, and comorbidities. Immunol Res. 2018;66:255-70.,1010 Pollmann R, Schmidt T, Eming R, Hertl M. Pemphigus: a Comprehensive Review on Pathogenesis, Clinical Presentation and Novel Therapeutic Approaches. Clin Rev Allergy Immunol. 2018;54:1-25,1212 Broussard KC, Leung TG, Moradi A, Thorne JE, Fine JD. Autoimmune bullous diseases with skin and eye involvement: Cicatricial pemphigoid, pemphigus vulgaris, and pemphigus paraneoplastica. Clin Dermatol. 2016;34:205-13.,2828 Committee for Guidelines for the Management of Pemphigus Disease, Amagai M, Tanikawa A, Shimizu T, Hashimoto T, Ikeda S, et al. Japanese guidelines for the management of pemphigus. J Dermatol. 2014;41:471-86.
CONCLUSIONS
PV is a rare disease. It is difficult to compare published studies among adjuvant drugs in terms of their efficacy in the control of PV, prevention of relapse, side effects, morbidity, and mortality, due to differences in study design, study populations, and the doses and combinations of the drugs. Primarily, this obstacle is attributed to the lack of randomized controlled clinical trials.
Recent systematic reviews and meta-analyses have concluded that systemic corticosteroids (prednisone or prednisolone) should form the basis of PV treatment but have not generated a consensus on the best initial corticosteroid dose or adjuvant drug. 2424 Atzmony L, Hodak E, Leshem YA, Rosenbaum O, Gdalevich M, Anhalt GJ, et al. The role of adjuvant therapy in pemphigus: A systematic review and meta-analysis. J Am Acad Dermatol. 2015;73:264-71.,3232 Martin LK, Werth VP, Villaneuva EV, Murrell DF. A systematic review of randomized controlled trials for pemphigus vulgaris and pemphigus foliaceus. J Am Acad Dermatol. 2011;64:903-8.,5454 Zhao CY, Murrell DF. Pemphigus Vulgaris: An Evidence-Based Treatment Update. Drugs. 2015;75:271-84.,7777 Atzmony L, Hodak E, Gdalevich M, Rosenbaum O, Mimouni D. Treatment of Pemphigus Vulgaris and Pemphigus Foliaceus: A Systematic Review and Meta- Analysis. Am J Clin Dermatol. 2014;15:503-15. Some studies have compared doses of prednisolone and corticosteroids IV versus placebo; AZA versus MMF; and other adjuvant therapies, such as methotrexate, cyclosporine, cyclophosphamide, and IVIG at high doses. 3232 Martin LK, Werth VP, Villaneuva EV, Murrell DF. A systematic review of randomized controlled trials for pemphigus vulgaris and pemphigus foliaceus. J Am Acad Dermatol. 2011;64:903-8.,7474 Martin LK, Werth V, Villanueva E, Segall J, Murrell DF. Interventions for pemphigus vulgaris and pemphigus foliaceus. Cochrane Database Syst Rev. 2009;CD006263. Although there is no definitive support from the literature, the combination of systemic corticosteroids (prednisolone 1-1.5mg/kg/day) and corticosteroid-sparing adjuvant drugs, mainly AZA and MMF, is considered the first-line standard therapy for PV by most groups. 1616 Hertl M, Jedlickova H, Karpati S, Marinovic B, Uzun S, Yayli S, et al. Pemphigus. S2 guideline for diagnosis and treatment-guided by the European Dermatology Forum (EDF) in cooperation with the European Academy of Dermatology and Venereology (EADV). J Eur Acad Dermatol Venereol. 2015;29:405-14. Several authors and expert groups have recommended rituximab as a first-line treatment for PV. 1818 Ruocco E, Wolf R, Ruocco V, Brunetti G, Romano F, Lo Schiavo A. Pemphigus: Associations and management guidelines: Facts and controversies. Clin Dermatol. 2013;31:382-90.,3636 Huang A, Madan RK, Levitt J. Future therapies for pemphigus vulgaris:Rituximab and beyond. J Am Acad Dermatol. 2016;74:746-53.,3838 Joly P, Maho-Vaillant M, Prost-Squarcioni C, Hebert V, Houivet E, Calbo S, et al., First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): a prospective, multicentre, parallel-group, open-label randomised trial. Lancet. 2017;389:2031-40
39 Ingen-Housz-Oro S, Valeyrie-Allanore L, Cosnes A, Ortonne N, Hüe S, Paul M, et al. First-line treatment of pemphigus vulgaris with a combination of rituximab and high-potency topical corticosteroids. JAMA Dermatol. 2015;151:200-3.-4040 Tavakolpour S, Mahmoudi H, Balighi K, Abedini R, Daneshpazhooh M. Sixteen-year history of rituximab therapy for 1085 pemphigus vulgaris patients: A systematic review. Int Immunopharmacol. 2018;54:131-8,4242 Heelan K, Al-Mohammedi F, Smith MJ, Knowles S, Lansang P, Walsh S, et al. Durable remission of pemphigus with a fixed-dose rituximab protocol. JAMA Dermatol. 2014;150:703-8.,4343 Kanwar AJ, Vinay K, Sawatkar GU, Dogra S, Minz RW, Shear NH, et al. Clinical and immunological outcomes of high and low dose rituximab treatments in pemphigus patients:a randomized comparative observer blinded study. Br J Dermatol. 2014;170:1341-9.,4949 Graves JE, Nunley K, Heffernan MP. Off-label uses of biologics in dermatology: rituximab, omalizumab, infliximab, etanercept, adalimumab, efalizumab, and alefacept (part 2 of 2). J Am Acad Dermatol. 2007;56:e55-79.
50 Lunardon L, Tsai KJ, Propert KJ, Fett N, Stanley JR, Werth VP, et al. Adjuvant rituximab therapy of pemphigus: a single-center experience with 31 patients. Arch Dermatol. 2012;148:1031-6.
51 Murrell DF, Peña S, Joly P, et al.Diagnosis and Management of Pemphigus: recommendations by an International Panel of Experts. J Am Acad Dermatol. 2018. pii: S0190-9622(18)30207-X [Epub ahead of print]-5252 Leshem YA, Hodak E, David M, Anhalt GJ, Mimouni D. Successful treatment of pemphigus with biweekly 1-g infusions of rituximab: a retrospective study of 47 patients. J Am Acad Dermatol. 2013;68:404-11.
PEMPHIGUS FOLIACEUS
INTRODUCTION
Pemphigus foliaceus (PF) is an autoimmune bullous disease in which IgG4 autoantibodies are directed against desmoglein-1 ectodomains in the desmosomal structures of the superficial layers of the epidermis, causing the separation of keratinocytes (acantholysis) and cleavage and the formation of flaccid vesicles. Lesions develop in seborrheic areas and can disseminate but do not compromise the mucous membranes. Cazenave (or classical) pemphigus foliaceus, endemic pemphigus foliaceus (or ‘fogo selvagem’ [FSENT]), pemphigus erythematosus (or Senear-Usher syndrome), and pemphigus herpetiformis are variants of pemphigus foliaceus. FS differentiates itself from the classical form, based on its epidemiology-it compromises young adults from rural areas of the geographic region of FS, with a family history of the disease. 99 Bystryn JC, Rudolph JL. Pemphigus. Lancet. 2005;366:61-73.,7878 Diaz LA, Sampaio SA, Rivitti EA, Martins CR, Cunha PR, Lombardi C, et al. Endemic pemphigus foliaceus (fogo selvagem). I. Clinical features and immunopathology. J Am Acad Dermatol. 1989;20:657-69.
79 Amagai M. Pemphigus. In: Bolognia JL, Lorizzo JO, Schaffer JV, editors. Dermatology. 2nd ed. Vol. 1. China: Elsevier Saunders; 2012. p.461-74.
80 Hans-Filho G, Cunha PR. Fogo Selvagem (Endemic Pemphigus Foliaceus). In: Tyring SK, Lupi O, Hengge UR. editors. Tropical Dermatology. 2nd ed. Edinburgh: Elsevier; 2017. p. 427-32.-8181 Hans-Filho G, Aoki V, Rivitti E, Eaton DP, Lin MS, Diaz LA. Endemic pemphigus foliaceus (fogo selvagem) - 1998. The Cooperative Group on Fogo Selvagem Research. Clin Dermatol. 1999;17:225-35.
EPIDEMIOLOGY
PF is less frequent than pemphigus vulgaris (PV) (incidence 0.1 to 0.5/1055 Pan M, Liu X, Zheng J. The pathogenic role of autoantibodies in pemphigus vulgaris. Clin Exp Dermatol. 2011;36:703-7.), except in areas of South America, North Africa, and Turkey. In rural areas in Brazil, the ratio of FS to PV can reach 17:1, and in the Terena indigenous reserve (Aldeia Limão Verde) in Mato Grosso do Sul, the prevalence is 3.4%. Most FS patients come from midwestern Brazil and its northwest colonies, and once the disease has developed, its incidence decreases. 7878 Diaz LA, Sampaio SA, Rivitti EA, Martins CR, Cunha PR, Lombardi C, et al. Endemic pemphigus foliaceus (fogo selvagem). I. Clinical features and immunopathology. J Am Acad Dermatol. 1989;20:657-69.,99 Bystryn JC, Rudolph JL. Pemphigus. Lancet. 2005;366:61-73.,8282 Counter CE. The disease called wildfire. Arch Dermatol. 1959;80:391-7.
83 Diaz LA, Sampaio SA, Rivitti EA, Martins CR, Cunha PR, Lombardi C, et al. Endemic pemphigus foliaceus (Fogo selvagem): II. Current and historic epidemiologic studies. J Invest Dermatol. 1989;92:4-12.
84 Robledo MA, Prada S, Jaramillo D, Leon W. South American pemphigus: study of an epidemic in El Bagre and Nechi, Colombia 1982 to 1986. Br J Dermatol. 1988;118:737-44.
