Abstract:
Bullous pemphigoid, mucous membrane pemphigoid and epidermolysis bullosa acquisita are subepidermal autoimmune blistering diseases whose antigenic target is located at the basement membrane zone. Mucous membrane pemphigoid and epidermolysis bullosa acquisita can evolve with cicatricial mucosal involvement, leading to respiratory, ocular and/or digestive sequelae with important morbidity. For each of these dermatoses, a literature review covering all therapeutic options was performed. A flowchart, based on the experience and joint discussion among the authors of this consensus, was constructed to provide treatment orientation for these diseases in Brazil. In summary, in the localized, low-risk or non-severe forms, drugs that have immunomodulatory action such as dapsone, doxycycline among others may be a therapeutic option. Topical treatment with corticosteroids or immunomodulators may also be used. Systemic corticosteroid therapy continues to be the treatment of choice for severe forms, especially those involving ocular, laryngeal-pharyngeal and/or esophageal mucosal involvement, as may occur in mucous membrane pemphigoid and epidermolysis bullosa acquisita. Several immunosuppressants are used as adjuvant alternatives. In severe and recalcitrant cases, intravenous immunoglobulin is an alternative that, while expensive, may be used. Immunobiological drugs such as rituximab are promising drugs in this area. Omalizumab has been used in bullous pemphigoid.
Keywords:
Autoimmunity; Basement membrane; Skin diseases
BULLOUS PEMPHIGOID
INTRODUCTION
Bullous pemphigoid (BP) accounts for 80% of subepidermal bullous dermatoses. It typically presents as generalized pruritus and tense bullous eruptions and may be associated with a 20% to 40% mortality rate. It primarily affects those aged above 70 years, and its annual incidence is estimated to be at least 13 to 62 new cases per million persons, rising significantly after 60 years old.11 Aoki V, Maruta W, Santi CG. Dermatoses Vesicobolhosas Autoimunes. In: Belda Jr. W, Di Chiacchio N, Criado PR. Tratado de Dermatologia. 2. ed. São Paulo: Atheneu; 2014. p. 417-28.
2 Alpsoy E, Akman-Karakas A, Uzun S. Geographic variations in epidemiology of two autoimmune bullous diseases: pemphigus and bullous pemphigoid. Arch Dermatol Res. 2015;307:291-8.-33 Langan SM, Smeeth L, Hubbard R, Fleming KM, Smith CJ, West J. Bullous pemphigoid and pemphigus vulgaris-incidence and mortality in the UK: population based cohort study. BMJ. 2008;337:a180.
ETIOPATHOGENESIS
In the etiopathogenesis of BP, IgG autoantibodies are produced against antigens of the basement membrane zone (BMZ)-180kDa and 230kDa -collagen XVII, COL17 or BPAg2, and BPAg1, respectively. BP180 is the most relevant antigenic determinant, whereas BP230 appears to be more related to cytoskeletal function and signaling of the dermoepidermal transition. The involvement of mast cells and IgE in the development of BP lesions has recently been described.44 Christophoridis S, Büdinger L, Borradori L, Hunziker T, Merk HF, Hertl M. IgG, IgA and IgE autoantibodies against the ectodomain of BP180 in patients with bullous and cicatricial pemphigoid and linear IgA bullous dermatosis. Br J Dermatol. 2000;143:349-55.
5 Dimson OG, Giudice GJ, Fu CL, Van den Bergh F, Warren SJ, Janson MM, et al. Identification of a potential effector function for IgE autoantibodies in the organ-specific autoimmune disease bullous pemphigoid. J Invest Dermatol. 2003;120:784-8.
6 Fang H, Zhang Y, Li N, Wang G, Liu Z. The Autoimmune Skin Disease Bullous Pemphigoid: The Role of Mast Cells in Autoantibody-Induced Tissue Injury. Front Immunol. 2018;9:407
7 Freire PC, Muñoz CH, Stingl G. IgE autoreactivity in bullous pemphigoid: eosinophils and mast cells as major targets of pathogenic immune reactants. Br J Dermatol. 2017;177:1644-53.
8 Hammers CM, Stanley JR. Mechanisms of Disease: Pemphigus and Bullous Pemphigoid. Annu Rev Pathol. 2016;11:175-97.
9 Hammers CM, Payne AS. Clinical significance of immunoglobulin E in bullous pemphigoid. Br J Dermatol. 2017;177:13-4.
10 Hashimoto T. Induced autoimmune bullous diseases. Br J Dermatol. 2017;176:304-5
11 Hashimoto T, Tsuruta D, Ishii N. Immunoglobulin E Autoantibodies in Bullous Pemphigoid Detected by Immunoglobulin E Enzyme-Linked Immunosorbent Assays. JAMA Dermatol. 2017;153:15-7.
12 Hashimoto T, Ohzono A, Teye K, Numata S, Hiroyasu S, Tsuruta D, et al. Detection of IgE autoantibodies to BP180 and BP230 and their relationship to clinical features in bullous pemphigoid. Br J Dermatol. 2017;177:141-51.
13 Liu Z, Chen L, Zhang C, Xiang LF. Circulating anti-bullous pemphigoid 180 autoantibody can be detected in a wide clinical spectrum: A cross-sectional study. J Am Acad Dermatol. 2018. pii: S0190-9622(18)32067-X
14 Liu Y, Li L, Xia Y. BP180 Is Critical in the Autoimmunity of Bullous Pemphigoid. Front Immunol. 2017;8:1752
15 Lin L, Hwang BJ, Culton DA, Li N, Burette S, Koller BH, et al. Eosinophils Mediate Tissue Injury in the Autoimmune Skin Disease Bullous Pemphigoid. J Invest Dermatol. 2018;138:1032-43.
16 Messingham KN, Holahan HM, Frydman AS, Fullenkamp C, Srikantha R, Fairley JA. Human eosinophils express the high affinity IgE receptor, FceRI, in bullous pemphigoid. PLoS One. 2014;9:e107725.
17 Moriuchi R, Nishie W, Ujiie H, Natsuga K, Shimizu H. In vivo analysis of IgE autoantibodies in bullous pemphigoid: a study of 100 cases. J Dermatol Sci. 2015;78:21-5
18 Nishie W. Update on the pathogenesis of bullous pemphigoid: an autoantibody- mediated blistering disease targeting collagen XVII. J Dermatol Sci. 2014;73:179-86-1919 Saniklidou AH, Tighe PJ, Fairclough LC, Todd I. IgE autoantibodies and their association with the disease activity and phenotype in bullous pemphigoid: a systematic review. Arch Dermatol Res. 2018;310:11-28.
The association of malignancies with BP is likely to be related to the incidence of both diseases in the elderly, although some reports have suggested an increase in the frequency of certain cancers, such as those in the digestive tract, lung, and bladder and lymphoproliferative diseases.2020 Ren Z, Hsu DY, Brieva J, Silverberg NB, Langan SM, Silverberg JI. Hospitalization, inpatient burden and comorbidities associated with bullous pemphigoid in the U.S.A. Br J Dermatol. 2017;176:87-99.,2121 Lucariello RJ, Villablanca SE, Mascaró JM Jr, Reichel M. Association between bullous pemphigoid and malignancy: A meta-analysis. Australas J Dermatol. 2018. doi: 10.1111/ajd.12764.
https://doi.org/10.1111/ajd.12764....
BP is rarely described in patients with inflammatory or autoimmune diseases. In certain patients, it appears to be triggered by trauma, burns, radiotherapy, or irradiation by ultraviolet rays. BP has also been observed in association with psoriasis and lichen planus.2222 Lo Schiavo A, Ruocco E, Brancaccio G, Caccavale S, Ruocco V, Wolf R. Bullous pemphigoid: etiology, pathogenesis, and inducing factors: facts and controversies. Clin Dermatol. 2013;31:391-9.
In some patients, systemic medications may lead to the development of BP, including diuretics (furosemide), antibiotics (amoxicillin, ciprofloxacin), potassium iodide, and captopril. The mechanism by which drugs induce BP has not been determined.2323 Bernard P, Borradori. Pemphigoid Group. In: Bolognia J.L, Jorizzo JL, Schaffer JV, editors. Dermatology. 3rd ed. Philadelphia, Pa.: Elsevier Saunders; 2012: p. 475-90.
Of greater interest, BP has been associated with neurological diseases, such as dementia, stroke, multiple sclerosis, epilepsy, and Parkinson disease, but the underlying pathophysiological mechanism is not completely understood. It is possible that the BP antigens BPA1 and BPA2 act as autoreactive antigens in the central nervous system and in tegument.2424 Bastuji-Garin S, Joly P, Lemordant P, Sparsa A, Bedane C, Delaporte E, et al. Risk factors for bullous pemphigoid in the elderly: a prospective case-control study. J Invest Dermatol. 2011;131:637-43.
25 Jedlickova H, Hlubinka M, Pavlik T, Semradova V, Budinska E, Vlasin Z.. Bullous pemphigoid and internal diseases - A case-control study. Eur J Dermatol. 2010;20:96-101.
26 Cordel N, Chosidow O, Hellot MF, Delaporte E, Lok C, Vaillant L, et al. Neurological disorders in patients with bullous pemphigoid. Dermatology. 2007;215:187-91.
27 Lai YC, Yew YW, Lambert WC. Bullous pemphigoid and its association with neurological diseases: a systematic review and meta-analysis. J Eur Acad Dermatol Venereol. 2016;30:2007-15.
28 Tarazona MJ, Mota AN, Gripp AC, Unterstell N, Bressan AL. Bullous pemphigoid and neurological disease: statistics from a dermatology service. An Bras Dermatol. 2015;90:280-2.
29 Teixeira VB, Cabral R, Brites MM, Vieira R, Figueiredo A. Bullous pemphigoid and comorbidities: a case-control study in Portuguese patients. An Bras Dermatol. 2014;89:274-8.
30 Taghipour K, Chi CC, Bhogal B, Groves RW, Venning V, Wojnarowska F. Immunopathological characteristics of patients with bullous pemphigoid and neurological disease. J Eur Acad Dermatol Venereol. 2014;28:569-73.
31 Vernal S, Julio T, Cruz F, Turatti A, Ishii N, Hashimoto T, et al. Bullous Pemphigoid Associated with Ischemic Cerebrovascular Accident and Dementia: Exclusive Blistering Lesions on the Upper Hemiparetic Limb. Acta Dermatovenerol Croat. 2018;26:179-82.
32 Milani-Nejad N, Zhang M, Kaffenberger J. The association between bullous pemphigoid and neurological disorders: a systematic review. Eur J Dermatol. 2017;27:472-81.
33 Försti AK, Huilaja L, Schmidt E, Tasanen K. Neurological and psychiatric associations in bullous pemphigoid-more than skin deep? Exp Dermatol. 2017;26:1228-34.
34 Ali A, Hu L, Zhao F, Qiu W, Wang P, Ma X, et al. BPAG1, a distinctive role in skin and neurological diseases. Semin Cell Dev Biol. 2017;69:34-96
35 Kokkonen N, Herukka SK, Huilaja L, Kokki M, Koivisto AM, Hartikainen P, et al. Increased Levels of the Bullous Pemphigoid BP180 Autoantibody Are Associated with More Severe Dementia in Alzheimer's Disease. J Invest Dermatol. 2017;137:71-6.-3636 Julio TA, Vernal S, Massaro JD, Silva MC, Donadi EA, Moriguti JC, et al. Biological predictors shared by dementia and bullous pemphigoid patients point out a cross-antigenicity between BP180/BP230 brain and skin isoforms. Immunol Res. 2018;66:567-76.
The HLA class II DQB1*03:01 allele is more prevalent in Caucasian patients with BP than in the general population and has also been associated with neurological diseases.3737 Chagury AA, Sennes LU, Gil JM, Kalil J, Rodrigues H, Rosales CB, et al. HLA-C*17, DQB1*03:01, DQA1*01:03 and DQA1*05:05 Alleles Associated to Bullous Pemphigoid in Brazilian Population. Ann Dermatol. 2018;30:8-12.,3838 Amber KT, Zikry J, Hertl M. A multi-hit hypothesis of bullous pemphigoid and associated neurological disease: Is HLA-DQB1*03:01, a potential link between immune privileged antigen exposure and epitope spreading? HLA. 2017;89:127-34
CLINICAL CONDITION
The tegumentary manifestations of BP are extremely polymorphic. In the non-blistering phase, the signs and symptoms are usually nonspecific, presenting as isolated pruritus, sometimes intractable or associated with excoriations, papules, or urticarial lesions, which may persist from weeks to months. Vesicles or blisters on apparently normal or erythematous basis, in addition to infiltrated papules and plaques that occasionally assume annular and figurative patterns, can characterize the bullous phase of BP. The blisters are smooth, with diameters ranging from 1 to 4cm, containing clear fluid, leaving crusted and eroded areas. The lesions often have a symmetrical distribution, predominating in the limb flexures and chest. Localized involvement is uncommon.3131 Vernal S, Julio T, Cruz F, Turatti A, Ishii N, Hashimoto T, et al. Bullous Pemphigoid Associated with Ischemic Cerebrovascular Accident and Dementia: Exclusive Blistering Lesions on the Upper Hemiparetic Limb. Acta Dermatovenerol Croat. 2018;26:179-82. Oral involvement is observed between 10% and 30% of affected patients. The eyes, nose, pharynx, esophagus, and anogenital regions are rarely affected.11 Aoki V, Maruta W, Santi CG. Dermatoses Vesicobolhosas Autoimunes. In: Belda Jr. W, Di Chiacchio N, Criado PR. Tratado de Dermatologia. 2. ed. São Paulo: Atheneu; 2014. p. 417-28.,1818 Nishie W. Update on the pathogenesis of bullous pemphigoid: an autoantibody- mediated blistering disease targeting collagen XVII. J Dermatol Sci. 2014;73:179-86,2222 Lo Schiavo A, Ruocco E, Brancaccio G, Caccavale S, Ruocco V, Wolf R. Bullous pemphigoid: etiology, pathogenesis, and inducing factors: facts and controversies. Clin Dermatol. 2013;31:391-9.
23 Bernard P, Borradori. Pemphigoid Group. In: Bolognia J.L, Jorizzo JL, Schaffer JV, editors. Dermatology. 3rd ed. Philadelphia, Pa.: Elsevier Saunders; 2012: p. 475-90.-2424 Bastuji-Garin S, Joly P, Lemordant P, Sparsa A, Bedane C, Delaporte E, et al. Risk factors for bullous pemphigoid in the elderly: a prospective case-control study. J Invest Dermatol. 2011;131:637-43.,3939 Schmidt E, Zillikens D. Pemphigoid diseases. Lancet. 2013;381:320-32.
LABORATORY DIAGNOSIS
The diagnosis of BP is based on clinical characteristics and, more importantly, direct immunofluorescence (DIF) and indirect immunofluorescence (IIF) findings by microscopy; the presence of antibodies against the BP180 peptide by enzyme-linked immunosorbent assay (ELISA) confirms the diagnosis. In most cases, immunofluorescence is essential and sufficient for the correct classification of subepidermal bullous dermatoses. Nevertheless, particularly in patients with negative DIF and IIF findings, additional immunoblotting assays are useful and required to demonstrate the presence of autoantibodies in the patient’s serum, based on disease target antigens (BP180 and BP230). In the absence of the onset of bullous lesions, such as in the early stages or atypical variants of the disease, the diagnosis of BP depends solely on positive results by DIF or IIF.3939 Schmidt E, Zillikens D. Pemphigoid diseases. Lancet. 2013;381:320-32.
40 Arbache ST, Nogueira TG, Delgado L, Miyamoto D, Aoki V. Immunofluorescence testing in the diagnosis of autoimmune blistering diseases: overview of 10-year experience. An Bras Dermatol. 2014;89:885-9.-4141 Ishii K. Importance of serological tests in diagnosis of autoimmune blistering diseases. J Dermatol. 2015;42:3-10.