85 Campos JA. Pênfigo foliáceo (fogo selvagem). Aspectos clínicos e epidemiológicos. São Paulo: Companhia Melhoramentos; 1942. 127p.-8686 Bastuji-Garin S, Souissi R, Blum L, Turki H, Nouira R, Jomaa B, et al. Comparative epidemiologic of pemphigus in Tunisia and France: unusual incidence of pemphigus foliaceus in young Tunisian women. J Invest Dermatol. 1995;104:302-5.
ETIOPATHOGENESIS
The etiology of FS shares similarities with those of vector-borne diseases, such as Chagas disease and leishmaniasis. The predominant black fly in areas of FS is Simulium nigrimanum. Autoantibodies from FS patients recognize the salivary protein LJM11 from the phlebotomide Lutzomyia longipalpis, and mice that have been immunized with LJM11 produce anti-Dsg1. 8686 Bastuji-Garin S, Souissi R, Blum L, Turki H, Nouira R, Jomaa B, et al. Comparative epidemiologic of pemphigus in Tunisia and France: unusual incidence of pemphigus foliaceus in young Tunisian women. J Invest Dermatol. 1995;104:302-5.
87 Hans-Filho G, dos Santos V, Katayama JH, Aoki V, Rivitti EA, Sampaio SA, et al. An active focus of high prevalence of fogo selvagem on an Amerindian reservation in Brazil. Cooperative Group on Fogo Selvagem Research. J Invest Dermatol. 1996;107:68-75.
88 Aoki V, Millikan RC, Rivitti EA, Hans-Filho G, Eaton DP, Warren SJ, et al. Environmental risk factors in endemic pemphigus foliaceus (fogo selvagem). J Investig Dermatol Symp Proc. 2004;9:34-40.
89 Diaz LA, Arteaga LA, Hilario-Vargas J, Valenzuela JG, Li N, Warren S, et al. Anti-desmoglein-1 antibodies in onchocerciasis, leishmaniasis and Chagas disease suggest a possible etiological link to fogo selvagem. J Invest Dermatol. 2004;123:1045-51.-9090 Rivitti EA, Aoki V, Hans-Filho G, Vargas JH, Diaz LA. Fogo Selvagem (endemic pemphigus foliaceus). In: Tyring SK, Lupi O, Hengge UR. Tropical Dermatology. China: Elsevier Churchill Livingstone; 2006. p.421-7.
Drugs - Thiol drugs can induce PF and, less frequently, PV, of which penicillamine and captopril are the most common. Other drugs include penicillins, cephalosporins, enalapril, rifampicin, and non-hormonal anti-inflammatory drugs.9191 Qian Y, Jeong JS, Maldonado M, Valenzuela JG, Gomes R, Teixeira C, et al. Cutting Edge: Brazilian Pemphigus Foliaceus Anti-Desmoglein 1 Autoantibodies Cross- React with Sand Fly Salivary LJM11 Antigen. J Immunol. 2012;189:1535-9.,9292 Marsden RA, Vanhegan RI, Walshe M, Hill H, Mowat AG. Pemphigus foliaceus induced by penicillamine. Br Med J. 1976;2:1423-4.
Genetic - FS can occur in genetically related relatives (~20%) and is associated with the expression of certain DRB1 alleles (*0404, *1402, *1406) of the type II major histocompatibility complex.9393 Brenner S, Goldberg I. Drug-induced pemphigus. Clin Dermatol. 2011;29:455-7,9494 Moraes ME, Fernandez-Vina M, Lazaro A, Diaz LA, Filho GH, Friedman H, et al. An epitope in the third hypervariable region of the DRB1 gene is involved in the susceptibility to endemic pemphigus foliaceus (fogo selvagem) in three different Brazilian populations. Tissue Antigens. 1997;49:35-40.
Autoantigen - Desmoglein-1 is a 160-kD transmembrane glycoprotein of the cadherin family (calcium-dependent cell adhesion molecules) in desmosomes and has 5 ectodomains, EC1-5. Based on the desmoglein compensation theory, Dsg-1 expression is more intense in the upper portions of the epidermis, whereas in PV, Dsg-3 predominates in the lower layers of the epidermis and in mucous membranes. 99 Bystryn JC, Rudolph JL. Pemphigus. Lancet. 2005;366:61-73.,9595 Piovezan BZ, Petzl-Erler ML. Both qualitative and quantitative genetic variation of MHC class II molecules may influence susceptibility to autoimmune diseases: The case of endemic pemphigus foliaceus. Hum Immunol. 2013;74:1134-40
96 Amagai M, Klaus-Kovtun V, Stanley JR. Autoantibodies against a novel epithelial cadherin in pemphigus vulgaris, a disease of cell adhesion. Cell. 1991;67:869-77.
97 Aoki V, Rivitti EA, Diaz LA; Cooperative Group on Fogo Selvagem Research. Update on fogo selvagem, an endemic form of pemphigus foliaceus. J Dermatol. 2015;42:18-26.
98 Di Zenzo G, Zambruno G, Borradori L. Endemic Pemphigus Foliaceus: Towards Understanding Autoimmune Mechanisms of Disease Development. J Invest Dermatol. 2012;132:2499-502.
99 Amagai M, Karpati S, Prussick R, Klaus-Kovtun V, Stanley JR. Autoantibodies against the amino-terminal cadherin-like binding domain of pemphigus vulgaris antigen are pathogenic. J Clin Invest. 1992;90:919-26.
100 Evangelista F, Roth AJ, Prisayanh P, Temple BR, Li N, Qian Y, et al. Pathogenic IgG4 autoantibodies from endemic pemphigus foliaceus recognize a desmoglein-1 conformational epitope. J Autoimmun. 2018;89:171-85.
101 Culton DA, Qian Y, Li N, Rubenstein D, Aoki V, Filhio GH, et al. Advances in pemphigus and its endemic pemphigus foliaceus (Fogo Selvagem) phenotype: A paradigm of human autoimmunity. J Autoimmun. 2008;31:311-24.-102102 Hans-Filho G, Aoki V, Bittner NRH, Bittner GC. Fogo selvagem: endemic pemphigus foliaceus. An Bras Dermatol. 2018;93:638-50.
Autoantibodies - Autoantibodies that trigger the disease belong to the IgG4 subclass. Normal controls in endemic areas have low levels of IgG1 and IgG4, but in FS patients, the IgG1 levels are the same but IgG4 is significantly elevated by up to 19-fold. IgG4 is pathogenic in the mouse model of FS. In the preclinical phase, IgG1 autoantibodies are directed against the EC5 ectodomains of Dsg-1, and in the disease phase, IgG4 autoantibodies react against EC1-2-a phenomenon that is known as epitope spreading. 99 Bystryn JC, Rudolph JL. Pemphigus. Lancet. 2005;366:61-73.,8282 Counter CE. The disease called wildfire. Arch Dermatol. 1959;80:391-7.,8787 Hans-Filho G, dos Santos V, Katayama JH, Aoki V, Rivitti EA, Sampaio SA, et al. An active focus of high prevalence of fogo selvagem on an Amerindian reservation in Brazil. Cooperative Group on Fogo Selvagem Research. J Invest Dermatol. 1996;107:68-75.,100100 Evangelista F, Roth AJ, Prisayanh P, Temple BR, Li N, Qian Y, et al. Pathogenic IgG4 autoantibodies from endemic pemphigus foliaceus recognize a desmoglein-1 conformational epitope. J Autoimmun. 2018;89:171-85.
101 Culton DA, Qian Y, Li N, Rubenstein D, Aoki V, Filhio GH, et al. Advances in pemphigus and its endemic pemphigus foliaceus (Fogo Selvagem) phenotype: A paradigm of human autoimmunity. J Autoimmun. 2008;31:311-24.
102 Hans-Filho G, Aoki V, Bittner NRH, Bittner GC. Fogo selvagem: endemic pemphigus foliaceus. An Bras Dermatol. 2018;93:638-50.
103 Waschke J. The desmosome and pemphigus. Histochem Cell Biol. 2008;130:21-54
104 Beutner EH, Prigenzi LS, Hale W, Leme Cde A, Bier OG. Immunofluorescent studies of autoantibodies to intercellular areas of epithelia in Brazilian Pemphigus Foliaceus. Proc Soc Exp Biol Med. 1968;127:81-6.
105 Sitaru C, Zillikens D. Mechanisms of blister induction by autoantibodies. Exp Dermatol. 2005;14:861-75.
106 van der Wier G, Jonkman MF, Pas HH, Diercks GF. Ultrastructure of acantholysis in pemphigus foliaceus re-examined from the current perspective. Br J Dermatol. 2012;167:1265-71.
107 Roscoe JT, Diaz L, Sampaio SA, Castro RM, Labib RS, Takahashi Y, et al. Brazilian pemphigus foliaceus autoantibodies are pathogenic to BALB/c mice by passive transfer. J Invest Dermatol. 1985;85:538-41.
108 Warren SJ, Lin MS, Giudice GJ, Hoffmann RG, Hans-Filho G, Aoki V, et al. The prevalence of antibodies against desmoglein 1 in endemic pemphigus foliaceus in Brazil. N Engl J Med. 2000;343:23-30.
109 Warren SJ, Arteaga LA, Rivitti EA, Aoki V, Hans-Filho G, Qaqish BF, et al. The Role of Subclass Switching in the Pathogenesis of Endemic Pemphigus Foliaceus. J Invest Dermatol. 2003;120:104-8.
110 Li N, Aoki V, Hans-Filho G, Rivitti EA, Diaz LA. The role of intramolecular epitope spreading in the pathogenesis of endemic pemphigus foliaceus (fogo selvagem). J Exp Med. 2003;197:1501-10.
111 Yoshida K, Ishii K, Shimizu A, Yokouchi M, Amagai M, Shiraishi K, et al. Non-pathogenic pemphigus foliaceus (PF) IgG acts synergistically with a directly pathogenic PF IgG to incrase blistering by p38MAPK-dependent desmoglein 1 clustering. J Dermatol Sci. 2017;85:197-207.
112 Grando SA. Pemphigus autoimmunity: hypotheses and realities. Autoimmunity. 2012;45:7-35.-113113 Diaz LA, Prisayanh PS, Dasher DA, Li N, Evangelista F, Aoki V, et al. The IgM anti-desmoglein 1 response distinguishes Brazilian pemphigus foliaceus (fogo selvagem) from other forms of pemphigus. J Invest Dermatol. 2008;128:667-75,44 Mahoney MG, Wang Z, Rothenberger K, Koch PJ, Amagai M, Stanley JR. Explanations for the clinical and microscopic localization of lesions in pemphigus foliaceus and vulgaris. J Clin Invest. 1999;103:461-8.
Interleukins - Many proinflammatory and anti-inflammatory chemokines and cytokines are produced in the immune response in PF. Although their function in the disease and its pathophysiology remains unknown, they tend to generate a Th2 response, with increased levels of IL-6, TNF-α, and IL-22.114114 Qaqish BF, Prisayanh P, Qian Y, Andraca E, Li N, Aoki V, et al. Development of na IgG4-based predictor of endemic pemphigus foliaceus (fogo selvagem). J Invest Dermatol. 2009;129:110-8.
115 Hilario-Vargas J, Dasher DA, Li N, Aoki V, Hans-Filho G, dos Santos V, et al. Prevalence of anti-desmogleina-3 antibodies in endemic regions of fogo selvagem in Brazil. J Invest Dermatol. 2006;126:2044-8.
116 Rocha-Rodrigues DB, Paschoini G, Pereira SA, dos Reis MA, Teixeira Vde P, Rodrigues Júnior V. High levels of interleukin-1 in patiens with endemic pemphigus foliaceus. Clin Diagn Lab Immunol. 2003;10:741-3.
117 Chriguer RS, Roselino AM, de Castro M. Glucocorticoid sensitivity and proinflammatory cytokines pattern in pemphigus. J Clin Immunol. 2012;32:786-93.-118118 López-Robles E, Avalos-Díaz E, Vega-Memije E, Hojyo-Tomoka T, Villalobos R, Fraire S, et al. TNF-a end IL-6 are mediator in the blisterin process of pemphigus. Int J Dermatol. 2001;40:185-8.
CLINICAL CONDITION
The clinical manifestations of FS, classical Cazenave PF, and drug-induced pemphigus are similar. 8080 Hans-Filho G, Cunha PR. Fogo Selvagem (Endemic Pemphigus Foliaceus). In: Tyring SK, Lupi O, Hengge UR. editors. Tropical Dermatology. 2nd ed. Edinburgh: Elsevier; 2017. p. 427-32.,8989 Diaz LA, Arteaga LA, Hilario-Vargas J, Valenzuela JG, Li N, Warren S, et al. Anti-desmoglein-1 antibodies in onchocerciasis, leishmaniasis and Chagas disease suggest a possible etiological link to fogo selvagem. J Invest Dermatol. 2004;123:1045-51.,119119 Timóteo RP, Silva MV, da Silva DAA, Catarino JDS, Alves FHC, Rodrigues Júnior V, et al. Cytokine and chemokines alterations in the form of pemphigus folicaceus (fogo selvagem). Front Immunol. 2017;8:978.,120120 Arakawa M, Dainichi T, Yasumoto S, Hashimoto T. Lesional Th 17 cells in pemphigus vulgaris and pemphigus foliaceus. J Dermatol Sci. 2009;53:228-31.
Forme fruste - Forme frustes are superficial and flaccid vesicular blisters that rupture and develop erythematous/erosive/crusted areas in seborrheic areas, such as the scalp, face (it can be distributed in the shape of a butterfly wing), and upper and central regions of the chest. The number of lesions varies. Nikolsky sign is present.
Disseminated form - This form can be invasive bullous or bullous exfoliative, when the number of lesions intensifies in seborrheic areas and spreads throughout the chest and limbs. Eventually, it can progress to exfoliative erythroderma.
Pemphigus herpetiformis - This condition can be seen in PF and PV. It manifests as urticarial plaques and vesicles in a herpetiform or annular distribution, with pruritus. 7979 Amagai M. Pemphigus. In: Bolognia JL, Lorizzo JO, Schaffer JV, editors. Dermatology. 2nd ed. Vol. 1. China: Elsevier Saunders; 2012. p.461-74.,8080 Hans-Filho G, Cunha PR. Fogo Selvagem (Endemic Pemphigus Foliaceus). In: Tyring SK, Lupi O, Hengge UR. editors. Tropical Dermatology. 2nd ed. Edinburgh: Elsevier; 2017. p. 427-32.,110110 Li N, Aoki V, Hans-Filho G, Rivitti EA, Diaz LA. The role of intramolecular epitope spreading in the pathogenesis of endemic pemphigus foliaceus (fogo selvagem). J Exp Med. 2003;197:1501-10.
Pemphigus erythematosus or Senear-Usher syndrome: Forme frustes, with lesions localizing predominantly to the malar regions, concomitant with laboratory findings of systemic lupus erythematosus.7979 Amagai M. Pemphigus. In: Bolognia JL, Lorizzo JO, Schaffer JV, editors. Dermatology. 2nd ed. Vol. 1. China: Elsevier Saunders; 2012. p.461-74.,8080 Hans-Filho G, Cunha PR. Fogo Selvagem (Endemic Pemphigus Foliaceus). In: Tyring SK, Lupi O, Hengge UR. editors. Tropical Dermatology. 2nd ed. Edinburgh: Elsevier; 2017. p. 427-32.
Neonatal pemphigus foliaceus is rarer than neonatal PV, due to the predominance of Dsg-3 compared with Dsg-1 in the newborn’s skin. Mothers of these newborns usually have disseminated disease and high titers of anti-Dsg1 autoantibodies. 121121 Juneja M. Nikolsky's sign revisited. Journal of Oral Science. 2008;50:213-4.
122 James KA, Culton DA, Diaz LA. Diagnosis and clinical features of pemphigus foliaceus. Dermatol Clin. 2011;29:405-12.
123 Avalos-Díaz E, Olague-Marchan M, López-Swiderski A, Herrera-Esparza R, Díaz LA. Transplacental passage of maternal pemphigus foliaceus autoantibodies induces neonatal pemphigus. J Am Acad Dermatol. 2000;43:1130-4.-124124 Zhao CY, Chiang YZ, Murrell DF. Neonatal Autoimmune Blistering Disease: A Systematic Review. Pediatr Dermatol. 2016;33:367-74.
In the differential diagnosis, seborrheic dermatitis, impetigo, chronic cutaneous lupus erythematosus, subacute cutaneous lupus erythematosus, IgA pemphigus, and the pemphigus variant of non-IgA subcorneal pustular dermatosis should be considered. In the evaluation of patients with erythroderma that is to be clarified, immunological examinations are recommended to rule out PF. 7878 Diaz LA, Sampaio SA, Rivitti EA, Martins CR, Cunha PR, Lombardi C, et al. Endemic pemphigus foliaceus (fogo selvagem). I. Clinical features and immunopathology. J Am Acad Dermatol. 1989;20:657-69.,99 Bystryn JC, Rudolph JL. Pemphigus. Lancet. 2005;366:61-73.,8181 Hans-Filho G, Aoki V, Rivitti E, Eaton DP, Lin MS, Diaz LA. Endemic pemphigus foliaceus (fogo selvagem) - 1998. The Cooperative Group on Fogo Selvagem Research. Clin Dermatol. 1999;17:225-35.
LABORATORY DIAGNOSIS
To confirm the diagnosis of any autoimmune bullous disease, clinical, histopathological, and immunological criteria are required. 99 Bystryn JC, Rudolph JL. Pemphigus. Lancet. 2005;366:61-73.,8989 Diaz LA, Arteaga LA, Hilario-Vargas J, Valenzuela JG, Li N, Warren S, et al. Anti-desmoglein-1 antibodies in onchocerciasis, leishmaniasis and Chagas disease suggest a possible etiological link to fogo selvagem. J Invest Dermatol. 2004;123:1045-51.,125125 Lin L, Zeng X, Chen Q. Pemphigus and pregnancy. Saudi Med J. 2015;36:1033-8.
126 Lorente Lavirgen AI, Bernabeu-Wittel J, Dominguez-Cruz J, Conejo-Mir J. Neonatal pemphigus foliaceus. J Pediatr. 2012;161:768.-127127 Payne AS, Stanley JR. Pemphigus. In: Goldsmith LA, Katz SI, Gilchrest BA, Peller AS, Lefflell DJ, Wolf K, editors. Fitzpatrick's Dermatology in General Medicine. 8th ed. Vol 1. New York: McGraw Hill; 2012. p.586.
Histopathology - In PF, cleavage below the stratum corneum is observed with the presence of acantholytic keratinocytes in or adjacent to the granulosa layer, and occasional neutrophils are seen. In the dermis, a mixed inflammatory infiltrate is observed with eosinophils and neutrophils; eosinophils are more prevalent in drug-induced PF. A biopsy for histopathology should be performed at the vesicle/blister or recent erosion edge, with a 4-mm punch.
Direct immunofluorescence (DIF) - A biopsy sample should be collected from seemingly normal perilesional skin. IgG and C3 deposition on the surface of keratinocytes throughout the epidermis is noted, although it might be concentrated in the upper layers in certain cases.
Indirect immunofluorescence (IIF) - More than 80% of patients have IIF-detectable IgG autoantibodies that correlate with disease activity and extent. Neither DIF nor IIF distinguishes PF from PV.
Enzyme-linked immunosorbent assay (ELISA) - Commercially available recombinant Dsg-1 is used as the antigen and has high sensitivity and specificity-higher than IIF. This technique can aid in monitoring disease activity and the response to treatment.
Other techniques include immunoblotting and immunoprecipitation, which are difficult to execute and are restricted to research services.