In most patients, DIF of non-impaired perilesional skin will demonstrate linear and continuous IgG or C3 deposition along the BMZ. Furthermore, the salt-split skin (SSS) IIF technique, when treating normal human skin with 1 M NaCl, followed by incubation with the patient’s serum, may be useful in distinguishing BP from other subepidermal bullous autoimmune diseases, primarily epidermolysis bullosa acquisita and bullous lupus erythematosus.4040 Arbache ST, Nogueira TG, Delgado L, Miyamoto D, Aoki V. Immunofluorescence testing in the diagnosis of autoimmune blistering diseases: overview of 10-year experience. An Bras Dermatol. 2014;89:885-9.,4141 Ishii K. Importance of serological tests in diagnosis of autoimmune blistering diseases. J Dermatol. 2015;42:3-10. In BP, immune deposits are found on the epidermal side or on both sides of the cleavage (dermal and epidermal). In 60% to 80% of patients, IgG autoantibodies and, less frequently, IgA and IgE are circulating; these autoantibodies also often bind to the cleavage on the epidermal side.2323 Bernard P, Borradori. Pemphigoid Group. In: Bolognia J.L, Jorizzo JL, Schaffer JV, editors. Dermatology. 3rd ed. Philadelphia, Pa.: Elsevier Saunders; 2012: p. 475-90.
ELISA using recombinant proteins that contain specific regions of BP antigens (eg, BP180 domain NC16A and the C-terminal portion of BP180 or BP230) is useful for the BP diagnosis. The determination of antibodies against BP180 and BP230 may be necessary to confirm the diagnosis; rarely, BP patients have antibodies against desmogleins-ie, pemphigus antigens.3131 Vernal S, Julio T, Cruz F, Turatti A, Ishii N, Hashimoto T, et al. Bullous Pemphigoid Associated with Ischemic Cerebrovascular Accident and Dementia: Exclusive Blistering Lesions on the Upper Hemiparetic Limb. Acta Dermatovenerol Croat. 2018;26:179-82.,4141 Ishii K. Importance of serological tests in diagnosis of autoimmune blistering diseases. J Dermatol. 2015;42:3-10.,4242 Julio T, Vernal S, Turatti A, Roselino AM. Anti-desmogleins autoantibodies detected by ELISA and blotting in bullous pemphigoid: what do they mean? Int J Dermatol. 2018;57:124-7 Serology may also be used for therapeutic follow-up, especially at the time of treatment discontinuation.4343 Feliciani C, Joly P, Jonkman MF, Zambruno G, Zillikens D, Ioannides D, et al. Management of bullous pemphigoid: the European Dermatology Forum consensus in collaboration with the European Academy of Dermatology and Venereology. Br J Dermatol. 2015;172:867-77.
TREATMENT
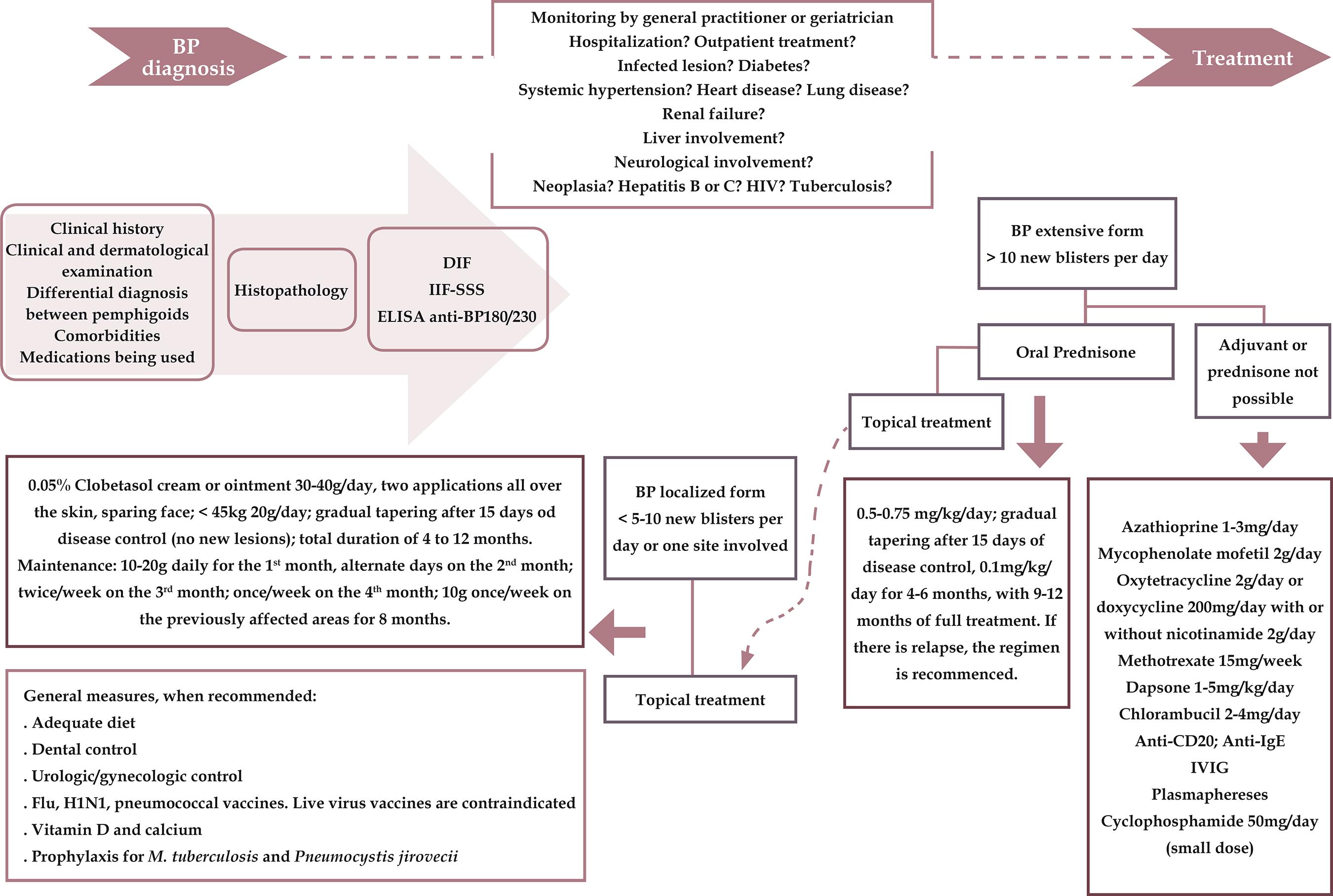
The treatment of BP is based more on clinical experience than on controlled studies. For the extensive form, systemic corticosteroids are the main option, wherein a regimen of oral prednisone of 0.5 to 0.75mg/kg/day is recommended. Large controlled studies have reported the value of potent topical corticosteroids, which also appear to control generalized BP with similar efficacy as systemic corticosteroids and with few side effects.2323 Bernard P, Borradori. Pemphigoid Group. In: Bolognia J.L, Jorizzo JL, Schaffer JV, editors. Dermatology. 3rd ed. Philadelphia, Pa.: Elsevier Saunders; 2012: p. 475-90.,4343 Feliciani C, Joly P, Jonkman MF, Zambruno G, Zillikens D, Ioannides D, et al. Management of bullous pemphigoid: the European Dermatology Forum consensus in collaboration with the European Academy of Dermatology and Venereology. Br J Dermatol. 2015;172:867-77. However, the refractory nature of some patients to corticosteroids merits further study.4444 Brulefert A, Le Jan S, Plée J, Durlach A, Bernard P, Antonicelli F, et al. Variation of the epidermal expression of glucocorticoid receptor-beta as potential predictive marker of bullous pemphigoid outcome. Exp Dermatol. 2017;26:1261-6.,4545 Kubin ME, Hellberg L, Palatsi R. Glucocorticoids: The mode of action in bullous pemphigoid. Exp Dermatol. 2017;26:1253-60.
Eventually, pulse therapy with methylprednisone may be required to control the disease more rapidly. The use of immunosuppressive drugs has been discussed; certain groups advocate their use only as second-line therapy, when monotherapy with corticosteroids fails or if there is any contraindication. The most frequently used agents are azathioprine, mycophenolate mofetil, methotrexate, chlorambucil, and cyclophosphamide.2323 Bernard P, Borradori. Pemphigoid Group. In: Bolognia J.L, Jorizzo JL, Schaffer JV, editors. Dermatology. 3rd ed. Philadelphia, Pa.: Elsevier Saunders; 2012: p. 475-90.,4343 Feliciani C, Joly P, Jonkman MF, Zambruno G, Zillikens D, Ioannides D, et al. Management of bullous pemphigoid: the European Dermatology Forum consensus in collaboration with the European Academy of Dermatology and Venereology. Br J Dermatol. 2015;172:867-77. Low doses of cyclophosphamide may have benefits.4646 Gual A, Iranzo P, Mascaró JM Jr. Treatment of bullous pemphigoid with low- dose oral cyclophosphamide: a case series of 20 patients. J Eur Acad Dermatol Venereol. 2014;28:814-8. The combination of nicotinamide and minocycline or tetracycline can be used as a therapeutic alternative.
Dapsone can be administered, especially when there is mucosal involvement, and the value of topical immunomodulators, such as tacrolimus, should be confirmed.2323 Bernard P, Borradori. Pemphigoid Group. In: Bolognia J.L, Jorizzo JL, Schaffer JV, editors. Dermatology. 3rd ed. Philadelphia, Pa.: Elsevier Saunders; 2012: p. 475-90.,4343 Feliciani C, Joly P, Jonkman MF, Zambruno G, Zillikens D, Ioannides D, et al. Management of bullous pemphigoid: the European Dermatology Forum consensus in collaboration with the European Academy of Dermatology and Venereology. Br J Dermatol. 2015;172:867-77.,4747 Schmidt E, Spindler V, Eming R, Amagai M, Antonicelli F, Baines JF, et al. Meeting Report of the Pathogenesis of Pemphigus and Pemphigoid Meeting in Munich, September 2016. J Invest Dermatol. 2017;137:1199-203. In cases of treatment resistance, intravenous immunoglobulin, plasmapheresis, or anti-CD20 immunotherapy (rituximab) may be prescribed. Based on the immunopathogenesis of BP, new compounds are being proposed.4747 Schmidt E, Spindler V, Eming R, Amagai M, Antonicelli F, Baines JF, et al. Meeting Report of the Pathogenesis of Pemphigus and Pemphigoid Meeting in Munich, September 2016. J Invest Dermatol. 2017;137:1199-203.
48 Shetty S, Ahmed AR. Treatment of bullous pemphigoid with rituximab: critical analysis of the current literature. J Drugs Dermatol. 2013 ;12:672-7.-4949 Ludwig RJ, Kalies K, Köhl J, Zillikens D, Schmidt E. Emerging treatments for pemphigoid diseases. Trends Mol Med. 2013;19:501-12.
Recently, a European consensus was developed for the treatment of BP.4343 Feliciani C, Joly P, Jonkman MF, Zambruno G, Zillikens D, Ioannides D, et al. Management of bullous pemphigoid: the European Dermatology Forum consensus in collaboration with the European Academy of Dermatology and Venereology. Br J Dermatol. 2015;172:867-77. The figure 1 that is presented here depicts this consensus with modifications, in addition to the revisions that have been incorporated for the treatment of BP. 4343 Feliciani C, Joly P, Jonkman MF, Zambruno G, Zillikens D, Ioannides D, et al. Management of bullous pemphigoid: the European Dermatology Forum consensus in collaboration with the European Academy of Dermatology and Venereology. Br J Dermatol. 2015;172:867-77.
44 Brulefert A, Le Jan S, Plée J, Durlach A, Bernard P, Antonicelli F, et al. Variation of the epidermal expression of glucocorticoid receptor-beta as potential predictive marker of bullous pemphigoid outcome. Exp Dermatol. 2017;26:1261-6.
45 Kubin ME, Hellberg L, Palatsi R. Glucocorticoids: The mode of action in bullous pemphigoid. Exp Dermatol. 2017;26:1253-60.
46 Gual A, Iranzo P, Mascaró JM Jr. Treatment of bullous pemphigoid with low- dose oral cyclophosphamide: a case series of 20 patients. J Eur Acad Dermatol Venereol. 2014;28:814-8.
47 Schmidt E, Spindler V, Eming R, Amagai M, Antonicelli F, Baines JF, et al. Meeting Report of the Pathogenesis of Pemphigus and Pemphigoid Meeting in Munich, September 2016. J Invest Dermatol. 2017;137:1199-203.
48 Shetty S, Ahmed AR. Treatment of bullous pemphigoid with rituximab: critical analysis of the current literature. J Drugs Dermatol. 2013 ;12:672-7.-4949 Ludwig RJ, Kalies K, Köhl J, Zillikens D, Schmidt E. Emerging treatments for pemphigoid diseases. Trends Mol Med. 2013;19:501-12.
MUCOUS MEMBRANE PEMPHIGOID
INTRODUCTION
Mucous membrane pemphigoid (MMP), also known as cicatricial pemphigoid, refers to a group of chronic autoimmune diseases that predominantly affect the mucous membranes and, occasionally, skin.5050 Lazarova Z, Yancey K. Cicatricial pemphigoid: immunopathogenesis and treatment. Derm Ther. 2002;15:382-8. It is part of a group of autoimmune dermatoses that present with subepidermal bullous lesions, characterized by the formation of antibodies against structures of the basement membrane zone (BMZ). In MMP, antibodies bind more frequently to BP-180 and laminin 332 (laminin 5).5151 Kridin K. Subepidermal autoimmune bullous diseases: overview, epidemiology, and associations. Immunol Res. 2018;66:6-17. Scarring of lesions on the mucous membranes is common-hence, the term “cicatricial”-which may lead to decreased visual acuity, blindness, supraglottic stenosis with hoarseness, or airway obstruction.5050 Lazarova Z, Yancey K. Cicatricial pemphigoid: immunopathogenesis and treatment. Derm Ther. 2002;15:382-8.
EPIDEMIOLOGY
MMP usually has a late onset, between age 60 and 80 years. Women appear to be more commonly affected than men, from 1.5 to 2 times more frequently, with no racial or geographic preference. European data show an incidence of approximately 1:1,000,000 people annually. The initial signs and symptoms may be subtle and non-specific, with irreversible and often debilitating consequences. It is associated with high morbidity and mortality if treatment is not initiated early and aggressively.5252 Kourosh AS, Yancey KB. Pathogenesis of mucous membrane pemphigoid. Dermatol Clin. 2011;29:479-84
ETIOPATHOGENESIS
MMP is characterized by the linear deposition of IgG, IgA, or C3 along the epithelial basement membrane area.53 Current evidence suggests that this process develops as a consequence of the loss of immune tolerance to structural proteins of the epidermal basement membrane, culminating in the development of circulating autoantibodies that bind to the epidermal basement membrane, causing inflammation and weakening the adhesion of the overlying epidermis.5252 Kourosh AS, Yancey KB. Pathogenesis of mucous membrane pemphigoid. Dermatol Clin. 2011;29:479-84
Studies have shown that the target antigens in the epithelial basement membrane area include bullous pemphigoid antigen 1 (BP230), bullous pemphigoid antigen 2 (BP180), laminin 5 (α3, β3, γ2 chains), laminin 6 (α3 chain), type VII collagen, and the β4 integrin subunit.5353 Chan LS, Ahmed AR, Anhalt GJ, Bernauer W, Cooper KD, Elder MJ, et al. The first international consensus on mucous membrane pemphigoid: definition, diagnostic criteria, pathogenic factors, medical treatment, and prognostic indicators. Arch Dermatol. 2002;138:370-9. BP180 is a transmembrane protein that crosses the lamina lucida and protrudes into the dense lamina of the epidermal basement membrane zone; it is usually the target antigen in approximately 70% of MMP patients. Serum from patients with generalized and ocular MMP recognizes the β-4 integrin subunit, whereas serum from those with oral MMP recognizes the α-6 integrin subunit.
The pathogenicity of anti-laminin 332 has been documented in vivo. Passive transfer of anti-laminin 332 IgG to mice induces subepidermal blistering on the skin and mucous membranes, mimicking the clinical, histopathological, and immunological characteristics of patients with MMP.5454 Kasperkiewicz M, Zillikens D, Schmidt E. Pemphigoid diseases: pathogenesis, diagnosis, and treatment. Autoimmunity. 2012;45:55-70. Fibroblasts also appear to be activated, secondary to the production of cytokines, such as transforming growth factor beta, which is known to induce fibrosis. The collagen that is produced can lead to scars. Several studies have shown a predominance of CD4 T lymphocyte and Langerhans cell infiltrates in the conjunctiva of patients with MMP, indicating the involvement of cellular immunity in its pathogenesis. Studies have linked MMP and the human leukocyte antigen (HLA) class II HLA-DQB1 * 0301 allele.5555 Xu HH, Werth VP, Parisi E, Sollecito TP. Mucous Membrane Pemphigoid. Dent Clin North Am. 2013;57:611-30.
CLINICAL CONDITION
MPP can affect the mucous membrane from several sites, occasionally with skin involvement. It is a chronic, progressive condition that most often affects the oral mucosa (85% of patients), followed by the ocular conjunctiva (65%), nasal mucosa (20-40%), skin (25-30%), anogenital region and pharynx (20%), larynx (5-15%), and esophagus (5-15%). There is a wide range in the variability and severity of presentations among patients, who can present with localized and generalized involvement. Those who present with only oral mucosal or skin involvement with a lower tendency toward scarring are defined as “low risk,” whereas in patients who are at “high risk,” the disease occurs in general ocular, esophageal, laryngeal, nasopharyngeal, and anogenital mucosae. The propensity for generating scars in these sites is associated with a poor prognosis, despite treatment.5656 Chan LS. Mucous membrane pemphigoid. Clin Dermatol. 2001;19:703-11.
Involvement of the oral mucosa typically manifests as erythematous plaques and erosions that are covered by pseudomembranes, most commonly located on the gum and palate and less frequently on the lips, tongue, and cheek mucosa. Gingival lesions are often described as desquamative gingivitis, also found in lichen planus and pemphigus vulgaris. The most common ocular lesions are conjunctival inflammation and erosions, forniceal shortening with formation of symblepharon, trichiasis, entropion, and cornea neovascularization, which may result in blindness. The most frequently affected skin areas are the scalp, face, neck, and upper chest. Lesions usually present as erythematous plaques, with the formation of recurrent blisters and erosions and consequent atrophic scarring.5555 Xu HH, Werth VP, Parisi E, Sollecito TP. Mucous Membrane Pemphigoid. Dent Clin North Am. 2013;57:611-30. Scarring of the laryngeal mucosa can lead to hoarseness, cough, dyspnea, and even acute respiratory failure. Scarring of the anogenital mucosa usually presents as blisters, erosions, and scars and can significantly affect a patient’s quality of life.56 Esophageal involvement in MMP is rare and is observed in patients with disseminated disease. The most common changes that occur are multiple membranes and esophageal constriction.5555 Xu HH, Werth VP, Parisi E, Sollecito TP. Mucous Membrane Pemphigoid. Dent Clin North Am. 2013;57:611-30.,5656 Chan LS. Mucous membrane pemphigoid. Clin Dermatol. 2001;19:703-11.
A diagnosis of MMP should be considered in patients with bullous lesions or erosions that compromise predominantly mucous membranes. The main differential diagnoses in MMP include bullous pemphigoid, epidermolysis bullosa acquisita, and linear IgA dermatosis. When there is oral involvement, MMP lesions should be differentiated from pemphigus vulgaris, bullous pemphigoid, erythema multiforme, and erosive lichen planus. Ocular cicatricial lesions in MMP should be distinguished from the scarring lesions of severe chronic infectious conjunctivitis, ophthalmic solution-induced ocular pseudopemphigoid, and scarring lesions in more severe cases of Stevens-Johnson syndrome and toxic epidermal necrolysis. Generalized skin bullous lesions are impossible to distinguish from those in bullous pemphigoid; however, the predominance of mucosal lesions, associated with a tendency toward scar formation, favors a diagnosis of MMP.5353 Chan LS, Ahmed AR, Anhalt GJ, Bernauer W, Cooper KD, Elder MJ, et al. The first international consensus on mucous membrane pemphigoid: definition, diagnostic criteria, pathogenic factors, medical treatment, and prognostic indicators. Arch Dermatol. 2002;138:370-9.,5555 Xu HH, Werth VP, Parisi E, Sollecito TP. Mucous Membrane Pemphigoid. Dent Clin North Am. 2013;57:611-30.
LABORATORY DIAGNOSIS
The diagnosis of MMP is based on the patient’s clinical presentation and histopathological characteristics. In these cases, tissue biopsy is extremely important, because the epithelium of MMP patients tends to move easily, rendering the sample inadequate if it is not performed correctly. 5656 Chan LS. Mucous membrane pemphigoid. Clin Dermatol. 2001;19:703-11.
Histopathology
Histopathology by hematoxylin-eosin staining typically reveals the presence of subepithelial blisters with or without significant inflammatory infiltrates, which may be composed of eosinophils, lymphocytes, and neutrophils, similar to the changes that are observed in other forms of pemphigoid. A second sample should be obtained from perilesional tissue that is adjacent to a new vesicle to complement the findings by microscopy.5757 Parisi E, Raghavendra S, Werth VP, Sollecito TP. Modification to the approach of the diagnosis of mucous membrane pemphigoid: A case report and literature review. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2003;95:182-6.
Direct immunofluorescence
Direct immunofluorescence of perilesional skin or mucosa shows continuous deposits of IgG, IgA, or C3 on the BMZ. Detection of these immune deposits establishes the disease’s autoimmune nature, and immune deposits that are characteristic of the BMZ distinguish MMP from other diseases with mucocutaneous involvement.5353 Chan LS, Ahmed AR, Anhalt GJ, Bernauer W, Cooper KD, Elder MJ, et al. The first international consensus on mucous membrane pemphigoid: definition, diagnostic criteria, pathogenic factors, medical treatment, and prognostic indicators. Arch Dermatol. 2002;138:370-9.
Indirect immunofluorescence
Indirect immunofluorescence is used to detect circulating antibodies in patient serum. To determine the BMZ components that are targeted by autoantibodies, a procedure, known as salt-split, is performed. This process results in separation of the epithelium from the connective tissue at the site of the lamina lucida, exposing BMZ antigens and resulting in greater sensitivity for the detection of serum binding antibodies that are directed against the substrate.5757 Parisi E, Raghavendra S, Werth VP, Sollecito TP. Modification to the approach of the diagnosis of mucous membrane pemphigoid: A case report and literature review. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2003;95:182-6. After separation of the layers, the epithelial side (“ceiling” pattern, classic in bullous pemphigoid), the dermis side (“floor” pattern, classic in epidermolysis bullosa acquisita), or their combination (ceiling and floor pattern) can be visualized, reflecting the various autoantigens that are recognized by these autoantibodies.5353 Chan LS, Ahmed AR, Anhalt GJ, Bernauer W, Cooper KD, Elder MJ, et al. The first international consensus on mucous membrane pemphigoid: definition, diagnostic criteria, pathogenic factors, medical treatment, and prognostic indicators. Arch Dermatol. 2002;138:370-9.
TREATMENT
MMP is a chronic, rare autoimmune bullous disease that can have a significant impact on a patient’s quality of life. Careful clinical examination of the skin and all mucosal surfaces should be performed, and complaints that are related to other systems should be investigated, such as visual impairment, epistaxis, hoarseness, cough, dysphagia, weight loss, dysuria, and rectal bleeding. The aim is to detect cutaneous and mucosal involvement early, due to the risk of the chronic inflammatory processes evolving, causing tissue destruction, scarring, and functional limitation.
A multidisciplinary approach is often required to minimize the risk of the adverse consequences of this disease. Collaboration between dermatologists, dentists, ophthalmologists, otolaryngologists, urologists, intensivists, and gastroenterologists can contribute to a better therapeutic outcome. The main goals of treatment are to improve symptoms, halt disease progression, and prevent the adverse sequelae of chronic inflammation and tissue scarring. Selection of the appropriate treatment depends on several factors, including the site that is involved, disease severity, and its progression.5050 Lazarova Z, Yancey K. Cicatricial pemphigoid: immunopathogenesis and treatment. Derm Ther. 2002;15:382-8.,5353 Chan LS, Ahmed AR, Anhalt GJ, Bernauer W, Cooper KD, Elder MJ, et al. The first international consensus on mucous membrane pemphigoid: definition, diagnostic criteria, pathogenic factors, medical treatment, and prognostic indicators. Arch Dermatol. 2002;138:370-9.
Patients can be divided into a low-risk group, with lesions that are restricted to the oral mucosa, and a high-risk group, with ocular, pharyngeal, laryngeal, esophageal, and genital lesions. The treatment for the low-risk group is conservative, prioritizing topical treatment when possible, whereas in the high-risk group, treatment should be aggressive, with early initiation of systemic agents.5353 Chan LS, Ahmed AR, Anhalt GJ, Bernauer W, Cooper KD, Elder MJ, et al. The first international consensus on mucous membrane pemphigoid: definition, diagnostic criteria, pathogenic factors, medical treatment, and prognostic indicators. Arch Dermatol. 2002;138:370-9.,5858 Barbosa Ldo N, Silva RS, Verardino GC, Gripp AC, Alves Mde F. Mucous membrane pemphigoid with severe esophageal stricture. An Bras Dermatol. 2011;86:565-8.
Due to the absence of large, multicenter, randomized, and controlled clinical trials for this disease group, the treatment strategy is based on expert experience and literature reports.5353 Chan LS, Ahmed AR, Anhalt GJ, Bernauer W, Cooper KD, Elder MJ, et al. The first international consensus on mucous membrane pemphigoid: definition, diagnostic criteria, pathogenic factors, medical treatment, and prognostic indicators. Arch Dermatol. 2002;138:370-9.,5959 Murrell DF, Marinovic B, Caux F, Prost C, Ahmed R, Wozniak K, et al. Definitions and outcome measures for mucous membrane pemphigoid: recommendations of an international panel of experts. J Am Acad Dermatol. 2015;72:168-74.
Topical treatment
Corticosteroids
Moderate- to high-potency topical corticosteroids are the first-line treatment for low-risk patients who have the disease limited to the oral mucosa with or without cutaneous involvement. A conservative approach is recommended, because there is less risk of scarring in these regions. These agents may also be useful as adjunctive therapy in the most severe cases, constituting important components of the therapeutic arsenal. Corticosteroid gels, ointments, or elixirs can be used 2 to 3 times a day. Gels are applied more easily and better tolerated in the oral cavity. It is important to guide the patient to dry the mucosa before application, rub the medication gently at the site for 30 seconds, and refrain from eating or drinking for 30 minutes. One of the applications should be performed before bedtime, because oral secretions are diminished during sleep. This step may allow the medication to remain longer in the treated area.6060 Knudson RM, Kalaaji AN, Bruce AJ. The management of mucous membrane pemphigoid and pemphigus. Dermatol Ther. 2010;23:268-80.
Alternative methods of administering corticosteroids include trays that are made by dentists to provide medicines under occlusion, for patients with gingival involvement. Trays should be inserted into the dental arch and held for 10 to 20 minutes.6060 Knudson RM, Kalaaji AN, Bruce AJ. The management of mucous membrane pemphigoid and pemphigus. Dermatol Ther. 2010;23:268-80.,6161 Gonzalez-Moles MA, Ruiz-Avila I, Rodriguez-Archilla A, Morales-Garcia P, Mesa-Aguado F, Bascones-Martinez A, et al. Treatment of severe erosive gingival lesions by topical application of clobetasol propionate in custom trays. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2003;95:688-92.
If the patient does not respond adequately to topical therapy, injections of intralesional corticosteroids, which have had some success, may be used. The application should be superficial, just below the erosions; if it is deeper, it may increase the risk of mucosal atrophy. Injection of triamcinolone hexacetonide at 5 to 10mg/ml every 2 to 4 weeks is recommended. The total dose that is administered on a per-session basis should not exceed 20mg.5353 Chan LS, Ahmed AR, Anhalt GJ, Bernauer W, Cooper KD, Elder MJ, et al. The first international consensus on mucous membrane pemphigoid: definition, diagnostic criteria, pathogenic factors, medical treatment, and prognostic indicators. Arch Dermatol. 2002;138:370-9.,6060 Knudson RM, Kalaaji AN, Bruce AJ. The management of mucous membrane pemphigoid and pemphigus. Dermatol Ther. 2010;23:268-80.
Some adverse effects of topical corticosteroids are known, such as mucosal atrophy, oropharyngeal candidiasis, and reactivation of herpes simplex virus. Usually, patients improve in several weeks with the use of topical corticosteroids. As improvement occurs, the patient can gradually decrease the frequency of applications and substitute them for less potent agents, avoiding mucosal atrophy. If treatment discontinuation is not possible due to relapses, the patient should be maintained with the lowest effective topical or systemic maintenance regimen.6060 Knudson RM, Kalaaji AN, Bruce AJ. The management of mucous membrane pemphigoid and pemphigus. Dermatol Ther. 2010;23:268-80.,6262 Gilbert SC. Management and prevention of recurrent herpes labialis in immunocompetent patients. Herpes. 2007;14:56-61.,6363 Cernik C, Gallina K, Brodell RT. The treatment of herpes simplex infections: an evidence-based review. Arch Intern Med. 2008;168:1137-44.
Tacrolimus
Topical tacrolimus, a calcineurin inhibitor, appears to be effective in patients with localized oral disease, effecting complete remission within 2 to 3 months of treatment. However, topical corticosteroids are more effective. The high cost of tacrolimus in relation to many topical corticosteroids makes it difficult for some patients to use this therapy. Tacrolimus is generally indicated for patients who experience relapse of lesions as the application frequency and potency of the corticosteroids are reduced. A maintenance regimen, in which corticosteroid treatment is alternated with topical tacrolimus, may help keep the patient in remission and reduce the risk of the long-term side effects of corticosteroids. Topical tacrolimus 0.1% ointment should be initially applied twice daily, according to the patient’s tolerance, because a transient burning sensation may occur soon after application.6464 Assmann T, Becker J, Ruzicka T, Megahed M. Topical tacrolimus for oral cicatricial pemphigoid. Clin Exp Dermatol. 2004;29:674-6.
65 Suresh L, Martinez Calixto LE, Radfar L. Successful treatment of mucous membrane pemphigoid with tacrolimus. Spec Care Dentist. 2006;26:66-70.-6666 Lee HY, Blazek C, Beltraminelli H, Borradori L.. Oral mucous membrane pemphigoid: complete response to topical tacrolimus. Acta Derm Venereol. 2011;91:604-5.
Oral care
When there is oral cavity involvement, the patient needs to be encouraged to exercise good hygiene. Frequent toothbrushing is indicated using soft-bristled brushes, proper technique, toothpaste that does not contain sodium lauryl sulfate, alcohol-free mouthwash, and flossing to avoid plaque build-up, acceleration of dental caries, and loss of gums and teeth. Topical anesthetics (e.g., lidocaine) may be used to reduce pain by applying them to lesions before meals, brushing, dental procedures, and other occasions. Mild debridement of necrotic mucosal tissue can be performed to prevent infection. Oral cavity trauma using ill-fitting dentures can lead to the formation of new erosions. Thus, regular dental evaluations are imperative in patient care.6060 Knudson RM, Kalaaji AN, Bruce AJ. The management of mucous membrane pemphigoid and pemphigus. Dermatol Ther. 2010;23:268-80.,6767 Kourosh AS, Yancey KB. Therapeutic approaches to patients with mucous membrane pemphigoid. Dermatol Clin. 2011;29:637-41.
Extraoral care
In addition to pharmacological treatment, other interventions may be useful in the management of patients with extraoral involvement. Early referral to specialists is essential to prevent scars and sequelae in these regions. When there is nasal involvement, lavage with saline, application of emollients, and the use of corticosteroids in spray form may be useful. Esophageal involvement justifies a liquid-pasty diet, measures for preventing gastroesophageal reflux, and dilatation of stenosis. When there is involvement of the anogenital region, corticosteroids, and even topical calcineurin inhibitors, can be indicated. The patient should be guided to consume a laxative diet that is rich in water and fiber intake for digestion and stool excretion.6060 Knudson RM, Kalaaji AN, Bruce AJ. The management of mucous membrane pemphigoid and pemphigus. Dermatol Ther. 2010;23:268-80.,6767 Kourosh AS, Yancey KB. Therapeutic approaches to patients with mucous membrane pemphigoid. Dermatol Clin. 2011;29:637-41.