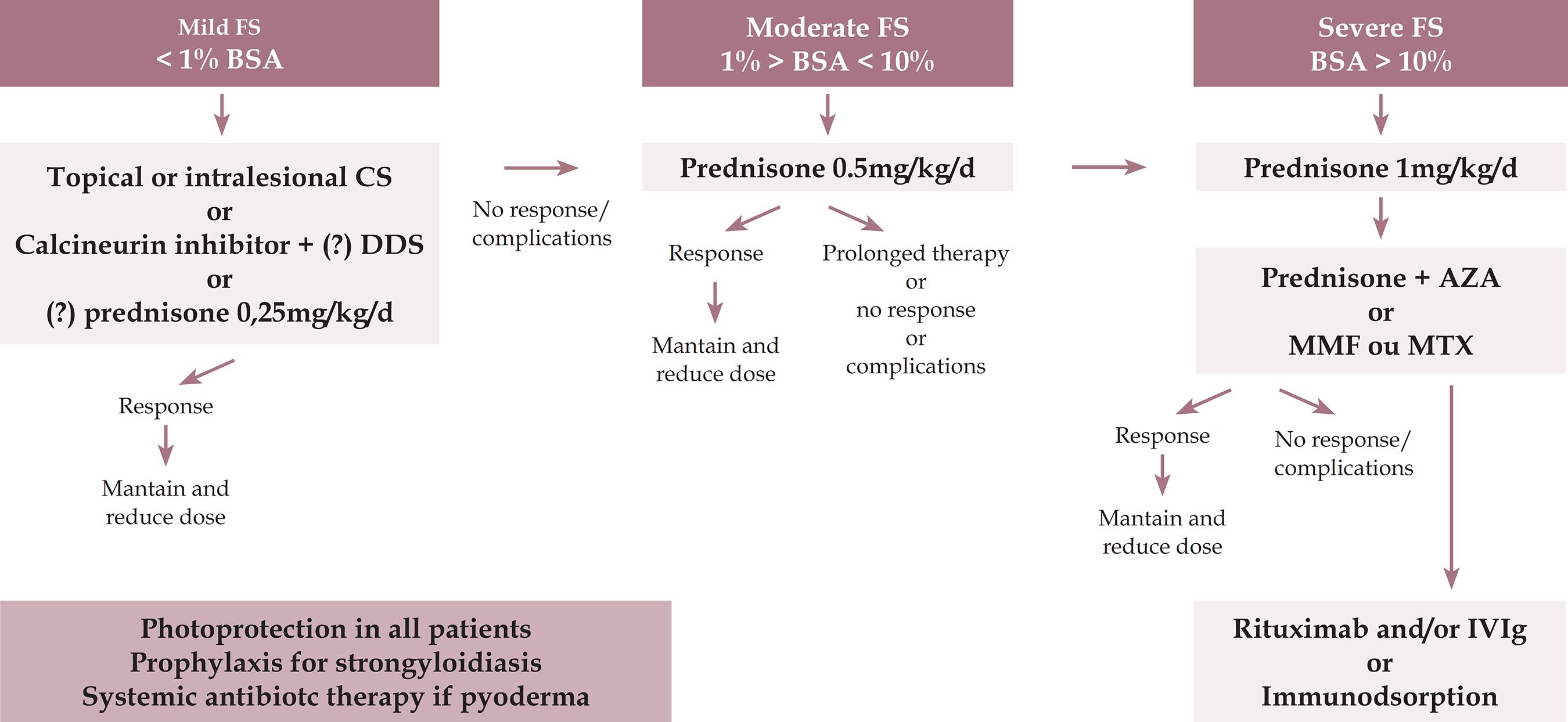
TREATMENT
Treatment is indicated from the outset of symptoms in PF, even if the clinical manifestation is mild. The goal is to induce rapid control of the disease and complete remission, minimizing treatment-related adverse effects. 101101 Culton DA, Qian Y, Li N, Rubenstein D, Aoki V, Filhio GH, et al. Advances in pemphigus and its endemic pemphigus foliaceus (Fogo Selvagem) phenotype: A paradigm of human autoimmunity. J Autoimmun. 2008;31:311-24.,128128 Brenner S, Bialy-Golan A, Ruocco V. Drug-induced pemphigus. Clin Dermatol. 1998;16:393-7.
129 Arbache ST, Nogueira TG, Delgado L, Miyamoto D, Aoki V. Immunofluorescence testing in the diagnosis of autoimmune blistering diseases: overview of 10-year experience. An Bras Dermatol. 2014;89:885-9.
130 Kasperkiewicz M, Schmidt E, Zillikens D.. Current therapy of the pemphigus group. Clin Dermatol. 2012;30:84-94.
131 Mutasim DF. Management of autoimmune bullous diseases: Pharmacology and therapeutics. J Am Acad Dermatol. 2004;51:859-77.
132 Schmidt E, Zillikens D. Immunoadsorption in dermatology. Arch Dermatol Res. 2010;302:241-53.
133 Uptodate.com [Internet]. Hertl M, Geller S. Inicial management of pemphigus vulgaris and pemphigus foliaceus. UpToDate Mar 2017. [cited 2018 Jun 08]. Available from: https://www.uptodate.com/contents/initial-management-of-pemphigus-vulgaris-and-pemphigus-foliaceus
https://www.uptodate.com/contents/initia...
-134134 Karnofsky DA, Burchenal JH. The Clinical Evaluation of Chemotherapeutic Agents in Cancer. In: MacLeod CM, editor. Evaluation of Chemotherapeutic Agents. New York: Columbia Univ Press; 1949. p. 196.
Considering the serious side effects of high and prolonged doses of systemic corticosteroid therapy, systemic non-steroidal immunomodulatory medication is recommended, especially with azathioprine, mycophenolate mofetil, or methotrexate, the most important of which is azathioprine. For severe and refractory cases, rituximab, intravenous immunoglobulin (IVIG), and eventually cyclophosphamide can be indicated.
Risk factors and comorbidities should always be investigated, in addition to the recent use of drugs with the potential to induce pemphigus. Regarding indications for hospital admission, it is suggested that the clinical condition be evaluated using the Karnofsky index-a 100% value refers to a normal person, with no evidence of disease; hospitalization is indicated when this index is 50% or less (50% refers to a patient requiring considerable help, often medical and/or specialized care, and 40% refers to a disabled patient who requires special and comprehensive care).
In deciding which regimen to prescribe for a patient with FS, the extent of pemphigus should be determined. A practical method would be to consider up to 1% involvement as a mild clinical form, up to 10% as moderate, and over 10% of compromised body area (FS active lesions) as severe, with 1% representing the sum of the area of FS lesions, corresponding approximately to the hand palmar area. In some departments, a protocol is included to verify extent and severity of the lesions using the PAAS (Pemphigus Area and Activity Score) and ABSIS (Autoimmune Bullous Skin Disorder Intensity Score) scores. 135135 Daniel BS, Hertl M, Werth VP, Eming R, Murrell DF. Severity score indexes for blistering diseases. Clin Dermatol. 2012;30:108-13.,136136 Sebaratnam DF, Murrell DF. Objective scoring systems for disease activity in autoimmune bullous disease. Dermatol Clin. 2011;29:515-20, xi.
Prior to initiation of therapy, complete blood count, creatinine, sodium, potassium, transaminases, gamma-glutamyl transferase (γGT), alkaline phosphatase, total proteins and protein fractions, fasting glycemia, serology for hepatitis B and C and human immunodeficiency virus (HIV), and chest x-ray should be examined. Optional recommendations include: ruling out IgA deficiency prior to IVIG; measuring thiopurine methyltransferase activity before azathioprine; abdominal ultrasonography (optional), PPD, or Quantiferon if the risk for tuberculosis is high; measuring activity of glucose-6-phosphate dehydrogenase (G6PD), bilirubins, and reticulocytes prior to dapsone; β-HCG (human chorionic gonadotrophin) to exclude pregnancy; bone densitometry before corticotherapy; and an ophthalmological evaluation to rule out glaucoma and cataract. Regardless of the parasitological stool examination, preventive treatment of strongyloidiasis is recommended, as is systemic antibiotic therapy if pyoderma is present.
In localized forms, with a limited number of lesions (up to 1% of body area), topical (moderate to high potency) or intralesional corticosteroid therapy (triamcinolone acetonide 2 to 3mg/ml) is used. Associated with topical therapy, one can prescribe dapsone 50 to 100mg/day. In small series, dapsone monotherapy at higher doses (200-300mg/d) has been suggested to be effective in the initial treatment of certain patients with PF. In nonresponsive patients, low dosages of prednisone (up to 0.25mg/kg/d) may be employed.
Systemic corticosteroid therapy (prednisone/prednisolone) is prescribed when topical treatment does not control the disease or if the cutaneous condition worsens, as evidence by an increase in lesion number, at a dosage of 0.5mg/kg/day.
In severe disseminated forms (above 10% of body area), the dosage of prednisone/prednisolone is 1mg/kg/day. Systemic corticosteroid therapy remains the most widely used, recognized, and established treatment option, due to its high efficacy and rapid control. Equivalent doses of triamcinolone may be administered in FS-resistant patients.
Considering that the use of prolonged and high-dose systemic corticosteroids can lead to severe or even fatal adverse effects, it is necessary in patients with disseminated disease to administer a combination of corticoid-sparing drugs (adjuvants) early, including methotrexate, azathioprine, and mycophenolate mofetil. According to recent publications, the ideal combination is deflazacort and azathioprine. Factors that should be considered in the choice of adjuvant are its availability, cost, and side effects.
No statistically significant difference has been observed in the time to disease control between higher and lower doses of systemic corticosteroids. There are also no differences in the average side effects. Thus, treatment is started at smaller doses, increasing the dose until clinical control of the disease is attained.