Systemic treatment
Antibiotics
Antibiotics, such as dapsone, other sulfonamides, and tetracyclines with or without nicotinamide, may be effective in the treatment of MMP. Dapsone is often used as first-line therapy to control localized conditions that have not responded to local therapy or extensive, slowly progressing conditions that benefit from systemic agents. If there is a partial response, systemic corticosteroids or immunosuppressive agents may be combined. Local therapy should be maintained as an adjunct.
Most patients respond to dosages of dapsone of between 50 and 200mg per day. Therapy should be started at a low dose, 25 to 50mg per day, and gradually increased (25 to 50mg per week), depending on the patient’s tolerance and age.6060 Knudson RM, Kalaaji AN, Bruce AJ. The management of mucous membrane pemphigoid and pemphigus. Dermatol Ther. 2010;23:268-80.,6868 Arash A, Shirin L. The management of oral mucous membrane pemphigoid with dapsone and topical corticosteroid. J Oral Pathol Med. 2008;37:341-4.,6969 Gürcan HM, Ahmed AR. Efficacy of dapsone in the treatment of pemphigus and pemphigoid: analysis of current data. Am J Clin Dermatol. 2009;10:383-96. Due to the potential for severe hematological side effects, laboratory monitoring is required for patients who receive dapsone. A complete differential blood count and hepatic function and renal function tests should be requested prior to the initiation of therapy, with the differential blood count repeated weekly during the first month of treatment and twice monthly for the following 2 months. Before initiating dapsone, patients should be tested for glucose-6-phosphate dehydrogenase (G6PD) deficiency. A reduction of 1 to 2g/dL of hemoglobin is often observed in dapsone users with normal G6PD levels. Provided that there are no significant comorbidities, patients tolerate it well.6060 Knudson RM, Kalaaji AN, Bruce AJ. The management of mucous membrane pemphigoid and pemphigus. Dermatol Ther. 2010;23:268-80.,7070 Hegarty AM, Ormond M, Sweeney M, Hodgson T. Dapsone efficacy and adverse events in the management of mucous membrane pemphigoid. Eur J Dermatol. 2010;20:223-4.
If the disease is not adequately controlled, tetracycline (1 to 2g per day) or doxycycline 100mg/day and nicotinamide (2 to 3g per day) may be used. The adverse effects of tetracycline include gastrointestinal discomfort and phototoxicity.5959 Murrell DF, Marinovic B, Caux F, Prost C, Ahmed R, Wozniak K, et al. Definitions and outcome measures for mucous membrane pemphigoid: recommendations of an international panel of experts. J Am Acad Dermatol. 2015;72:168-74.,6060 Knudson RM, Kalaaji AN, Bruce AJ. The management of mucous membrane pemphigoid and pemphigus. Dermatol Ther. 2010;23:268-80.
Systemic corticosteroids
Systemic corticosteroids are the first-line treatment for patients with severe, extensive, rapidly progressing MMP and those who do not respond to initial therapies. The dosage may vary depending on the patient’s clinical context, and it can be started at 0.5mg/kg/day for moderate conditions, reaching 1-2mg/kg/day for more severe conditions. It should be administered in a single morning dose and maintained until the disease is controlled, with healing of all lesions and no emergence of new lesions. A rapid response is usually observed when treatment is started. Pulse therapy with methylprednisolone (at 500mg to 1g/day for 3 days) has been frequently employed with good results, effecting a clinical response more rapidly. Yet, treatment may be necessary over a long period, accompanied by the risk of adverse effects of prolonged corticosteroid therapy.
Steroid-sparing immunosuppressive agents, such as azathioprine, mycophenolate mofetil, cyclophosphamide, and methotrexate, may be combined early to help maintain disease control and allow a gradual reduction in corticosteroids. Decreases of 5 to 10mg per week, over several weeks, are recommended. These immunosuppressive agents are maintained over the long term, whereas corticosteroids are tapered over 6 to 12 months. The ultimate goal is to administer monotherapy with an immunosuppressive agent. Topical therapy can also be maintained as adjuvant treatment throughout this regimen.
A rigorous clinical monitoring protocol is necessary for all patients during therapy. Treatment strategies are recommended to protect patients from the many adverse effects of prolonged corticotherapy. These approaches include the use of an H2 blocker or proton pump inhibitor for gastric ulcer prophylaxis and bisphosphonate supplementation (daily, weekly, or monthly), calcium (1000 to 1500mg daily), and vitamin D (800 to 1000IU per day) for prophylaxis of osteoporosis and strongyloidiasis before immunosuppression.6060 Knudson RM, Kalaaji AN, Bruce AJ. The management of mucous membrane pemphigoid and pemphigus. Dermatol Ther. 2010;23:268-80.,6767 Kourosh AS, Yancey KB. Therapeutic approaches to patients with mucous membrane pemphigoid. Dermatol Clin. 2011;29:637-41.
Immunosuppressants
Azathioprine
Azathioprine is one of the choices for adjuvant therapy in MMP, as long as the disease is not progressing rapidly or is threatening vision. A delay in its introduction, up to 8 weeks, limits its initial use as monotherapy. The dose of azathioprine (1-3 mg/kg/day) should be individualized, based on the activity of thiopurine methyltransferase (TPMT), an enzyme that is involved in drug metabolism. The main side effects of azathioprine include hepatotoxicity, hypersensitivity syndrome, and neutropenia. Regular laboratory monitoring should be performed for early detection of adverse effects.6060 Knudson RM, Kalaaji AN, Bruce AJ. The management of mucous membrane pemphigoid and pemphigus. Dermatol Ther. 2010;23:268-80.,6767 Kourosh AS, Yancey KB. Therapeutic approaches to patients with mucous membrane pemphigoid. Dermatol Clin. 2011;29:637-41.
Mycophenolate mofetil
Mycophenolate mofetil is an immunosuppressive agent that reversibly inhibits inosine monophosphate dehydrogenase. Unlike other cell lines, T and B lymphocytes depend on this pathway for purine synthesis, which is essential for cell proliferation. Its use has had good results in disease control, with a favorable safety profile and limited side effects compared with other immunosuppressants, such as azathioprine and methotrexate.7171 Megahed M, Schmiedeberg S, Becker J, Ruzicka T. Treatment of cicatricial pemphigoid with mycophenolate mofetil as a steroid-sparing agent. J Am Acad Dermatol. 2001;45:256-9.,7272 Nottage JM, Hammersmith KM, Murchison AP, Felipe AF, Penne R, Raber I. Treatment of mucous membrane pemphigoid with mycophenolate mofetil. Cornea. 2013;32:810-5.
Treatment with low doses should be started by gradually, increasing the dose according to the clinical effect and the appearance of adverse effects; in certain cases, it will be necessary to switch to mycophenolate sodium. The daily dosage can reach 2 to 3g/day, divided into 2 doses. Once disease control is achieved and maintained for several months, mycophenolate mofetil can be reduced gradually and even discontinued.6767 Kourosh AS, Yancey KB. Therapeutic approaches to patients with mucous membrane pemphigoid. Dermatol Clin. 2011;29:637-41.
The most common side effects of mycophenolate mofetil are gastrointestinal complaints. But the cytopenias are the major concerns and require regular monitoring. A complete blood count with differential can be obtained at baseline and every two weeks during the first two to three months of therapy, then once monthly within the first year, and every three months thereafter. 7373 Perlis C, Pan TD, McDonald CJ. Cytotoxic agents. In: Wolverton SE, editor. Comprehensive Dermatologic Drug Therapy. 2nd ed. Philadelphia: Elsevier Inc.; 2007. p.197.
Methotrexate
Although the use of methotrexate is simple and although dermatologists are familiar with its administration, it is not commonly used as adjuvant therapy in mucous membrane pemphigoid. However, it can be considered as an option in patients with contraindications or those who have failed other adjuvant agents. The dose of methotrexate is more suitable for patients with mild to moderate disease. Low weekly dosages (10 to 17.5mg per week) have been effective and well tolerated.6060 Knudson RM, Kalaaji AN, Bruce AJ. The management of mucous membrane pemphigoid and pemphigus. Dermatol Ther. 2010;23:268-80.,7474 McCluskey P, Chang JH, Singh R, Wakefield D. Methotrexate therapy for ocular cicatricial pemphigoid. Ophthalmology. 2004;111:796-801. Folic acid supplementation helps prevent gastrointestinal adverse effects and elevated transaminase levels. Patients should be monitored regularly by laboratory testing, adjusting the dose as necessary.7575 Neff AG, Turner M, Mutasim DF. Treatment strategies in mucous membrane pemphigoid. Ther Clin Risk Manag. 2008;4:617-26.,7676 Prey S, Paul C. Effect of folic or folinic acid supplementation on methotrexate- associated safety and efficacy in inflammatory disease: a systematic review. Br J Dermatol. 2009;160:622-8.
Cyclophosphamide
Cyclophosphamide is a rapid and effective therapeutic agent and is reserved for patients with severe, rapidly progressing disease or those who have not responded to other immunosuppressive agents. It can be administered alone (1-2mg/kg/day) or in combination with corticosteroids in high doses (prednisone 1-1.5mg/ kg per day).5353 Chan LS, Ahmed AR, Anhalt GJ, Bernauer W, Cooper KD, Elder MJ, et al. The first international consensus on mucous membrane pemphigoid: definition, diagnostic criteria, pathogenic factors, medical treatment, and prognostic indicators. Arch Dermatol. 2002;138:370-9. In some cases, more aggressive treatment with pulse therapy is required at 1g/day or 10-15mg/kg/day, in combination with dexamethasone 100mg on the first day, followed by dexamethasone only on the other 2 days; this cycle may be repeated every 26 days, depending on symptomatology. Regular laboratory monitoring includes a complete differential blood count and urinalysis. Due to its potential toxicity, cyclophosphamide should be used as a short-term therapy and replaced with an alternative adjuvant after disease control has been achieved.6060 Knudson RM, Kalaaji AN, Bruce AJ. The management of mucous membrane pemphigoid and pemphigus. Dermatol Ther. 2010;23:268-80.,7777 Munyangango EM, Le Roux-Villet C, Doan S, Pascal F, Soued I, Alexandre M, et al. Oral cyclophosphamide without corticosteroids to treat mucous membrane pemphigoid. Br J Dermatol. 2013;168:381-90.
Immunoglobulin
Intravenous immunoglobulin (IVIG) is an immunomodulatory agent that is composed of polyclonal antibodies and derived from the plasma of a large group of healthy donors. Its mechanism of action is unclear; it appears to regulate the immune system in various ways. It is not a first-line treatment, but it may be beneficial for patients with severely debilitating, potentially fatal, and rapidly progressing disease. Patients who do not respond to conventional therapy, discontinue it due to severe side effects, or have contraindications are also candidates for IVIG. Immunosuppressive agents increase the risk of the reactivation of chronic infections, such as HBV and HCV. Therapeutic combination with IVIG reduces the risk of reactivation and contributes to clinical improvement.7878 Tavakolpour S. The role of intravenous immunoglobulin in treatment of mucous membrane pemphigoid: A review of literature. J Res Med Sci. 2016;21:37.
IVIG has a relatively safe response and rapid efficacy compared with conventional treatments. The suggested dose is 2/kg per cycle. One cycle consists of the total dose divided into equal doses over a period of 3 to 5 days. The need for subsequent cycles should be assessed according to the clinical response.6060 Knudson RM, Kalaaji AN, Bruce AJ. The management of mucous membrane pemphigoid and pemphigus. Dermatol Ther. 2010;23:268-80. Treated patients have shown excellent results, with significantly shorter treatment durations, fewer relapses, higher remission rates, fewer adverse effects, and better quality-of-life assessments compared with conventional therapy.7979 Ahmed AR, Colón JE.. Comparison between intravenous immunoglobulin and conventional immunosuppressive therapy regimens in patients with severe oral pemphigoid: effects on disease progression in patients nonresponsive to dapsone therapy. Arch Dermatol. 2001;137:1181-9. Most patients do not have serious adverse effects, rendering the treatment safe. The major limitation of IVIG use is its high cost.6060 Knudson RM, Kalaaji AN, Bruce AJ. The management of mucous membrane pemphigoid and pemphigus. Dermatol Ther. 2010;23:268-80.,7878 Tavakolpour S. The role of intravenous immunoglobulin in treatment of mucous membrane pemphigoid: A review of literature. J Res Med Sci. 2016;21:37.
Rituximab
Rituximab is a chimeric monoclonal antibody (murine and human) that is directed against CD20 on pre-B and B lymphocytes, depleting these cells from circulation for 6 to 12 months. It is not a first-line treatment, due to the absence of confirmatory studies. However, it has been used successfully for the treatment of severe pulmonary arterial hypertension, especially in patients who have not responded to conventional therapy or have had serious side effects or contraindications. A rituximab cycle is administered at a dose of 375mg/m2, administered weekly for 4 consecutive weeks. IVIG, systemic corticosteroids, and other immunosuppressive agents are often given concomitantly with rituximab. After the induction cycle, long-term infusion therapy every 4 or 12 weeks may be performed or according to clinical need.6060 Knudson RM, Kalaaji AN, Bruce AJ. The management of mucous membrane pemphigoid and pemphigus. Dermatol Ther. 2010;23:268-80.,8080 Le Roux-Villet C, Prost-Squarcioni C, Alexandre M, Caux F, Pascal F, Doan S, et al. Rituximab for patients with refractory mucous membrane pemphigoid. Arch Dermatol. 2011;147:843-9.
The most common adverse effects are associated with infusion reactions, often related to the rate of administration and hypersensitivity reactions (fever, chills, bronchospasm, pruritus, and hypotension). Rituximab is contraindicated in pregnancy; patients of childbearing age should use effective contraception during its use and up to 12 months afterward.6060 Knudson RM, Kalaaji AN, Bruce AJ. The management of mucous membrane pemphigoid and pemphigus. Dermatol Ther. 2010;23:268-80.,8181 Bomm L, Fracaroli TS, Sodré JL, Bressan A, Gripp AC. Off-label use of rituximab in dermatology: pemphigus treatment. An Bras Dermatol. 2013;88:676-8.
Anti-TNF alpha agents
In addition to conventional treatments, anti-TNF alpha agents have been reported to be effective in mucous membrane pemphigoid, perhaps merited by the high serum TNF-alpha levels in patients. They are generally indicated for those who are in need of aggressive systemic treatment, do not respond to conventional therapy, or have severe side effects or contraindications. Several reports in the literature have demonstrated the efficacy of etanercept and infliximab. The clinical improvement after treatment with these agents supports the hypothesis that TNF-alpha plays an important role in the pathogenesis of mucous membrane pemphigoid.8282 Sacher C, Rubbert A, König C, Scharffetter-Kochanek K, Krieg T, Hunzelmann N. Treatment of recalcitrant cicatricial pemphigoid with the tumor necrosis factor alpha antagonist etanercept. J Am Acad Dermatol. 2002;46:113-5.
83 Canizares MJ, Smith DI, Conners MS, Maverick KJ, Heffernan MP. Successful treatment of mucous membrane pemphigoid with etanercept in 3 patients. Arch Dermatol. 2006;142:1457-61.-8484 Heffernan MP, Bentley DD. Successful treatment of mucous membrane pemphigoid with infliximab. Arch Dermatol. 2006;142:1268-70.
PROGNOSIS
Mucous membrane pemphigoid is a chronic, often devastating, but rarely fatal disease. Even when localized, it can have a tremendous negative impact on a patient’s quality of life. Its chronic inflammatory nature, tissue destruction, and scarring are responsible for the sequelae, which are often disabling. The therapeutic strategy will depend on the site, severity, and rate of progression. Patients with low risk (oral mucosa with or without cutaneous involvement) have a better prognosis and respond well to conservative approaches, whereas high-risk patients (ocular, nasopharyngeal, laryngeal, esophageal, and anogenital mucosa) have a worse prognosis, being poor responders and often experiencing scarring, despite treatment. In the latter, an early aggressive approach helps stop the inflammatory process and prevent scarring.5353 Chan LS, Ahmed AR, Anhalt GJ, Bernauer W, Cooper KD, Elder MJ, et al. The first international consensus on mucous membrane pemphigoid: definition, diagnostic criteria, pathogenic factors, medical treatment, and prognostic indicators. Arch Dermatol. 2002;138:370-9.,6060 Knudson RM, Kalaaji AN, Bruce AJ. The management of mucous membrane pemphigoid and pemphigus. Dermatol Ther. 2010;23:268-80.
CONCLUSION
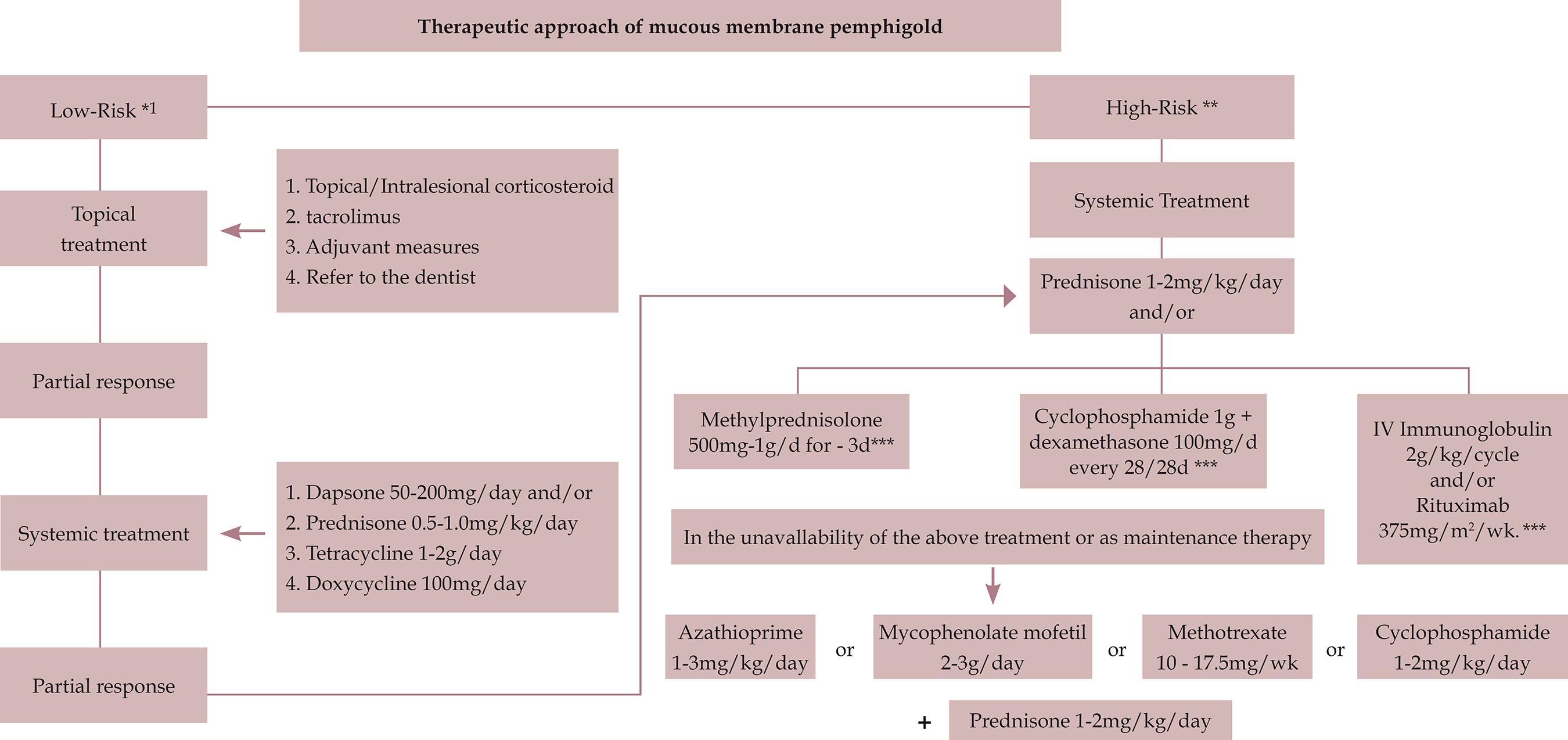
Our experience indicates that the approach should be as aggressive as possible for high-risk patients. Some therapeutic options can be employed, depending on the clinical context and their availability. Pulse therapy with methylprednisolone, cyclophosphamide in combination with dexamethasone, and IVIG with rituximab have effected good responses in our service. In cases of unavailability, contraindications, or serious side effects to these options, prednisone (1-2mg/kg/day) can be administered concomitantly with such immunosuppressants as azathioprine, mycophenolate mofetil, and methotrexate, as demonstrated in the treatment flowchart (Figure 2). We believe that long-term follow-up of these patients should be performed to detect relapses and initiate early therapeutic intervention, and previous pulses or cycles may be repeated.
Mucous membranes pemphigoid treatment algorithm.
* Low risk: disease limited to oral mucosa with or without cutaneous involvement
**High risk: ocular, pharyngeal, laryngeal, esophageal, and/or genital lesions
*** Prednisone at a dosage of 1-2mg/kg/day should be maintained between pulse therapy intervals
EPIDERMOLYSIS BULLOSA ACQUISITA
INTRODUCTION
Epidermolysis bullosa acquisita (EBA) is an autoimmune subepidermal bullous dermatosis that affects the skin and mucous membranes, characterized by autoantibodies that are directed against type VII collagen (COLVII). 8585 Lehman JS, Camilleri MJ, Gibson LE. Epidermolysis bullosa acquisita: concise review and practical considerations. Int J Dermatol. 2009;48:227-35;
EPIDEMIOLOGY
EBA is a rare disease, accounting for approximately 5% of autoimmune bullous diseases that target antigens in the basement membrane zone (BMZ), with an annual incidence varying between 0.08 to 0.5 cases per million individuals.86,87 EBA has no predilection for gender and can occur at any age, peaking in incidence between the fourth and fifth decades.8888 Kim JH, Kim SC. Epidermolysis bullosa acquisita. J Eur Acad Dermatol Venereol. 2013;27:1204-13,8989 Gupta R, Woodley DT, Chen M. Epidermolysis bullosa acquisita. Clin Dermatol. 2012;30:60-9.
PATHOPHYSIOLOGY
Recent evidence shows that IgG autoantibodies against epitopes in the non-collagenous domain of COLVII are relevant in the pathogenesis of EBA.9090 Kasperkiewicz M, Sadik CD, Bieber K, Ibrahim SM, Manz RA, Schmidt E, et al. Epidermolysis Bullosa Acquisita: From Pathophysiology to Novel Therapeutic Options. J Invest Dermatol. 2016;136:24-33 COLVII is the main component of anchoring fibrils in the sublamina densa of the BMZ. Therefore, the involvement of these structures results in the loss of dermoepidermal adhesion, with subsequent cutaneous fragility, vesicle and blister formation, and scarring.
CLINICAL PRESENTATION
EBA has a wide spectrum of clinical presentations and can affect the skin and mucous membranes. EBA has 5 main phenotypes.9191 Hashimoto T, Jin Z, Ishii N. Clinical and immunological studies for 105 Japanese seropositive patients of epidermolysis bullosa acquisita examined at Kurume University. Expert Rev Clin Immunol. 2016;12:895-902
-
The classical or mechanobullous form is observed in approximately one-third of EBA patients and is characterized by skin fragility in trauma-prone areas, with tense blisters, erosions, atrophic scars, milia formation, anonychia, and digital contracture, especially on the fingers.8888 Kim JH, Kim SC. Epidermolysis bullosa acquisita. J Eur Acad Dermatol Venereol. 2013;27:1204-13,9292 Ludwig RJ. Clinical presentation, pathogenesis, diagnosis, and treatment of epidermolysis bullosa acquisita. ISRN Dermatol. 2013;2013:812029.
-
The bullous pemphigoid-like variant presents as vesicles and blisters with erythematous or urticarial lesions, usually without skin fragility or milia formation, mainly in the extremities, trunk, and skin folds.9393 Chen M, Kim GH, Prakash L, Woodley DT. Epidermolysis bullosa acquisita: autoimmunity to anchoring fibril collagen. Autoimmunity. 2012;45:91-101
-
The cicatricial pemphigoid-like form presents with exclusive or predominant mucous involvement, affecting any stratified squamous cell epithelia, such as ocular, oral, nasal, laryngeal, esophageal, and genital mucosa.9494 Vorobyev A, Ludwig RJ, Schmidt E. Clinical features and diagnosis of epidermolysis bullosa acquisita. Expert Rev Clin Immunol. 2017;13:157-69.
-
The Brunsting-Perry pemphigoid-like variant is defined as a vesiculobullous eruption on the head and neck, which may eventually have oral involvement.9595 Asfour L, Chong H, Mee J, Groves R, Singh M. Epidermolysis Bullosa Acquisita (Brunsting-Perry Pemphigoid Variant) Localized to the Face and Diagnosed With Antigen Identification Using Skin Deficient in Type VII Collagen. Am J Dermatopathol. 2017;39:e90-e96.
-
The linear IgA bullous dermatosis-like form is characterized by edematous plaques with tense blisters and vesicules that can present in annular or polycyclic arrangement. In this form of EBA, the direct immunofluorescence demonstrates linear IgA deposition in the BMZ.9494 Vorobyev A, Ludwig RJ, Schmidt E. Clinical features and diagnosis of epidermolysis bullosa acquisita. Expert Rev Clin Immunol. 2017;13:157-69.
The differential diagnosis of EBA includes bullous systemic lupus erythematosus, porphyria cutanea tarda, and other subepidermal bullous dermatosis, as mentioned in the clinical variants.
DIAGNOSIS
The diagnosis of EBA comprises histopathological analysis and direct (DIF) and indirect (IIF) immunofluorescence, which allow the diagnosis of subepidermal bullous dermatosis. However, they are not always conclusive for the diagnosis of EBA. The definite diagnosis of EBA requires additional techniques that are only available in research centers.
The histopathological evaluation of injured skin in patients with EBA shows subepidermal cleavage, with varying degrees of inflammatory infiltrate. In the mechanobullous form, the inflammatory infiltrate is absent or minimal, whereas in the inflammatory forms of EBA, it is composed of neutrophils with variable numbers of eosinophils, monocytes, and lymphocytes.9494 Vorobyev A, Ludwig RJ, Schmidt E. Clinical features and diagnosis of epidermolysis bullosa acquisita. Expert Rev Clin Immunol. 2017;13:157-69.
DIF of perilesional skin reveals linear deposition of IgG and C3 along the BMZ. Occasionally, IgA and/or IgM may be present. Serration pattern analysis by DIF allows a differential diagnosis between EBA and bullous pemphigoid, which present as U and N patterns, respectively.9696 M Meijer JM, Atefi I, Diercks GFH, Vorobyev A, Zuiderveen J, Meijer HJ, et al. Serration pattern analysis for differentiating epidermolysis bullosa acquisita from other pemphigoid diseases. J Am Acad Dermatol. 2018;78:754-9.e6. IIF of normal human skin also identifies IgG deposits with a linear pattern in the BMZ. The salt-split skin technique, which produces BMZ cleavage in the lamina lucida, shows IgG deposition on the dermal side of the cleavage in EBA, whereas in bullous pemphigoid, this deposit is present on the epidermal or epidermal and dermal side of the cleavage.
TREATMENT
EBA is often refractory to various therapeutic modalities, which makes its long-term remission a challenge. Because it is a rare disease with several clinical presentations, there are no randomized clinical trials in the literature, compromising the selection of an ideal treatment. The inflammatory form of EBA apparently has a more favorable clinical response to conventional therapy with corticosteroids and corticosteroid-sparing agents than the mechanobullous form.9797 Mehren CR, Gniadecki R. Epidermolysis bullosa acquisita: current diagnosis and therapy. Dermatol Reports. 2011;3:e38.
I) Clinical and laboratory evaluation prior to immunosuppressive therapy
-
- Multidisciplinary evaluation for diagnosis of mucosal involvement:
-
- Ophthalmology
-
- Otolaryngology
-
- Gastroenterology
-
- Gynecology
-
-
- Evaluation of comorbidities:
-
- Systemic arterial hypertension
-
- Diabetes mellitus
-
- Bone evaluation (bone mineral densitometry)
-
- Inflammatory bowel disease
-
- Infectious diseases (hepatitis, Chagas disease, HIV, tuberculosis)
-
- Neoplasms
-
- Others
-
-
- Vaccination
-
- Stool examination
-
- Prophylaxis with ivermectin
-
-
- Pregnancy (pregnancy test and contraception)
II) General skin care
Local care should be encouraged for better disease control. Similar to inherited epidermolysis bullosa, the general care includes the prevention of local trauma and infection and the use of non-adherent dressings.9898 Pope E, Lara-Corrales I, Mellerio J, Martinez A, Schultz G, Burrell R, et al. A consensus approach to wound care in epidermolysis bullosa. J Am Acad Dermatol. 2012;67:904-17.
III) Systemic treatment
A) Introduction
A Cochrane systematic review found 11 non-randomized studies of treatment for EBA, involving interventions in 20 adults and 11 children.9999 Kirtschig G, Murrell D, Wojnarowska F, Khumalo N. Interventions for mucous membrane pemphigoid and epidermolysis bullosa acquisita. C Cochrane Database Syst Rev. 2003:CD004056. Adult patients were treated with various therapeutic modalities, including systemic corticosteroids, immunosuppressants, dapsone, colchicine, and intravenous immunoglobulin. Most children were treated with systemic corticosteroids and/or dapsone. The authors concluded that there is no recommendation for treating EBA, based on reliable evidence.
B) Medications
Corticosteroids
EBA patients, especially those of the mechanobullous form, usually do not experience a good therapeutic response to systemic corticosteroids, such as in other autoimmune blistering diseases.
Despite this limitation, systemic corticosteroids are still considered the first-line treatment for EBA, with usual dosages of 0.5-1.5mg/kg/day; they can be used as monotherapy in some cases of mild disease.100100 Ishii N, Hamada T, Dainichi T, Karashima T, Nakama T, Yasumoto S, et al. Epidermolysis bullosa acquisita: what's new? J Dermatol. 2010;37:220-30. A retrospective Spanish study of 12 patients with EBA showed that a dosage greater than 15mg/day is required to control disease activity.101101 Iranzo P, Herrero-González JE, Mascaró-Galy JM, Suárez-Fernández R, España A. Epidermolysis bullosa acquisita: a retrospective analysis of 12 patients evaluated in four tertiary hospitals in Spain. Br J Dermatol. 2014;171:1022-30 However, most patients require adjuvant treatment for better disease control or to avoid undesirable side effects of prolonged use of corticosteroids, such as obesity, hypertension, and osteoporosis. One study demonstrated that the use of methylprednisolone at a high dosage (> 8mg/day) led to earlier remission compared with low-dose use (≤ 8mg/day) and that some patients obtained a good response to pulse therapy with methylprednisolone (500mg for 3 days).102102 Kim JH, Kim YH, Kim SC. Epidermolysis bullosa acquisita: a retrospective clinical analysis of 30 cases. Acta Derm Venereol. 2011;91:307-12.