If no new lesions develop within 2 weeks, a reduction in corticosteroid from 0.25mg/kg every 2 weeks to 30mg/d is suggested, with subsequent reductions of 5mg every 2 weeks until 10mg/d is reached. Subsequently, the dosage is reduced by 2.5mg/wk until 0, and adjuvant therapy is maintained.
Immunosuppressive medications (first-line adjuvants)
The ideal adjuvants are azathioprine and mycophenolate mofetil, which have established corticoid-sparing effects. Azathioprine is used at 1-3mg/kg/d (beginning at 50mg/d, increasing progressively until the total daily dose is reached). The activity of thiopurine-methyl transferase should be measured, if possible, prior to treatment initiation, because if it is low, medication may compromise the bone marrow.
Mycophenolate mofetil, or mycophenolic acid, is administered at a dosage of 2g/d (starting dosage of 1g/d, with a gradual increase of 500mg/day to improve gastric tolerance). This drug is an excellent option but has a high cost. Another possible side effect of mycophenolate is pancytopenia.
Methotrexate is an interesting option, based on its low cost and wide availability. However, it is hepatotoxic. It is used at 7.5 to 25mg/week, administered over 1 or 2 consecutive days. After 24 hours, folic acid should be prescribed at a dose of 5mg. Alcohol, sulfamidic derivatives, and allopurinol are banned.
For less extensive forms, one can attempt dapsone 100mg/d or up to 1.5mg/kg/d, because it has also corticosteroid-sparing effects. However, glucose-6-phosphate dehydrogenase (G6PD) activity should be evaluated beforehand. Considering that pemphigus is an antibody-mediated disease, dapsone use is controversial.
Cyclophosphamide can also be used at 500mg IV as a bolus or 2mg/kg/d. It has corticosteroid-sparing effects, but the risk of sterility, hemorrhagic cystitis, and secondary malignant neoplasia should be considered.
The evidence indicates that the major benefit of adjuvant use is the corticoid-sparing effect-not in modifying the course of the disease. In addition, adjuvants reduce the risk of relapse without altering remission rates.
Newer and more relevant therapies
Rituximab (monoclonal anti-CD20) is indicated when the patient is refractory to conventional therapy or if prednisone is required at dosages of higher than 10mg/d in combination with an immunosuppressant for more than 6 months. It is administered as pulse therapy, preferably at a dose of 1 g IV, repeated after 15 days (rheumatoid arthritis protocol) or at 375mg/m2/week in 4 sessions (protocol for lymphoma). Lower doses are ineffective. If necessary, the protocol should be repeated in 6 months. Rituximab can be combined with prednisone in a regression scheme of up to 4 months or with an immunosuppressive agent (up to 12 months). Hypersensitivity to murine proteins should be excluded. Adverse effects include infections (up to 10%), reactions during infusion, and rarely Stevens-Johnson syndrome and progressive multifocal leukoence-phalopathy (potentially life-threatening complications).
Historically, monoclonal anti-CD20 has been used for treatment-refractory pemphigus and for patients with severe side effects to conventional immunosuppressive treatments. However, recent data indicate that the combination of rituximab and prednisone may be effective as an initial treatment for pemphigus, allowing the doses of prednisone to be reduced. It is associated with lower rates of side effects compared with high and prolonged doses of corticoid monotherapy.
In certain centers that specialize in the treatment of pemphigus (PV and PF), for moderate and severe clinical forms, rituximab, in combination with systemic corticosteroid therapy, has been used as initial and maintenance treatment in all patients.
Intravenous immunoglobulin: IVIG is indicated for very severe, refractory patients, those who present with significant adverse effects, and severe and disseminated forms of pemphigus that require a more rapid clinical response. Infused immunoglobulin appears to accelerate pathogenic catabolism of IgG autoantibodies. Doses of 2 to 3g/kg/cycle are recommended (cycle of 4 to 5 consecutive days) every 30 days. Systemic corticosteroid and adjuvant drugs are maintained and have been used in combination with rituximab. Aseptic meningitis is a rare side effect. IgA deficiency should be excluded before starting this treatment.
Future therapies
Immunoadsorption is a selective extracorporeal clearance technique that removes immunoglobulins, especially IgG1, IgG2, and IgG4. With this technique, circulating levels of immunoglobulins can be decreased by over 80%. It is another option for refractory or very severe patients and is available in advanced centers for autoimmune diseases. Monthly cycles of 4 consecutive days are performed, with 2.5 times the plasma volume being perfused per day. Contraindications include severe systemic infection, severe cardiovascular disease, extensive hemorrhagic diathesis, and the use of ACE inhibitors.
We present an algorithm for the treatment of fogo selvagem (see Figure 2)
Pemphigus foliaceus - treatment algorithm
FS - fogo selvagem; BSA - body surface area (1% means the sum of injured areas corresponding to the palm area); CS - corticosteroid; DDS - diamino-diphenil-sulfone or dapsone; AZA - azathioprine; MMF - mycophenolate mofetil; MTX - methotrexate; IVIg - intravenous immunoglobulin.
Maintenance after consolidation phase: CE should be reduced progressively, after disease control or the end of the consolidation phase, reducing the corticosteroid dose by approximately 25% every 2 weeks until 20mg/d and then slowly. If more than 3 lesions appear, return to the previous dose. If the disease recurs, return to the dose of the 2 previous phases. If it does not stabilize within 2 weeks, return to the starting dose. If the treatment is combined with an immunosuppressant, replace it or use rituximab, IVIG, or immunoadsorption. High levels of anti-Dsg-1 by ELISA indicate cutaneous relapses.
Clinical and laboratory monitoring: Clinical reassessments (skin and mucous membranes) should be performed every 2 weeks and, after control, monthly. Significant adverse effects include diabetes mellitus, systemic arterial hypertension, and heart failure due to Corticosteroid (CS) therapy; respiratory distress, anemia, and hepatitis (DDS, MTX); respiratory infections and hepatitis (CS, immunosuppressants); mental disorders (CS); myopathy, osteoporosis, avascular bone necrosis, glaucoma, and cataract (CS); and hematological abnormalities (immunosuppressants).
Vaccination: The use of adjuvant immunosuppressants and rituximab contraindicates vaccination with live virus.
Serological monitoring: Serology (IIF and/or ELISA) should be performed at the outset of treatment, after 3 months, and then according to disease progression.
Treatment discontinuation: Discontinuation is based primarily on clinical signs and may be accompanied by anti-Dsg ELISA and IIF. In some centers, negativity by IIF is recommended. Discontinue CS in patients with complete remission and minimal therapy, and discontinue adjuvants 6 to 12 months later.
EVOLUTION AND PROGNOSIS
Until the 1950s, the prognosis was poor, with a significant mortality rate due to cachexia and systemic infection. With the introduction of systemic corticosteroid therapy, this rate fell to 30%. By combining it with immunosuppressive adjuvants and recognizing and treating the adverse effects of systemic corticosteroids, the mortality rate was reduced to approximately 6%. PF without compromising mucous membranes has a better prognosis than pemphigus vulgaris. PF usually has a chronic evolution and it is necessary increase the posology of the medication when relapses occur. Approximately half of all patients experience remission but remain at risk of recurrence, even after years without treatment. The remaining patients keep their disease under control with low doses of therapy.
CONCLUSION
PF is an autoimmune, anti-cadherin-based, acantholytic bullous disease. In Brazil, its incidence is high, mainly in the midwest and northwest regions, and is called endemic PF or FS. This differs from classical pemphigus foliaceus, based on its epidemiology: it compromises young adults from the rural areas of fogo selvagem regions, with a family history. Anti-desmoglein 1 autoantibodies are directed against desmosomal structures, wherein the loss of adhesion of keratinocytes in the upper layers of the epidermis causes superficial blisters. Its etiology is multifactorial, including genetic, immunological, and environmental factors, primarily hematophagous bites, and it is recommended that drug exposure be assessed. Flaccid blisters rupture quickly, leaving erosion-crusted areas in seborrheic regions, and it should be differentiated from seborrheic dermatitis, actinic keratosis, and chronic lupus erythematosus.
The clinical presentation varies from localized to disseminated. Clinical suspicion should be confirmed with histopathological and immunological examinations, especially immunofluorescence. Its evolution is usually chronic, and the treatment varies according to the clinical condition, but it typically requires systemic corticosteroid therapy, in combination with adjuvant immunosuppressive therapy to decrease the adverse effects of corticosteroids. The disease, once controlled, remains stable with low doses of medication, and a significant proportion of patients experience remission.
-
*
Work conducted at the Sociedade Brasileira de Dermatologia, Rio de Janeiro (RJ), Brazil.
-
Financial support: None.
REFERENCES
-
1Kridin K, Sagi SZ, Bergman R. Mortality and cause of death in Israeli patients with pemphigus. Acta Derm Venereol. 2017;97:607-611.
-
2Hsu DY, Brieva J, Sinha AA, Langan SM, Silverberg JI. Comorbidities and inpatient mortality for pemphigus in the USA. Br J Dermatol. 2016;174:1290-8.
-
3Di Zenzo G, Amber KT, Sayar BS, Müller EJ, Borradori L. Immune response in pemphigus and beyond: progresses and emerging concepts. Semin Immunopathol. 2016;38:57-74.
-
4Mahoney MG, Wang Z, Rothenberger K, Koch PJ, Amagai M, Stanley JR. Explanations for the clinical and microscopic localization of lesions in pemphigus foliaceus and vulgaris. J Clin Invest. 1999;103:461-8.
-
5Pan M, Liu X, Zheng J. The pathogenic role of autoantibodies in pemphigus vulgaris. Clin Exp Dermatol. 2011;36:703-7.
-
6Joly P, Litrowski N. Pemphigus group (vulgaris, vegetans, foliaceus, herpetiformis, brasiliensis). Clin Dermatol. 2011;29(4):432-6.
-
7Amber KT, Staropoli P, Shiman MI, Elgart GW, Hertl M. Autoreactive T cells in the immune pathogenesis of pemphigus vulgaris. Exp Dermatol. 2013; 22:699-704.