Anti-inflammatory agents (dapsone, colchicine, tetracycline)
The main anti-inflammatory benefits of these medications are exerted by an antineutrophilic action.103103 Intong LR, Murrell DF. Management of epidermolysis bullosa acquisita. Dermatol Clin. 2011;29:643-7
Dapsone use has been described in several cases of EBA.101101 Iranzo P, Herrero-González JE, Mascaró-Galy JM, Suárez-Fernández R, España A. Epidermolysis bullosa acquisita: a retrospective analysis of 12 patients evaluated in four tertiary hospitals in Spain. Br J Dermatol. 2014;171:1022-30,102102 Kim JH, Kim YH, Kim SC. Epidermolysis bullosa acquisita: a retrospective clinical analysis of 30 cases. Acta Derm Venereol. 2011;91:307-12.,104104 Delgado L, Aoki V, Santi C, Gabbi T, Sotto M, Maruta C. Clinical and immunopathological evaluation of epidermolysis bullosa acquisita. Clin Exp Dermatol. 2011;36:12-8 The commonly used dosages range from 25-100mg/ day or 1-2mg/kg/day, with a favorable response being reported in adult and pediatric cases.105105 Gürcan HM, Ahmed AR. Current concepts in the treatment of epidermolysis bullosa acquisita. Expert Opin Pharmacother. 2011 ;12:1259-68. It should be noted that unlike the condition in adults, EBA in childhood tends to have a better prognosis, usually with a good response to dapsone and systemic corticosteroid.106106 Goyal N, Rao R, Balachandran C, Pai S, Bhogal BS, Schmidt E, et al. Childhood Epidermolysis Bullosa Acquisita: Confirmation of Diagnosis by Skin Deficient in Type VII Collagen, Enzyme-linked Immunosorbent Assay, and Immunoblotting. Indian J Dermatol. 2016;61:329-32.,107107 Yang B, Wang C, Wang N, Pan F, Chen S, Zhou G, et al. Childhood epidermolysis bullosa acquisita: report of a Chinese case. Pediatr Dermatol. 2012;29:614-7. The proposed protocols for the treatment of EBA in the literature include dapsone as one of the first therapeutic options, combined with systemic corticosteroid therapy, primarily for the mild forms of EBA.100100 Ishii N, Hamada T, Dainichi T, Karashima T, Nakama T, Yasumoto S, et al. Epidermolysis bullosa acquisita: what's new? J Dermatol. 2010;37:220-30.,101101 Iranzo P, Herrero-González JE, Mascaró-Galy JM, Suárez-Fernández R, España A. Epidermolysis bullosa acquisita: a retrospective analysis of 12 patients evaluated in four tertiary hospitals in Spain. Br J Dermatol. 2014;171:1022-30 Common medication side effects include hemolysis and methemoglobinemia.108108 Mutasim DF. Autoimmune bullous dermatoses in the elderly: an update on pathophysiology, diagnosis and management. Drugs Aging. 2010;27:1-19.
The efficacy of colchicine in EBA was reported for the first time in 1989, with dosages ranging from 0.5-2mg/day.109109 D Dasgeb B, Kornreich D, McGuinn K, Okon L, Brownell I, Sackett DL. Colchicine: an ancient drug with novel applications. Br J Dermatol. 2018;178:350-6 Among adjuvant treatments, colchicine is considered a first-line agent and is frequently associated with systemic corticosteroids.100100 Ishii N, Hamada T, Dainichi T, Karashima T, Nakama T, Yasumoto S, et al. Epidermolysis bullosa acquisita: what's new? J Dermatol. 2010;37:220-30. Although it is less common, colchicine has also been reported as a monotherapy, yielding a good clinical response.110110 Adachi A, Komine M, Suzuki M, Murata S, Hirano T, Ishii N, et al. Oral colchicine monotherapy for epidermolysis bullosa acquisita: Mechanism of action and efficacy. J Dermatol. 2016;43:1389-91. However, its side effects, especially diarrhea, can limit its use, making it difficult to achieve a sufficient dose to control the disease in certain patients.111111 Culton DA, Diaz LA. Treatment of subepidermal immunobullous diseases. Clin Dermatol. 2012;30:95-102.
Minocycline is a broad-spectrum tetracycline that inhibits the recruitment of neutrophils and eosinophils as well as cytokine production.112112 Kawase K, Oshitani Y, Mizutani Y, Shu E, Fujine E, Seishima M. Inflammatory epidermolysis bullosa acquisita effectively treated with minocycline. Acta Derm Venereol. 2014;94:615-6. A Japanese study that evaluated the clinical and immunological aspects of 105 patients with EBA described minocycline as one of the most common therapeutic options, having been used in 16 patients; however, details regarding the therapeutic response were not specified.9191 Hashimoto T, Jin Z, Ishii N. Clinical and immunological studies for 105 Japanese seropositive patients of epidermolysis bullosa acquisita examined at Kurume University. Expert Rev Clin Immunol. 2016;12:895-902 A recent case report showed a patient with an inflammatory form of EBA who was treated with various therapeutic options, such as systemic corticosteroid, cyclosporine, and dapsone, responding to the combination of minocycline (200mg/day) and systemic corticosteroid.112112 Kawase K, Oshitani Y, Mizutani Y, Shu E, Fujine E, Seishima M. Inflammatory epidermolysis bullosa acquisita effectively treated with minocycline. Acta Derm Venereol. 2014;94:615-6. This study suggested that minocycline may be considered as a therapeutic option in EBA, mainly as an adjuvant in inflammatory forms of the disease.
Immunosuppressants (azathioprine, mycophenolate mofetil, cyclosporine, methotrexate)
These medications are commonly used as systemic corticosteroid-sparing agents.
Azathioprine has varying results in the literature.101101 Iranzo P, Herrero-González JE, Mascaró-Galy JM, Suárez-Fernández R, España A. Epidermolysis bullosa acquisita: a retrospective analysis of 12 patients evaluated in four tertiary hospitals in Spain. Br J Dermatol. 2014;171:1022-30,104104 Delgado L, Aoki V, Santi C, Gabbi T, Sotto M, Maruta C. Clinical and immunopathological evaluation of epidermolysis bullosa acquisita. Clin Exp Dermatol. 2011;36:12-8,113113 Tu J, Kumarasinghe PW. Epidermolysis bullosa acquisita with moderately severe Dysphagia due to esophageal strictures. Indian J Dermatol. 2011;56:224-7. Although its use is well established in pemphigus vulgaris, its application in EBA is less convincing, with other medications, such as dapsone and mycophenolate mofetil, being preferred more often.114114 Meurer M. Immunosuppressive therapy for autoimmune bullous diseases. Clin Dermatol. 2012;30:78-83.
Mycophenolate mofetil (MMF) is an immunosuppressive agent that alters the proliferation of B and T lymphocytes.111111 Culton DA, Diaz LA. Treatment of subepidermal immunobullous diseases. Clin Dermatol. 2012;30:95-102. In EBA, MMF has been reported to be effective in some refractory cases.105105 Gürcan HM, Ahmed AR. Current concepts in the treatment of epidermolysis bullosa acquisita. Expert Opin Pharmacother. 2011 ;12:1259-68. A recent study described 4 patients with EBA who were treated with MMF at 2 to 3g/day, with success.115115 Sami N. Mycophenolate mofetil (MMF) in the treatment of epidermolysis bullosa acquisita (EBA) long-term follow-up. JAAD Case Rep. 2015;1:321-3. One of the limiting factors of its use in daily practice is its high cost.
Cyclosporine is usually used at doses of 4-9mg/kg, leading to disease improvement in recalcitrant cases.103103 Intong LR, Murrell DF. Management of epidermolysis bullosa acquisita. Dermatol Clin. 2011;29:643-7,105105 Gürcan HM, Ahmed AR. Current concepts in the treatment of epidermolysis bullosa acquisita. Expert Opin Pharmacother. 2011 ;12:1259-68. A report of 2 patients with the rare association of EBA and acquired hemophilia A demonstrated a good therapeutic response with prednisone and cyclosporine, with no adverse effects.116116 Yan TM, He CX, Hua BL, Li L, Jin HZ, Liu YH, et al. Coexistence of acquired hemophilia A and epidermolysis bullosa acquisita: Two case reports and published work review. J Dermatol. 2017;44:76-9. In addition, cyclosporine is considered a relatively safer immunosuppressive drug in pregnancy.114114 Meurer M. Immunosuppressive therapy for autoimmune bullous diseases. Clin Dermatol. 2012;30:78-83.
Methotrexate is a folic acid analogue and antimetabolite that inhibits the synthesis of DNA and RNA, impairing lymphocyte function.108108 Mutasim DF. Autoimmune bullous dermatoses in the elderly: an update on pathophysiology, diagnosis and management. Drugs Aging. 2010;27:1-19. There are no reports of monotherapy in patients with EBA and no data that documents its effectiveness as a systemic corticosteroid-sparing agent in EBA, reserving its use for refractory cases of the disease.9292 Ludwig RJ. Clinical presentation, pathogenesis, diagnosis, and treatment of epidermolysis bullosa acquisita. ISRN Dermatol. 2013;2013:812029.
Intravenous immunoglobulin (IVIG)
IVIG modulates the autoimmune response, reducing and neutralizing antibodies.117117 Mosqueira CB, Furlani Lde A, Xavier AF, Cunha PR, Galvão AM. Intravenous immunoglobulin for treatment of severe acquired bullous epidermolysis refractory to conventional immunosuppressive therapy. An Bras Dermatol. 2010;85:521-4. It is usually administered at 2g/kg/ cycle over 3 to 5 days as monotherapy or in combination with other drugs, such as systemic corticosteroid and dapsone, with good response in patients with recalcitrant disease.103103 Intong LR, Murrell DF. Management of epidermolysis bullosa acquisita. Dermatol Clin. 2011;29:643-7,117117 Mosqueira CB, Furlani Lde A, Xavier AF, Cunha PR, Galvão AM. Intravenous immunoglobulin for treatment of severe acquired bullous epidermolysis refractory to conventional immunosuppressive therapy. An Bras Dermatol. 2010;85:521-4. One long-term study evaluated 10 patients with EBA that were resistant to conventional therapy after IVIG administration, resulting in a satisfactory response in all cases.118118 Ahmed AR, Gürcan HM. Treatment of epidermolysis bullosa acquisita with intravenous immunoglobulin in patients non-responsive to conventional therapy: clinical outcome and post-treatment long-term follow-up. J Eur Acad Dermatol Venereol. 2012;26:1074-83. Patients received 16-31 IVIG cycles over 30-52 months, allowing adjuvant medications (prednisone, dapsone, and others) and, later, IVIG to be withdrawn. No disease recurrence was observed at a mean follow-up of 53.9 months after discontinuation of IVIG, suggesting that this drug leads to prolonged remission of EBA. Immunoglobulin was also administered subcutaneously in a patient with severe and resistant disease, which can represent an alternative in cases with difficult venous puncture.119119 Tayal U, Burton J, Dash C, Wojnarowska F, Chapel H. Subcutaneous immunoglobulin therapy for immunomodulation in a patient with severe epidermolysis bullosa acquisita. Clin Immunol. 2008;129:518-9.
Extracorporeal photopheresis (ECP)
ECP is an immunomodulatory therapy that was initially used in cutaneous T cell lymphoma that consists on the separation of a peripheral blood leukocyte/lymphocyte-enriched cellular fraction, extracorporeal treatment of 8-MOP/UVA cells, and subsequent reinfusion of cells into the patient.120120 Sanli H, Akay BN, Ayyildiz E, Anadolu R, Ilhan O. Remission of severe autoimmune bullous disorders induced by long-term extracorporeal photochemotherapy. Transfus Apher Sci. 2010;43:353-9. Its main effect in EBA appears to be associated with inhibiting the production of pathogenic autoantibodies by B lymphocytes and producing regulatory T cells.9797 Mehren CR, Gniadecki R. Epidermolysis bullosa acquisita: current diagnosis and therapy. Dermatol Reports. 2011;3:e38.
ECP has proven to be efficient in inducing partial to complete remission in patients with recalcitrant EBA and is considered a well-tolerated therapy, with promising results.121121 Baroudjian B, Le Roux-Villet C, Bréchignac S, Alexandre M, Caux F, Prost-Squarcioni C, et al. Long-term efficacy of extracorporeal photochemotherapy in a patient with refractory epidermolysis bullosa acquisita. Eur J Dermatol. 2012;22:795-7.
122 Liszewski W, Omland SH, Gniadecki R. The successful use of extracorporeal photopheresis in a 12-year-old patient with refractory epidermolysis bullosa acquisita. Pediatr Dermatol. 2015;32:e60-1.-123123 Adamski J, Kinard T, Ipe T, Cooling L. Extracorporeal photopheresis for the treatment of autoimmune diseases. Transfus Apher Sci. 2015;52:171-82.
In the literature, a recent review revealed that 8 patients with severe and refractory EBA were treated with ECP, all of which progressed with clinical improvement.123123 Adamski J, Kinard T, Ipe T, Cooling L. Extracorporeal photopheresis for the treatment of autoimmune diseases. Transfus Apher Sci. 2015;52:171-82.
European guidelines on ECP states that such treatment can be used in severe and refractory cases of EBA.124124 Knobler R, Berlin G, Calzavara-Pinton P, Greinix H, Jaksch P, Laroche L, et al. Guidelines on the use of extracorporeal photopheresis. J Eur Acad Dermatol Venereol. 2014;28(Suppl 1):1-37
Rituximab
Rituximab is a chimeric anti-CD20 monoclonal antibody that causes B cell depletion through various mechanisms and is approved for the treatment of B cell lymphoma, rheumatoid arthritis, and granulomatosis with polyangiitis.125125 Lamberts A, Euverman HI, Terra JB, Jonkman MF, Horváth B. Effectiveness and Safety of Rituximab in Recalcitrant Pemphigoid Diseases. Front Immunol. 2018;9:248. In the context of autoimmune bullous diseases, its use was initially described for recalcitrant cases of pemphigus and, recently, as first-line treatment in combination with short term prednisone, being effective and potentially safer than a standard regimen of high doses of corticosteroids in patients with moderate to severe pemphigus.126126 Joly P, Maho-Vaillant M, Prost-Squarcioni C, Hebert V, Houivet E, Calbo S, et al. First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): a prospective, multicentre, parallel-group, open-label randomised trial. Lancet. 2017;389:2031-2040,127127 Hebert V, Joly P. Rituximab in pemphigus. Immunotherapy. 2018;10:27-37.
Rituximab use in subepidermal bullous diseases, however, is more limited. In EBA, a few promising case reports have been described, usually in patients with recalcitrant disease who have undergone several treatments before rituximab (such as systemic corticosteroid, dapsone, azathioprine, cyclosporine, mycophenolate, and cyclophosphamide), with an effective response.128128 Saha M, Cutler T, Bhogal B, Black MM, Groves RW. Refractory epidermolysis bullosa acquisita: successful treatment with rituximab. Clin Exp Dermatol. 2009;34:e979-80
129 Kim JH, Lee SE, Kim SC. Successful treatment of epidermolysis bullosa acquisita with rituximab therapy. J Dermatol. 2012;39:477-9.
130 McKinley SK, Huang JT, Tan J, Kroshinsky D, Gellis S. A case of recalcitrant epidermolysis bullosa acquisita responsive to rituximab therapy. Pediatr Dermatol. 2014;31:241-4-131131 Cavailhes A, Balme B, Gilbert D, Skowron F. Successful use of combined corticosteroids and rituximab in the treatment of recalcitrant epidermolysis bullosa acquisita. Ann Dermatol Venereol. 2009;136:795-9 The therapeutic scheme was usually 375mg/m2 weekly, administered over 4 weeks. Another proposed scheme is 1g, administered on Days 0 and 14.
Rituximab (375mg/m2 weekly, managed over 4 weeks), combined with IVIG (2g/kg/monthly cycle), was given to 4 patients who were resistant to conventional therapy, showing an effective and efficient response.132132 Oktem A, Akay BN, Boyvat A, Kundakci N, Erdem C, Bostanci S, et al. Long-term results of rituximab-intravenous immunoglobulin combination therapy in patients with epidermolysis bullosa acquisita resistant to conventional therapy. J Dermatolog Treat. 2017;28:50-4. In addition, rituximab combined with protein A immunoadsorption has, in some cases, resulted in favorable outcomes.133133 Kubisch I, Diessenbacher P, Schmidt E, Gollnick H, Leverkus M. Premonitory epidermolysis bullosa acquisita mimicking eyelid dermatitis: successful treatment with rituximab and protein A immunoapheresis. Am J Clin Dermatol. 2010;11:289-93.,134134 Kolesnik M, Becker E, Reinhold D, Ambach A, Heim MU, Gollnick H, et al. Treatment of severe autoimmune blistering skin diseases with combination of protein A immunoadsorption and rituximab: a protocol without initial high dose or pulse steroid medication. J Eur Acad Dermatol Venereol. 2014;28:771-80.
In the authors’ personal experience, 5 patients with EBA were treated with rituximab. All patients presented with mechanobullous form, severe mucosal impairment and were resistant to conventional therapy, including systemic corticosteroid therapy (1mg/kg/day), dapsone (100mg/day), cyclosporine (5mg/kg), azathioprine (150mg/day), and mycophenolate mofetil (3g/day). In most cases, rituximab was administered following the rheumatoid arthritis protocol, at 1g IV on Day 0 and 1g IV on Day 14. All patients needed a new drug cycle after 6 months. At present, 2 patients are in complete remission, 1 is in partial remission, and 2 experienced disease relapse after being in complete remission for 1 year.
IV) Classification of EBA severity
There is no standardized severity score for EBA; 2 recent classifications have been proposed in the literature:
Classification I:103103 Intong LR, Murrell DF. Management of epidermolysis bullosa acquisita. Dermatol Clin. 2011;29:643-7
-
-Mild EBA: < 5% of body surface affected, without mucosal involvement.
-
-Moderate EBA: 5% to 15% of body surface affected with at most 2 affected mucous membrane sites, not including ocular involvement.
-
-Severe EBA: > 5% of body surface affected and > 2 mucous membrane sites involved or ocular involvement.
In mild forms of EBA, use of oral steroids (0.5-1mg/kg/day) with additional colchicine and dapsone is suggested. Oral steroids (1-1.5mg/kg/day) with additional colchicine is recommended for moderate EBA. Treatment suggested for severe forms of EBA involves the same as in moderate EBA, with the addition of one or a combination of steroid pulses, cyclosporine, plasmapheresis, IVIg or rituximab.
Classification II:
-
-Non-severe EBA: absence of severe mucosal involvement (IIC ocular, laryngeal or esophageal) or diffuse cutaneous involvement.135135 Prost-Squarcioni C, Ingen-Housz-Oro S, Joly P, Bernard P, Bedane C; Centres de référence des maladies bulleuses auto-immunes. Société Française deDermatologie. Epidermolysis bullosa acquisita. Guidelines for the diagnosis and treatment. Centres deréférence des maladies bulleuses auto-immunes. Société Française de Dermatologie. Ann Dermatol Venereol. 2011;138:274-9.
-
-Severe EBA: presence of severe mucosal involvement - ocular, laryngeal, or esophageal.
In the non-severe localized form, topical corticosteroids are suggested, whereas in the non-severe, more diffuse form, the use of colchicine, dapsone, sulfasalazine, and topical corticosteroid is advised. In severe EBA, cyclosporine or IVIG is recommended; rituximab or ECF can be considered as an alternative. Systemic corticosteroid therapy is reserved for urgent cases, such as laryngeal edema.
The authors of this consensus propose the following classification of EBA severity:
-
-Non-severe EBA: up to 10% of total body surface involved without functional alteration (flexion contracture of fingers) and absence of severe mucosal involvement (ocular, laryngeal, or esophageal).
-
-Severe EBA: greater than 10% of total body surface area affected, and/or presence of functional alteration (flexion contracture of fingers), and/or severe mucosal involvement (ocular, laryngeal, or esophageal), or non-severe EBA that is unresponsive to the proposed treatment.
This classification was used for the treatment algorithm below.
V) Proposed treatment algorithm
In the non-severe form of EBA, the first treatment option suggested by the authors is colchicine (0.5-2mg/day) or dapsone (50-100mg/day). If a partial response occurs, prednisone should be added at 0.5-1mg/kg/day. If absence of clinical response and/or side effect, treatment should be replaced for prednisone (0.5-1mg/kg/day). (Figure 3)
In the severe form of EBA, the first treatment option suggested is IVIG (2g/kg over 5 days) in combination with oral systemic corticosteroid (1-1.5mg/kg/day) or pulse therapy with methylprednisolone (1g IV for 3 days). If partial or absence of response occurs, an immunosuppressant, such as mycophenolate mofetil (2-3g/day), cyclosporine (5mg/kg/day), or azathioprine (100-200mg/day), should be introduced. In refractory cases, rituximab is suggested, at the dosage of 1 g IV on Days 0 and 14 or 375mg/m2 weekly over 4 weeks, every 6 months. Immunosuppressive therapy (MMF, cyclosporine, or azathioprine) should be discontinued in case of no therapeutic response or side effects. In cases of partial response to such immunosuppressants, they may be maintained in combination with rituximab. However, one should consider the possible side effects of this association, especially infectious ones. The possibility of infectious prophylaxis should be considered.
Oral corticosteroids should be maintained at a dosage of 1mg/kg/day until the development of new lesions is interrupted and pre-existing lesions begin to heal (average of 4 weeks). Afterward, a reduction of 10mg every 2 weeks should be made until the dose reaches approximately 0.5mg/kg. Thereafter, reductions should be made by 10mg every 4 weeks, according to the patient’s clinical evolution. When the dosage reaches 7.5 to 5mg/day of prednisone, absolute care should be taken due to adrenal gland suppression. Prednisone at a dosage of 5mg/day should be replaced with hydrocortisone at 20mg at 8 hours and 10mg at 14 hours until the serum cortisol level exceeds 10µg/dL or, ideally, 18µg/dL. Attention should be given to stressful situations (such as surgeries and infections), in which the dose of hydrocortisone should be doubled.
In the case of pulse therapy, at the end of 3 days, corticosteroid therapy should be maintained through oral administration of prednisone at 0.5 to 1mg/kg/day.
EVOLUTION AND PROGNOSIS
EBA is a disease that can evolve with high morbidity due to its potential for scarring. The cutaneous involvement in EBA can progress with extensive erosions, atrophic scars, nail dystrophies, and fibrosis, with flexion contracture of fingers, limiting adequate mobility.
Different mucosal sites with stratified squamous epithelium may be affected, such as the conjunctival, upper airway, oral, esophageal, genital, and anal areas.
Ocular involvement in EBA is frequent, with various forms of presentation, including conjunctivitis, symblepharon, keratitis, subepithelial vesiculation, perforation, and corneal opacification.136136 Bauer JW, Schaeppi H, Metze D, Muss W, Pohla-Gubo G, Hametner R, et al. Ocular involvement in IgA-epidermolysis bullosa acquisita. B Br J Dermatol. 1999;141:887-92.,137137 Caux F, Kirtschig G, Lemarchand-Venencie F, Venencie PY, Hoang-Xuan T, Robin H, et al. IgA-epidermolysis bullosa acquisita in a child resulting in blindness. Br J Dermatol. 1997;137:270-5. In severe cases, blindness may occur.136136 Bauer JW, Schaeppi H, Metze D, Muss W, Pohla-Gubo G, Hametner R, et al. Ocular involvement in IgA-epidermolysis bullosa acquisita. B Br J Dermatol. 1999;141:887-92.
137 Caux F, Kirtschig G, Lemarchand-Venencie F, Venencie PY, Hoang-Xuan T, Robin H, et al. IgA-epidermolysis bullosa acquisita in a child resulting in blindness. Br J Dermatol. 1997;137:270-5.-138138 Lang PG Jr, Tapert MJ. Severe ocular involvement in a patient with epidermolysis bullosa acquisita. JJ Am Acad Dermatol. 1987;16:439-43. Upper airway involvement can occur, with formation of synechia, erosions, thickening of the epiglottis, and supraglottic stenosis.139139 Benton EC, Bhogal B, Oakley R, Groves RW. Beware the blistering patient with dysphonia. Clin Exp Dermatol. 2013;38:691-2. Some cases result in acute airway obstruction, requiring a tracheostomy.139139 Benton EC, Bhogal B, Oakley R, Groves RW. Beware the blistering patient with dysphonia. Clin Exp Dermatol. 2013;38:691-2.,140140 Clement M, Ratnesar P, Thirumoorthy T, McGrath J, Black MM. Epidermolysis bullosa acquisita--a case with upper airways obstruction requiring tracheostomy and responding to cyclosporin. Clin Exp Dermatol. 1993;18:548-51. Esophageal involvement may present as the formation of erosions and stenosis and dysphagia.113113 Tu J, Kumarasinghe PW. Epidermolysis bullosa acquisita with moderately severe Dysphagia due to esophageal strictures. Indian J Dermatol. 2011;56:224-7.,141141 Ishii N, Furumura M, Hamada T, Mori O, Ohzono A, Ueda A, et al. Oesophageal involvement in epidermolysis bullosa acquisita. Br J Dermatol. 2015;172:288-90. Sometimes, patients must undergo periodic esophageal dilations and, in extreme cases, gastrostomy.
Mucosal involvement may occur in a silent, chronic manner and progress to life-threatening complications.142142 Luke MC, Darling TN, Hsu R, Summers RM, Smith JA, Solomon BI, et al. Mucosal morbidity in patients with epidermolysis bullosa acquisita. Arch Dermatol. 1999;135:954-9. Such impairments worsen the patients’ prognosis, impacting their quality of life and requiring long-term clinical and laboratory follow-up.104104 Delgado L, Aoki V, Santi C, Gabbi T, Sotto M, Maruta C. Clinical and immunopathological evaluation of epidermolysis bullosa acquisita. Clin Exp Dermatol. 2011;36:12-8 In this context, multidisciplinary follow-up becomes mandatory for the early diagnosis and detection of subclinical lesions, thus preventing subsequent severe complications. In addition, mucosal involvement may render the disease more difficult to control, requiring aggressive immunosuppressive therapy.
CONCLUSION
EBA is a rare disease with a broad clinical spectrum, making the evaluation of therapeutic results difficult. Mild forms of the disease may have a favorable response to treatment with dapsone or colchicine. Severe forms, with mucosal involvement, often subclinical, may evolve with acute and/or chronic complications with respiratory, ocular, and digestive sequelae, requiring aggressive treatments. However, no proposed therapeutic regimen has proven to be the standard for severe EBA.
-
*
Work conducted at the Sociedade Brasileira de Dermatologia, Rio de Janeiro (RJ), Brazil.
-
Financial support: None.
REFERENCES
-
1Aoki V, Maruta W, Santi CG. Dermatoses Vesicobolhosas Autoimunes. In: Belda Jr. W, Di Chiacchio N, Criado PR. Tratado de Dermatologia. 2. ed. São Paulo: Atheneu; 2014. p. 417-28.
-
2Alpsoy E, Akman-Karakas A, Uzun S. Geographic variations in epidemiology of two autoimmune bullous diseases: pemphigus and bullous pemphigoid. Arch Dermatol Res. 2015;307:291-8.
-
3Langan SM, Smeeth L, Hubbard R, Fleming KM, Smith CJ, West J. Bullous pemphigoid and pemphigus vulgaris-incidence and mortality in the UK: population based cohort study. BMJ. 2008;337:a180.
-
4Christophoridis S, Büdinger L, Borradori L, Hunziker T, Merk HF, Hertl M. IgG, IgA and IgE autoantibodies against the ectodomain of BP180 in patients with bullous and cicatricial pemphigoid and linear IgA bullous dermatosis. Br J Dermatol. 2000;143:349-55.
-
5Dimson OG, Giudice GJ, Fu CL, Van den Bergh F, Warren SJ, Janson MM, et al. Identification of a potential effector function for IgE autoantibodies in the organ-specific autoimmune disease bullous pemphigoid. J Invest Dermatol. 2003;120:784-8.
-
6Fang H, Zhang Y, Li N, Wang G, Liu Z. The Autoimmune Skin Disease Bullous Pemphigoid: The Role of Mast Cells in Autoantibody-Induced Tissue Injury. Front Immunol. 2018;9:407
-
7Freire PC, Muñoz CH, Stingl G. IgE autoreactivity in bullous pemphigoid: eosinophils and mast cells as major targets of pathogenic immune reactants. Br J Dermatol. 2017;177:1644-53.
-
8Hammers CM, Stanley JR. Mechanisms of Disease: Pemphigus and Bullous Pemphigoid. Annu Rev Pathol. 2016;11:175-97.
-
9Hammers CM, Payne AS. Clinical significance of immunoglobulin E in bullous pemphigoid. Br J Dermatol. 2017;177:13-4.
-
10Hashimoto T. Induced autoimmune bullous diseases. Br J Dermatol. 2017;176:304-5
-
11Hashimoto T, Tsuruta D, Ishii N. Immunoglobulin E Autoantibodies in Bullous Pemphigoid Detected by Immunoglobulin E Enzyme-Linked Immunosorbent Assays. JAMA Dermatol. 2017;153:15-7.
-
12Hashimoto T, Ohzono A, Teye K, Numata S, Hiroyasu S, Tsuruta D, et al. Detection of IgE autoantibodies to BP180 and BP230 and their relationship to clinical features in bullous pemphigoid. Br J Dermatol. 2017;177:141-51.
-
13Liu Z, Chen L, Zhang C, Xiang LF. Circulating anti-bullous pemphigoid 180 autoantibody can be detected in a wide clinical spectrum: A cross-sectional study. J Am Acad Dermatol. 2018. pii: S0190-9622(18)32067-X
-
14Liu Y, Li L, Xia Y. BP180 Is Critical in the Autoimmunity of Bullous Pemphigoid. Front Immunol. 2017;8:1752
-
15Lin L, Hwang BJ, Culton DA, Li N, Burette S, Koller BH, et al. Eosinophils Mediate Tissue Injury in the Autoimmune Skin Disease Bullous Pemphigoid. J Invest Dermatol. 2018;138:1032-43.
-
16Messingham KN, Holahan HM, Frydman AS, Fullenkamp C, Srikantha R, Fairley JA. Human eosinophils express the high affinity IgE receptor, FceRI, in bullous pemphigoid. PLoS One. 2014;9:e107725.
-
17Moriuchi R, Nishie W, Ujiie H, Natsuga K, Shimizu H. In vivo analysis of IgE autoantibodies in bullous pemphigoid: a study of 100 cases. J Dermatol Sci. 2015;78:21-5
-
18Nishie W. Update on the pathogenesis of bullous pemphigoid: an autoantibody- mediated blistering disease targeting collagen XVII. J Dermatol Sci. 2014;73:179-86
-
19Saniklidou AH, Tighe PJ, Fairclough LC, Todd I. IgE autoantibodies and their association with the disease activity and phenotype in bullous pemphigoid: a systematic review. Arch Dermatol Res. 2018;310:11-28.
-
20Ren Z, Hsu DY, Brieva J, Silverberg NB, Langan SM, Silverberg JI. Hospitalization, inpatient burden and comorbidities associated with bullous pemphigoid in the U.S.A. Br J Dermatol. 2017;176:87-99.
-
21Lucariello RJ, Villablanca SE, Mascaró JM Jr, Reichel M. Association between bullous pemphigoid and malignancy: A meta-analysis. Australas J Dermatol. 2018. doi: 10.1111/ajd.12764.
» https://doi.org/10.1111/ajd.12764. -
22Lo Schiavo A, Ruocco E, Brancaccio G, Caccavale S, Ruocco V, Wolf R. Bullous pemphigoid: etiology, pathogenesis, and inducing factors: facts and controversies. Clin Dermatol. 2013;31:391-9.
-
23Bernard P, Borradori. Pemphigoid Group. In: Bolognia J.L, Jorizzo JL, Schaffer JV, editors. Dermatology. 3rd ed. Philadelphia, Pa.: Elsevier Saunders; 2012: p. 475-90.
-
24Bastuji-Garin S, Joly P, Lemordant P, Sparsa A, Bedane C, Delaporte E, et al. Risk factors for bullous pemphigoid in the elderly: a prospective case-control study. J Invest Dermatol. 2011;131:637-43.
-
25Jedlickova H, Hlubinka M, Pavlik T, Semradova V, Budinska E, Vlasin Z.. Bullous pemphigoid and internal diseases - A case-control study. Eur J Dermatol. 2010;20:96-101.
-
26Cordel N, Chosidow O, Hellot MF, Delaporte E, Lok C, Vaillant L, et al. Neurological disorders in patients with bullous pemphigoid. Dermatology. 2007;215:187-91.