-
8Kridin K. Pemphigus group: overview, epidemiology, mortality, and comorbidities. Immunol Res. 2018;66:255-70.
-
9Bystryn JC, Rudolph JL. Pemphigus. Lancet. 2005;366:61-73.
-
10Pollmann R, Schmidt T, Eming R, Hertl M. Pemphigus: a Comprehensive Review on Pathogenesis, Clinical Presentation and Novel Therapeutic Approaches. Clin Rev Allergy Immunol. 2018;54:1-25
-
11Hammers CM, Stanley JR. Mechanisms of disease: pemphigus and bullous pemphigoid. Annu Rev Pathol. 2016 May 23;11:175-97.
-
12Broussard KC, Leung TG, Moradi A, Thorne JE, Fine JD. Autoimmune bullous diseases with skin and eye involvement: Cicatricial pemphigoid, pemphigus vulgaris, and pemphigus paraneoplastica. Clin Dermatol. 2016;34:205-13.
-
13Becker BA, Gaspari AA. Pemphigus vulgaris and vegetans. Dermatol Clin. 1993;11:429-52.
-
14Meurer M. Immunosuppressive therapy for autoimmune bullous diseases. Clin Dermatol. 2012;30:78-83.
-
15Bystryn JC, Steinman NM. The adjuvant therapy of pemphigus. An update. Arch Dermatol. 1996;132:203-12.
-
16Hertl M, Jedlickova H, Karpati S, Marinovic B, Uzun S, Yayli S, et al. Pemphigus. S2 guideline for diagnosis and treatment-guided by the European Dermatology Forum (EDF) in cooperation with the European Academy of Dermatology and Venereology (EADV). J Eur Acad Dermatol Venereol. 2015;29:405-14.
-
17Tavakolpour S. Current and future treatment options for pemphigus: Is it time to move towards more effective treatments? Int Immunopharmacol. 2017;53:133-42.
-
18Ruocco E, Wolf R, Ruocco V, Brunetti G, Romano F, Lo Schiavo A. Pemphigus: Associations and management guidelines: Facts and controversies. Clin Dermatol. 2013;31:382-90.
-
19Harman KE, Brown D, Exton LS, Groves RW, Hampton PJ, Mohd Mustapa MF, et al. British Association of Dermatologists' guidelines for the management of pemphigus vulgaris 2017. Br J Dermatol. 2017;177:1170-201
-
20Rao PN, Lakshmi TS. Pulse therapy and its modifications in pemphigus: a six year study. Indian J Dermatol Venereol Leprol. 2003;69:329-33.
-
21Chams-Davatchi C, Esmaili N, Daneshpazhooh M, Valikhani M, Balighi K, Hallaji Z, et al. Randomized controlled open-label trial of four treatment regimens for pemphigus vulgaris. J Am Acad Dermatol. 2007;57:622-8.
-
22Ahmed AR, Moy R. Death in pemphigus. J Am Acad Dermatol. 1982;7:221-8.
-
23Frew JW, Martin LK, Murrell DF. Evidence-based treatments in pemphigus vulgaris and pemphigus foliaceus. Dermatol Clin. 2011;29:599-606.
-
24Atzmony L, Hodak E, Leshem YA, Rosenbaum O, Gdalevich M, Anhalt GJ, et al. The role of adjuvant therapy in pemphigus: A systematic review and meta-analysis. J Am Acad Dermatol. 2015;73:264-71.
-
25Maltzman JS, Koretzky GA. Azathioprine: old drug, new actions. J Clin Invest. 2003;111:1122-4.
-
26Chams-Davatchi C, Mortazavizadeh A, Daneshpazhooh M, Davatchi F, Balighi K, Esmaili N, et al. Randomized double blind trial of prednisolone and azathioprine, vs. prednisolone and placebo, in the treatment of pemphigus vulgaris. J Eur Acad Dermatol Venereol. 2013;27:1285-92.
-
27Chaidemenos G, Apalla Z, Koussidou T, Papagarifallou I, Ioannides D. High dose oral prednisone vs. prednisone plus azathioprine for the treatment of oral pemphigus: a retrospective, bi-centre, comparative study. J Eur Acad Dermatol Venereol. 2011;25:206-10.
-
28Committee for Guidelines for the Management of Pemphigus Disease, Amagai M, Tanikawa A, Shimizu T, Hashimoto T, Ikeda S, et al. Japanese guidelines for the management of pemphigus. J Dermatol. 2014;41:471-86.
-
29Olejarz W, Bryk D, Zapolska-Downar D. Mycophenolate mofetil-a new atheropreventive drug? Acta Pol Pharm. 2014;71:353-61.
-
30Cholera M, Chainani-Wu N. Management of Pemphigus Vulgaris. Adv Ther. 2016;33:910-58.
-
31Vyas N, Patel NS, Cohen GF. Mycophenolate mofetil as a first-line steroid sparing agent in the treatment of pemphigus vulgaris. J Drugs Dermatol. 2013;12:210-6.
-
32Martin LK, Werth VP, Villaneuva EV, Murrell DF. A systematic review of randomized controlled trials for pemphigus vulgaris and pemphigus foliaceus. J Am Acad Dermatol. 2011;64:903-8.
-
33Beissert S, Mimouni D, Kanwar AJ, Solomons N, Kalia V, Anhalt GJ. Treating pemphigus vulgaris with prednisone and mycophenolate mofetil: a multicenter, randomized, placebo controlled trial. J Invest Dermatol. 2010;130:2041-8.
-
34Ahmed AR, Spigelman Z, Cavacini LA, Posner MR. Treatment of pemphigus vulgaris with rituximab and intravenous immuneglobulin. N Engl J Med. 2006;355:1772-9.
-
35Eming R, Nagel A, Wolff-Franke S, Podstawa E, Debus D, Hertl M. Rituximab exerts a dual effect in pemphigus vulgaris. J Invest Dermatol. 2008;128:2850-8.
-
36Huang A, Madan RK, Levitt J. Future therapies for pemphigus vulgaris:Rituximab and beyond. J Am Acad Dermatol. 2016;74:746-53.
-
37Wang HH, Liu CW, Li YC, Huang YC. Efficacy of rituximab for pemphigus: a systematic review and meta-analysis of different regimens. Acta Derm Venereol. 2015;95:928-32.
-
38Joly P, Maho-Vaillant M, Prost-Squarcioni C, Hebert V, Houivet E, Calbo S, et al., First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): a prospective, multicentre, parallel-group, open-label randomised trial. Lancet. 2017;389:2031-40
-
39Ingen-Housz-Oro S, Valeyrie-Allanore L, Cosnes A, Ortonne N, Hüe S, Paul M, et al. First-line treatment of pemphigus vulgaris with a combination of rituximab and high-potency topical corticosteroids. JAMA Dermatol. 2015;151:200-3.
-
40Tavakolpour S, Mahmoudi H, Balighi K, Abedini R, Daneshpazhooh M. Sixteen-year history of rituximab therapy for 1085 pemphigus vulgaris patients: A systematic review. Int Immunopharmacol. 2018;54:131-8
-
41Cianchini G, Lupi F, Masini C, Corona R, Puddu P, De Pità O. Therapy with rituximab for autoimmune pemphigus: results from a single-center observational study on 42 cases with long-term follow-up. J Am Acad Dermatol. 2012;67:617-22.
-
42Heelan K, Al-Mohammedi F, Smith MJ, Knowles S, Lansang P, Walsh S, et al. Durable remission of pemphigus with a fixed-dose rituximab protocol. JAMA Dermatol. 2014;150:703-8.
-
43Kanwar AJ, Vinay K, Sawatkar GU, Dogra S, Minz RW, Shear NH, et al. Clinical and immunological outcomes of high and low dose rituximab treatments in pemphigus patients:a randomized comparative observer blinded study. Br J Dermatol. 2014;170:1341-9.
-
44Amber KT, Hertl M. An assessment of treatment history and its association with clinical outcomes and relapse in 155 pemphigus patients with response to a single cycle of rituximab. J Eur Acad Dermatol Venereol. 2015;29:777-82.
-
45Joly P, Mouquet H, Roujeau JC, D'Incan M, Gilbert D, Jacquot S, et al. A single cycle of rituximab for the treatment of severe pemphigus. N Engl J Med. 2007;357:545-52.
-
46Londhe PJ, Kalyanpad Y, Khopkar US. Intermediate doses of rituximab used as adjuvant therapy in refractory pemphigus. Indian J Dermatol Venereol Leprol. 2014;80:300-5.
-
47Ahmed AR, Nguyen T, Kaveri S, Spigelman ZS. First line treatment of pemphigus vulgaris with a novel protocol in patients with contraindications to systemic corticosteroids and immunosuppressive agents: preliminary retrospective study with a seven year follow-up. Int Immunopharmacol. 2016;34:25-31.
-
48Behzad M, Möbs C, Kneisel A, Möller M, Hoyer J, Hertl M, et al. Combined treatment with immunoadsorption and rituximab leads to fast and prolonged clinical remission in difficult-to-treat pemphigus vulgaris. Br J Dermatol. 2012;166:844-52.
-
49Graves JE, Nunley K, Heffernan MP. Off-label uses of biologics in dermatology: rituximab, omalizumab, infliximab, etanercept, adalimumab, efalizumab, and alefacept (part 2 of 2). J Am Acad Dermatol. 2007;56:e55-79.
-
50Lunardon L, Tsai KJ, Propert KJ, Fett N, Stanley JR, Werth VP, et al. Adjuvant rituximab therapy of pemphigus: a single-center experience with 31 patients. Arch Dermatol. 2012;148:1031-6.
-
51Murrell DF, Peña S, Joly P, et al.Diagnosis and Management of Pemphigus: recommendations by an International Panel of Experts. J Am Acad Dermatol. 2018. pii: S0190-9622(18)30207-X [Epub ahead of print]
-
52Leshem YA, Hodak E, David M, Anhalt GJ, Mimouni D. Successful treatment of pemphigus with biweekly 1-g infusions of rituximab: a retrospective study of 47 patients. J Am Acad Dermatol. 2013;68:404-11.
-
53Shaik F, Botha J, Aboobaker J, Mosam A. Corticosteroid /cyclophosphamide pulse treatment in south African patients with pemphigus. Clin Exp Dermatol. 2010;35:245-50.
-
54Zhao CY, Murrell DF. Pemphigus Vulgaris: An Evidence-Based Treatment Update. Drugs. 2015;75:271-84.
-
55Sharma VK, Khandpur S. Evaluation of cyclophosphamide pulse therapy as an adjuvant to oral corticosteroid in the management of pemphigus vulgaris. Clin Exp Dermatol. 2013;38:659-64.
-
56Rose E, Wever S, Zilliken D, Linse R, Haustein UF, Bröcker EB, et al. Intravenous dexamethasone-cyclophosphamide pulse therapy in comparison with oral methylprednisolone-azathioprine therapy in patients with pemphigus: results of a multicenter prospectively randomized study. J Dtsch Dermatol Ges. 2005;3:200-6.
-
57Sethy PK, Khandpur S, Sharma VK. Randomized open comparative trial of dexamethasone-cyclophosphamide pulse and daily oral cyclophosphamide versus cyclophosphamide pulse and daily oral prednisolone in pemphigus vulgaris. Indian J Dermatol Venereol Leprol. 2009;75:476-82.
-
58Parmar NV, Kanwar AJ, Minz RW, Parsad D, Vinay K, Tsuruta D, et al. Assessment of the therapeutic benefit of dexamethasone cyclophosphamide pulse versus only oral cyclophosphamide in phase II of the dexamethasone cyclophosphamide pulse therapy: a preliminary prospective randomized controlled study. Indian J Dermatol Venereol Leprol. 2013;79:70-6.
-
59Gürcan HM, Ahmed AR. Analysis of current data on the use of methotrexate in the treatment of pemphigus and pemphigoid. Br J Dermatol. 2009;161:723-31.
-
60Baum S, Greenberger S, Samuelov L, Solomon M, Lyakhovitsky A, Trau H, et al. Methotrexate is an effective and safe adjuvant therapy for pemphigus vulgaris. Eur J Dermatol. 2012;22:83-7.
-
61Tran KD, Wolverton JE, Soter NA. Methotrexate in the treatment of pemphigus vulgaris: experience in 23 patients. Br J Dermatol. 2013;169:916-21.
-
62Gürcan HM, Ahmed AR. Efficacy of dapsone in the treatment of pemphigus and pemphigoid: analysis of current data. Am J Clin Dermatol. 2009;10:383-96.
-
63Baum S, Debby A, Gilboa S, Trau H, Barzilai A. Efficacy of Dapsone in the treatment of pemphigus vulgaris: a single-center case study. Dermatology. 2016;232:578-85.
-
64Olszewska M, Kolacinska-Strasz Z, Sulej J, Labecka H, Cwikla J, Natorska U, et al. Efficacy and safety of cyclophosphamide, azathioprine, and cyclosporine (ciclosporin) as adjuvant drugs in pemphigus vulgaris. Am J Clin Dermatol. 2007;8:85-92.
-
65Chaigne B, Mouthon L. Mechanisms of action of intravenous immunoglobulin. Transfus Apher Sci. 2017;56:45-9.
-
66Svecova D. IVIG therapy in pemphigus vulgaris has corticosteroid-sparing and immunomodulatory effects. Australas J Dermatol. 2016;57:141-4.
-
67Amagai M, Ikeda S, Shimizu H, Iizuka H, Hanada K, Aiba S, et al. A randomized double-blind trial of intravenous immunoglobulin for pemphigus. J Am Acad Dermatol. 2009;60:595-603.
-
68Sinha AA, Hoffman MB, Janicke EC. Pemphigus vulgaris: approach to treatment. Eur J Dermatol. 2015;25:103-13.
-
69Eming R, Rech J, Barth S, Kalden JR, Schuler G, Harrer T, et al. Prolonged clinical remission of patients with severe pemphigus upon rapid removal of desmoglein- reactive autoantibodies by immunoadsorption. Dermatology. 2006;212:177-87.
-
70Meyersburg D, Schmidt E, Kasperkiewicz M, Zillikens D. Immunoadsorption in dermatology. Ther Apher Dial. 2012;16:311-20.
-
71Gach JE, Ilchyshyn A. Beneficial effects of topical tacrolimus on recalcitrant erosions of pemphigus vulgaris. Clin Exp Dermatol. 2004;29:271-2.
-
72Murrell DF, Dick S, Ahmed AR, Amagai M, Barnadas MA, Borradori L, et al. Consensus statement on definitions of disease, end points, and therapeutic response for pemphigus. J Am Acad Dermatol. 2008;58:1043-6.
-
73Czernik A, Bystryn JC. Kinetics of response to conventional treatment in patients with pemphigus vulgaris. Arch Dermatol. 2008;144:682-3.
-
74Martin LK, Werth V, Villanueva E, Segall J, Murrell DF. Interventions for pemphigus vulgaris and pemphigus foliaceus. Cochrane Database Syst Rev. 2009;CD006263.
-
75Herbst A, Bystryn JC. Patterns of remission in pemphigus vulgaris. J Am Acad Dermatol. 2000;42:422-7.
-
76Almugairen N, Hospital V, Bedane C, Duvert-Lehembre S, Picard D, Tronquoy AF, et al. Assessment of the rate of long-term complete remission off therapy in patients with pemphigus treated with different regimens including medium- and high-dose corticosteroids. J Am Acad Dermatol. 2013;69:583-8.
-
77Atzmony L, Hodak E, Gdalevich M, Rosenbaum O, Mimouni D. Treatment of Pemphigus Vulgaris and Pemphigus Foliaceus: A Systematic Review and Meta- Analysis. Am J Clin Dermatol. 2014;15:503-15.
-
78Diaz LA, Sampaio SA, Rivitti EA, Martins CR, Cunha PR, Lombardi C, et al. Endemic pemphigus foliaceus (fogo selvagem). I. Clinical features and immunopathology. J Am Acad Dermatol. 1989;20:657-69.
-
79Amagai M. Pemphigus. In: Bolognia JL, Lorizzo JO, Schaffer JV, editors. Dermatology. 2nd ed. Vol. 1. China: Elsevier Saunders; 2012. p.461-74.
-
80Hans-Filho G, Cunha PR. Fogo Selvagem (Endemic Pemphigus Foliaceus). In: Tyring SK, Lupi O, Hengge UR. editors. Tropical Dermatology. 2nd ed. Edinburgh: Elsevier; 2017. p. 427-32.
-
81Hans-Filho G, Aoki V, Rivitti E, Eaton DP, Lin MS, Diaz LA. Endemic pemphigus foliaceus (fogo selvagem) - 1998. The Cooperative Group on Fogo Selvagem Research. Clin Dermatol. 1999;17:225-35.
-
82Counter CE. The disease called wildfire. Arch Dermatol. 1959;80:391-7.
-
83Diaz LA, Sampaio SA, Rivitti EA, Martins CR, Cunha PR, Lombardi C, et al. Endemic pemphigus foliaceus (Fogo selvagem): II. Current and historic epidemiologic studies. J Invest Dermatol. 1989;92:4-12.
-
84Robledo MA, Prada S, Jaramillo D, Leon W. South American pemphigus: study of an epidemic in El Bagre and Nechi, Colombia 1982 to 1986. Br J Dermatol. 1988;118:737-44.
-
85Campos JA. Pênfigo foliáceo (fogo selvagem). Aspectos clínicos e epidemiológicos. São Paulo: Companhia Melhoramentos; 1942. 127p.
-
86Bastuji-Garin S, Souissi R, Blum L, Turki H, Nouira R, Jomaa B, et al. Comparative epidemiologic of pemphigus in Tunisia and France: unusual incidence of pemphigus foliaceus in young Tunisian women. J Invest Dermatol. 1995;104:302-5.
-
87Hans-Filho G, dos Santos V, Katayama JH, Aoki V, Rivitti EA, Sampaio SA, et al. An active focus of high prevalence of fogo selvagem on an Amerindian reservation in Brazil. Cooperative Group on Fogo Selvagem Research. J Invest Dermatol. 1996;107:68-75.
-
88Aoki V, Millikan RC, Rivitti EA, Hans-Filho G, Eaton DP, Warren SJ, et al. Environmental risk factors in endemic pemphigus foliaceus (fogo selvagem). J Investig Dermatol Symp Proc. 2004;9:34-40.
-
89Diaz LA, Arteaga LA, Hilario-Vargas J, Valenzuela JG, Li N, Warren S, et al. Anti-desmoglein-1 antibodies in onchocerciasis, leishmaniasis and Chagas disease suggest a possible etiological link to fogo selvagem. J Invest Dermatol. 2004;123:1045-51.
-
90Rivitti EA, Aoki V, Hans-Filho G, Vargas JH, Diaz LA. Fogo Selvagem (endemic pemphigus foliaceus). In: Tyring SK, Lupi O, Hengge UR. Tropical Dermatology. China: Elsevier Churchill Livingstone; 2006. p.421-7.
-
91Qian Y, Jeong JS, Maldonado M, Valenzuela JG, Gomes R, Teixeira C, et al. Cutting Edge: Brazilian Pemphigus Foliaceus Anti-Desmoglein 1 Autoantibodies Cross- React with Sand Fly Salivary LJM11 Antigen. J Immunol. 2012;189:1535-9.
-
92Marsden RA, Vanhegan RI, Walshe M, Hill H, Mowat AG. Pemphigus foliaceus induced by penicillamine. Br Med J. 1976;2:1423-4.
-
93Brenner S, Goldberg I. Drug-induced pemphigus. Clin Dermatol. 2011;29:455-7
-
94Moraes ME, Fernandez-Vina M, Lazaro A, Diaz LA, Filho GH, Friedman H, et al. An epitope in the third hypervariable region of the DRB1 gene is involved in the susceptibility to endemic pemphigus foliaceus (fogo selvagem) in three different Brazilian populations. Tissue Antigens. 1997;49:35-40.
-
95Piovezan BZ, Petzl-Erler ML. Both qualitative and quantitative genetic variation of MHC class II molecules may influence susceptibility to autoimmune diseases: The case of endemic pemphigus foliaceus. Hum Immunol. 2013;74:1134-40
-
96Amagai M, Klaus-Kovtun V, Stanley JR. Autoantibodies against a novel epithelial cadherin in pemphigus vulgaris, a disease of cell adhesion. Cell. 1991;67:869-77.
-
97Aoki V, Rivitti EA, Diaz LA; Cooperative Group on Fogo Selvagem Research. Update on fogo selvagem, an endemic form of pemphigus foliaceus. J Dermatol. 2015;42:18-26.
-
98Di Zenzo G, Zambruno G, Borradori L. Endemic Pemphigus Foliaceus: Towards Understanding Autoimmune Mechanisms of Disease Development. J Invest Dermatol. 2012;132:2499-502.
-
99Amagai M, Karpati S, Prussick R, Klaus-Kovtun V, Stanley JR. Autoantibodies against the amino-terminal cadherin-like binding domain of pemphigus vulgaris antigen are pathogenic. J Clin Invest. 1992;90:919-26.
-
100Evangelista F, Roth AJ, Prisayanh P, Temple BR, Li N, Qian Y, et al. Pathogenic IgG4 autoantibodies from endemic pemphigus foliaceus recognize a desmoglein-1 conformational epitope. J Autoimmun. 2018;89:171-85.
-
101Culton DA, Qian Y, Li N, Rubenstein D, Aoki V, Filhio GH, et al. Advances in pemphigus and its endemic pemphigus foliaceus (Fogo Selvagem) phenotype: A paradigm of human autoimmunity. J Autoimmun. 2008;31:311-24.
-
102Hans-Filho G, Aoki V, Bittner NRH, Bittner GC. Fogo selvagem: endemic pemphigus foliaceus. An Bras Dermatol. 2018;93:638-50.
-
103Waschke J. The desmosome and pemphigus. Histochem Cell Biol. 2008;130:21-54
-
104Beutner EH, Prigenzi LS, Hale W, Leme Cde A, Bier OG. Immunofluorescent studies of autoantibodies to intercellular areas of epithelia in Brazilian Pemphigus Foliaceus. Proc Soc Exp Biol Med. 1968;127:81-6.
-
105Sitaru C, Zillikens D. Mechanisms of blister induction by autoantibodies. Exp Dermatol. 2005;14:861-75.
-
106van der Wier G, Jonkman MF, Pas HH, Diercks GF. Ultrastructure of acantholysis in pemphigus foliaceus re-examined from the current perspective. Br J Dermatol. 2012;167:1265-71.
-
107Roscoe JT, Diaz L, Sampaio SA, Castro RM, Labib RS, Takahashi Y, et al. Brazilian pemphigus foliaceus autoantibodies are pathogenic to BALB/c mice by passive transfer. J Invest Dermatol. 1985;85:538-41.
-
108Warren SJ, Lin MS, Giudice GJ, Hoffmann RG, Hans-Filho G, Aoki V, et al. The prevalence of antibodies against desmoglein 1 in endemic pemphigus foliaceus in Brazil. N Engl J Med. 2000;343:23-30.
-
109Warren SJ, Arteaga LA, Rivitti EA, Aoki V, Hans-Filho G, Qaqish BF, et al. The Role of Subclass Switching in the Pathogenesis of Endemic Pemphigus Foliaceus. J Invest Dermatol. 2003;120:104-8.
-
110Li N, Aoki V, Hans-Filho G, Rivitti EA, Diaz LA. The role of intramolecular epitope spreading in the pathogenesis of endemic pemphigus foliaceus (fogo selvagem). J Exp Med. 2003;197:1501-10.
-
111Yoshida K, Ishii K, Shimizu A, Yokouchi M, Amagai M, Shiraishi K, et al. Non-pathogenic pemphigus foliaceus (PF) IgG acts synergistically with a directly pathogenic PF IgG to incrase blistering by p38MAPK-dependent desmoglein 1 clustering. J Dermatol Sci. 2017;85:197-207.
-
112Grando SA. Pemphigus autoimmunity: hypotheses and realities. Autoimmunity. 2012;45:7-35.
-
113Diaz LA, Prisayanh PS, Dasher DA, Li N, Evangelista F, Aoki V, et al. The IgM anti-desmoglein 1 response distinguishes Brazilian pemphigus foliaceus (fogo selvagem) from other forms of pemphigus. J Invest Dermatol. 2008;128:667-75
-
114Qaqish BF, Prisayanh P, Qian Y, Andraca E, Li N, Aoki V, et al. Development of na IgG4-based predictor of endemic pemphigus foliaceus (fogo selvagem). J Invest Dermatol. 2009;129:110-8.
-
115Hilario-Vargas J, Dasher DA, Li N, Aoki V, Hans-Filho G, dos Santos V, et al. Prevalence of anti-desmogleina-3 antibodies in endemic regions of fogo selvagem in Brazil. J Invest Dermatol. 2006;126:2044-8.
-
116Rocha-Rodrigues DB, Paschoini G, Pereira SA, dos Reis MA, Teixeira Vde P, Rodrigues Júnior V. High levels of interleukin-1 in patiens with endemic pemphigus foliaceus. Clin Diagn Lab Immunol. 2003;10:741-3.
-
117Chriguer RS, Roselino AM, de Castro M. Glucocorticoid sensitivity and proinflammatory cytokines pattern in pemphigus. J Clin Immunol. 2012;32:786-93.
-
118López-Robles E, Avalos-Díaz E, Vega-Memije E, Hojyo-Tomoka T, Villalobos R, Fraire S, et al. TNF-a end IL-6 are mediator in the blisterin process of pemphigus. Int J Dermatol. 2001;40:185-8.
-
119Timóteo RP, Silva MV, da Silva DAA, Catarino JDS, Alves FHC, Rodrigues Júnior V, et al. Cytokine and chemokines alterations in the form of pemphigus folicaceus (fogo selvagem). Front Immunol. 2017;8:978.
-
120Arakawa M, Dainichi T, Yasumoto S, Hashimoto T. Lesional Th 17 cells in pemphigus vulgaris and pemphigus foliaceus. J Dermatol Sci. 2009;53:228-31.
-
121Juneja M. Nikolsky's sign revisited. Journal of Oral Science. 2008;50:213-4.
-
122James KA, Culton DA, Diaz LA. Diagnosis and clinical features of pemphigus foliaceus. Dermatol Clin. 2011;29:405-12.
-
123Avalos-Díaz E, Olague-Marchan M, López-Swiderski A, Herrera-Esparza R, Díaz LA. Transplacental passage of maternal pemphigus foliaceus autoantibodies induces neonatal pemphigus. J Am Acad Dermatol. 2000;43:1130-4.
-
124Zhao CY, Chiang YZ, Murrell DF. Neonatal Autoimmune Blistering Disease: A Systematic Review. Pediatr Dermatol. 2016;33:367-74.
-
125Lin L, Zeng X, Chen Q. Pemphigus and pregnancy. Saudi Med J. 2015;36:1033-8.
-
126Lorente Lavirgen AI, Bernabeu-Wittel J, Dominguez-Cruz J, Conejo-Mir J. Neonatal pemphigus foliaceus. J Pediatr. 2012;161:768.
-
127Payne AS, Stanley JR. Pemphigus. In: Goldsmith LA, Katz SI, Gilchrest BA, Peller AS, Lefflell DJ, Wolf K, editors. Fitzpatrick's Dermatology in General Medicine. 8th ed. Vol 1. New York: McGraw Hill; 2012. p.586.
-
128Brenner S, Bialy-Golan A, Ruocco V. Drug-induced pemphigus. Clin Dermatol. 1998;16:393-7.
-
129Arbache ST, Nogueira TG, Delgado L, Miyamoto D, Aoki V. Immunofluorescence testing in the diagnosis of autoimmune blistering diseases: overview of 10-year experience. An Bras Dermatol. 2014;89:885-9.
-
130Kasperkiewicz M, Schmidt E, Zillikens D.. Current therapy of the pemphigus group. Clin Dermatol. 2012;30:84-94.
-
131Mutasim DF. Management of autoimmune bullous diseases: Pharmacology and therapeutics. J Am Acad Dermatol. 2004;51:859-77.
-
132Schmidt E, Zillikens D. Immunoadsorption in dermatology. Arch Dermatol Res. 2010;302:241-53.
-
133Uptodate.com [Internet]. Hertl M, Geller S. Inicial management of pemphigus vulgaris and pemphigus foliaceus. UpToDate Mar 2017. [cited 2018 Jun 08]. Available from: https://www.uptodate.com/contents/initial-management-of-pemphigus-vulgaris-and-pemphigus-foliaceus
» https://www.uptodate.com/contents/initial-management-of-pemphigus-vulgaris-and-pemphigus-foliaceus -
134Karnofsky DA, Burchenal JH. The Clinical Evaluation of Chemotherapeutic Agents in Cancer. In: MacLeod CM, editor. Evaluation of Chemotherapeutic Agents. New York: Columbia Univ Press; 1949. p. 196.
-
135Daniel BS, Hertl M, Werth VP, Eming R, Murrell DF. Severity score indexes for blistering diseases. Clin Dermatol. 2012;30:108-13.
-
136Sebaratnam DF, Murrell DF. Objective scoring systems for disease activity in autoimmune bullous disease. Dermatol Clin. 2011;29:515-20, xi.
Publication Dates
-
Publication in this collection
03 June 2019 -
Date of issue
Mar-Apr 2019
History
-
Received
14 Sept 2018 -
Accepted
30 Jan 2019