-
27Lai YC, Yew YW, Lambert WC. Bullous pemphigoid and its association with neurological diseases: a systematic review and meta-analysis. J Eur Acad Dermatol Venereol. 2016;30:2007-15.
-
28Tarazona MJ, Mota AN, Gripp AC, Unterstell N, Bressan AL. Bullous pemphigoid and neurological disease: statistics from a dermatology service. An Bras Dermatol. 2015;90:280-2.
-
29Teixeira VB, Cabral R, Brites MM, Vieira R, Figueiredo A. Bullous pemphigoid and comorbidities: a case-control study in Portuguese patients. An Bras Dermatol. 2014;89:274-8.
-
30Taghipour K, Chi CC, Bhogal B, Groves RW, Venning V, Wojnarowska F. Immunopathological characteristics of patients with bullous pemphigoid and neurological disease. J Eur Acad Dermatol Venereol. 2014;28:569-73.
-
31Vernal S, Julio T, Cruz F, Turatti A, Ishii N, Hashimoto T, et al. Bullous Pemphigoid Associated with Ischemic Cerebrovascular Accident and Dementia: Exclusive Blistering Lesions on the Upper Hemiparetic Limb. Acta Dermatovenerol Croat. 2018;26:179-82.
-
32Milani-Nejad N, Zhang M, Kaffenberger J. The association between bullous pemphigoid and neurological disorders: a systematic review. Eur J Dermatol. 2017;27:472-81.
-
33Försti AK, Huilaja L, Schmidt E, Tasanen K. Neurological and psychiatric associations in bullous pemphigoid-more than skin deep? Exp Dermatol. 2017;26:1228-34.
-
34Ali A, Hu L, Zhao F, Qiu W, Wang P, Ma X, et al. BPAG1, a distinctive role in skin and neurological diseases. Semin Cell Dev Biol. 2017;69:34-96
-
35Kokkonen N, Herukka SK, Huilaja L, Kokki M, Koivisto AM, Hartikainen P, et al. Increased Levels of the Bullous Pemphigoid BP180 Autoantibody Are Associated with More Severe Dementia in Alzheimer's Disease. J Invest Dermatol. 2017;137:71-6.
-
36Julio TA, Vernal S, Massaro JD, Silva MC, Donadi EA, Moriguti JC, et al. Biological predictors shared by dementia and bullous pemphigoid patients point out a cross-antigenicity between BP180/BP230 brain and skin isoforms. Immunol Res. 2018;66:567-76.
-
37Chagury AA, Sennes LU, Gil JM, Kalil J, Rodrigues H, Rosales CB, et al. HLA-C*17, DQB1*03:01, DQA1*01:03 and DQA1*05:05 Alleles Associated to Bullous Pemphigoid in Brazilian Population. Ann Dermatol. 2018;30:8-12.
-
38Amber KT, Zikry J, Hertl M. A multi-hit hypothesis of bullous pemphigoid and associated neurological disease: Is HLA-DQB1*03:01, a potential link between immune privileged antigen exposure and epitope spreading? HLA. 2017;89:127-34
-
39Schmidt E, Zillikens D. Pemphigoid diseases. Lancet. 2013;381:320-32.
-
40Arbache ST, Nogueira TG, Delgado L, Miyamoto D, Aoki V. Immunofluorescence testing in the diagnosis of autoimmune blistering diseases: overview of 10-year experience. An Bras Dermatol. 2014;89:885-9.
-
41Ishii K. Importance of serological tests in diagnosis of autoimmune blistering diseases. J Dermatol. 2015;42:3-10.
-
42Julio T, Vernal S, Turatti A, Roselino AM. Anti-desmogleins autoantibodies detected by ELISA and blotting in bullous pemphigoid: what do they mean? Int J Dermatol. 2018;57:124-7
-
43Feliciani C, Joly P, Jonkman MF, Zambruno G, Zillikens D, Ioannides D, et al. Management of bullous pemphigoid: the European Dermatology Forum consensus in collaboration with the European Academy of Dermatology and Venereology. Br J Dermatol. 2015;172:867-77.
-
44Brulefert A, Le Jan S, Plée J, Durlach A, Bernard P, Antonicelli F, et al. Variation of the epidermal expression of glucocorticoid receptor-beta as potential predictive marker of bullous pemphigoid outcome. Exp Dermatol. 2017;26:1261-6.
-
45Kubin ME, Hellberg L, Palatsi R. Glucocorticoids: The mode of action in bullous pemphigoid. Exp Dermatol. 2017;26:1253-60.
-
46Gual A, Iranzo P, Mascaró JM Jr. Treatment of bullous pemphigoid with low- dose oral cyclophosphamide: a case series of 20 patients. J Eur Acad Dermatol Venereol. 2014;28:814-8.
-
47Schmidt E, Spindler V, Eming R, Amagai M, Antonicelli F, Baines JF, et al. Meeting Report of the Pathogenesis of Pemphigus and Pemphigoid Meeting in Munich, September 2016. J Invest Dermatol. 2017;137:1199-203.
-
48Shetty S, Ahmed AR. Treatment of bullous pemphigoid with rituximab: critical analysis of the current literature. J Drugs Dermatol. 2013 ;12:672-7.
-
49Ludwig RJ, Kalies K, Köhl J, Zillikens D, Schmidt E. Emerging treatments for pemphigoid diseases. Trends Mol Med. 2013;19:501-12.
-
50Lazarova Z, Yancey K. Cicatricial pemphigoid: immunopathogenesis and treatment. Derm Ther. 2002;15:382-8.
-
51Kridin K. Subepidermal autoimmune bullous diseases: overview, epidemiology, and associations. Immunol Res. 2018;66:6-17.
-
52Kourosh AS, Yancey KB. Pathogenesis of mucous membrane pemphigoid. Dermatol Clin. 2011;29:479-84
-
53Chan LS, Ahmed AR, Anhalt GJ, Bernauer W, Cooper KD, Elder MJ, et al. The first international consensus on mucous membrane pemphigoid: definition, diagnostic criteria, pathogenic factors, medical treatment, and prognostic indicators. Arch Dermatol. 2002;138:370-9.
-
54Kasperkiewicz M, Zillikens D, Schmidt E. Pemphigoid diseases: pathogenesis, diagnosis, and treatment. Autoimmunity. 2012;45:55-70.
-
55Xu HH, Werth VP, Parisi E, Sollecito TP. Mucous Membrane Pemphigoid. Dent Clin North Am. 2013;57:611-30.
-
56Chan LS. Mucous membrane pemphigoid. Clin Dermatol. 2001;19:703-11.
-
57Parisi E, Raghavendra S, Werth VP, Sollecito TP. Modification to the approach of the diagnosis of mucous membrane pemphigoid: A case report and literature review. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2003;95:182-6.
-
58Barbosa Ldo N, Silva RS, Verardino GC, Gripp AC, Alves Mde F. Mucous membrane pemphigoid with severe esophageal stricture. An Bras Dermatol. 2011;86:565-8.
-
59Murrell DF, Marinovic B, Caux F, Prost C, Ahmed R, Wozniak K, et al. Definitions and outcome measures for mucous membrane pemphigoid: recommendations of an international panel of experts. J Am Acad Dermatol. 2015;72:168-74.
-
60Knudson RM, Kalaaji AN, Bruce AJ. The management of mucous membrane pemphigoid and pemphigus. Dermatol Ther. 2010;23:268-80.
-
61Gonzalez-Moles MA, Ruiz-Avila I, Rodriguez-Archilla A, Morales-Garcia P, Mesa-Aguado F, Bascones-Martinez A, et al. Treatment of severe erosive gingival lesions by topical application of clobetasol propionate in custom trays. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2003;95:688-92.
-
62Gilbert SC. Management and prevention of recurrent herpes labialis in immunocompetent patients. Herpes. 2007;14:56-61.
-
63Cernik C, Gallina K, Brodell RT. The treatment of herpes simplex infections: an evidence-based review. Arch Intern Med. 2008;168:1137-44.
-
64Assmann T, Becker J, Ruzicka T, Megahed M. Topical tacrolimus for oral cicatricial pemphigoid. Clin Exp Dermatol. 2004;29:674-6.
-
65Suresh L, Martinez Calixto LE, Radfar L. Successful treatment of mucous membrane pemphigoid with tacrolimus. Spec Care Dentist. 2006;26:66-70.
-
66Lee HY, Blazek C, Beltraminelli H, Borradori L.. Oral mucous membrane pemphigoid: complete response to topical tacrolimus. Acta Derm Venereol. 2011;91:604-5.
-
67Kourosh AS, Yancey KB. Therapeutic approaches to patients with mucous membrane pemphigoid. Dermatol Clin. 2011;29:637-41.
-
68Arash A, Shirin L. The management of oral mucous membrane pemphigoid with dapsone and topical corticosteroid. J Oral Pathol Med. 2008;37:341-4.
-
69Gürcan HM, Ahmed AR. Efficacy of dapsone in the treatment of pemphigus and pemphigoid: analysis of current data. Am J Clin Dermatol. 2009;10:383-96.
-
70Hegarty AM, Ormond M, Sweeney M, Hodgson T. Dapsone efficacy and adverse events in the management of mucous membrane pemphigoid. Eur J Dermatol. 2010;20:223-4.
-
71Megahed M, Schmiedeberg S, Becker J, Ruzicka T. Treatment of cicatricial pemphigoid with mycophenolate mofetil as a steroid-sparing agent. J Am Acad Dermatol. 2001;45:256-9.
-
72Nottage JM, Hammersmith KM, Murchison AP, Felipe AF, Penne R, Raber I. Treatment of mucous membrane pemphigoid with mycophenolate mofetil. Cornea. 2013;32:810-5.
-
73Perlis C, Pan TD, McDonald CJ. Cytotoxic agents. In: Wolverton SE, editor. Comprehensive Dermatologic Drug Therapy. 2nd ed. Philadelphia: Elsevier Inc.; 2007. p.197.
-
74McCluskey P, Chang JH, Singh R, Wakefield D. Methotrexate therapy for ocular cicatricial pemphigoid. Ophthalmology. 2004;111:796-801.
-
75Neff AG, Turner M, Mutasim DF. Treatment strategies in mucous membrane pemphigoid. Ther Clin Risk Manag. 2008;4:617-26.
-
76Prey S, Paul C. Effect of folic or folinic acid supplementation on methotrexate- associated safety and efficacy in inflammatory disease: a systematic review. Br J Dermatol. 2009;160:622-8.
-
77Munyangango EM, Le Roux-Villet C, Doan S, Pascal F, Soued I, Alexandre M, et al. Oral cyclophosphamide without corticosteroids to treat mucous membrane pemphigoid. Br J Dermatol. 2013;168:381-90.
-
78Tavakolpour S. The role of intravenous immunoglobulin in treatment of mucous membrane pemphigoid: A review of literature. J Res Med Sci. 2016;21:37.
-
79Ahmed AR, Colón JE.. Comparison between intravenous immunoglobulin and conventional immunosuppressive therapy regimens in patients with severe oral pemphigoid: effects on disease progression in patients nonresponsive to dapsone therapy. Arch Dermatol. 2001;137:1181-9.
-
80Le Roux-Villet C, Prost-Squarcioni C, Alexandre M, Caux F, Pascal F, Doan S, et al. Rituximab for patients with refractory mucous membrane pemphigoid. Arch Dermatol. 2011;147:843-9.
-
81Bomm L, Fracaroli TS, Sodré JL, Bressan A, Gripp AC. Off-label use of rituximab in dermatology: pemphigus treatment. An Bras Dermatol. 2013;88:676-8.
-
82Sacher C, Rubbert A, König C, Scharffetter-Kochanek K, Krieg T, Hunzelmann N. Treatment of recalcitrant cicatricial pemphigoid with the tumor necrosis factor alpha antagonist etanercept. J Am Acad Dermatol. 2002;46:113-5.
-
83Canizares MJ, Smith DI, Conners MS, Maverick KJ, Heffernan MP. Successful treatment of mucous membrane pemphigoid with etanercept in 3 patients. Arch Dermatol. 2006;142:1457-61.
-
84Heffernan MP, Bentley DD. Successful treatment of mucous membrane pemphigoid with infliximab. Arch Dermatol. 2006;142:1268-70.
-
85Lehman JS, Camilleri MJ, Gibson LE. Epidermolysis bullosa acquisita: concise review and practical considerations. Int J Dermatol. 2009;48:227-35;
-
86Zhu XJ, Niimi Y, Bystryn JC. Epidermolysis bullosa acquisita. Incidence in patients with basement membrane zone antibodies. Arch Dermatol. 1990;126:171-4.
-
87Kridin K. Subepidermal autoimmune bullous diseases: overview, epidemiology, and associations. Immunol Res. 2018;66:6-17.
-
88Kim JH, Kim SC. Epidermolysis bullosa acquisita. J Eur Acad Dermatol Venereol. 2013;27:1204-13
-
89Gupta R, Woodley DT, Chen M. Epidermolysis bullosa acquisita. Clin Dermatol. 2012;30:60-9.
-
90Kasperkiewicz M, Sadik CD, Bieber K, Ibrahim SM, Manz RA, Schmidt E, et al. Epidermolysis Bullosa Acquisita: From Pathophysiology to Novel Therapeutic Options. J Invest Dermatol. 2016;136:24-33
-
91Hashimoto T, Jin Z, Ishii N. Clinical and immunological studies for 105 Japanese seropositive patients of epidermolysis bullosa acquisita examined at Kurume University. Expert Rev Clin Immunol. 2016;12:895-902
-
92Ludwig RJ. Clinical presentation, pathogenesis, diagnosis, and treatment of epidermolysis bullosa acquisita. ISRN Dermatol. 2013;2013:812029.
-
93Chen M, Kim GH, Prakash L, Woodley DT. Epidermolysis bullosa acquisita: autoimmunity to anchoring fibril collagen. Autoimmunity. 2012;45:91-101
-
94Vorobyev A, Ludwig RJ, Schmidt E. Clinical features and diagnosis of epidermolysis bullosa acquisita. Expert Rev Clin Immunol. 2017;13:157-69.
-
95Asfour L, Chong H, Mee J, Groves R, Singh M. Epidermolysis Bullosa Acquisita (Brunsting-Perry Pemphigoid Variant) Localized to the Face and Diagnosed With Antigen Identification Using Skin Deficient in Type VII Collagen. Am J Dermatopathol. 2017;39:e90-e96.
-
96M Meijer JM, Atefi I, Diercks GFH, Vorobyev A, Zuiderveen J, Meijer HJ, et al. Serration pattern analysis for differentiating epidermolysis bullosa acquisita from other pemphigoid diseases. J Am Acad Dermatol. 2018;78:754-9.e6.
-
97Mehren CR, Gniadecki R. Epidermolysis bullosa acquisita: current diagnosis and therapy. Dermatol Reports. 2011;3:e38.
-
98Pope E, Lara-Corrales I, Mellerio J, Martinez A, Schultz G, Burrell R, et al. A consensus approach to wound care in epidermolysis bullosa. J Am Acad Dermatol. 2012;67:904-17.
-
99Kirtschig G, Murrell D, Wojnarowska F, Khumalo N. Interventions for mucous membrane pemphigoid and epidermolysis bullosa acquisita. C Cochrane Database Syst Rev. 2003:CD004056.
-
100Ishii N, Hamada T, Dainichi T, Karashima T, Nakama T, Yasumoto S, et al. Epidermolysis bullosa acquisita: what's new? J Dermatol. 2010;37:220-30.
-
101Iranzo P, Herrero-González JE, Mascaró-Galy JM, Suárez-Fernández R, España A. Epidermolysis bullosa acquisita: a retrospective analysis of 12 patients evaluated in four tertiary hospitals in Spain. Br J Dermatol. 2014;171:1022-30
-
102Kim JH, Kim YH, Kim SC. Epidermolysis bullosa acquisita: a retrospective clinical analysis of 30 cases. Acta Derm Venereol. 2011;91:307-12.
-
103Intong LR, Murrell DF. Management of epidermolysis bullosa acquisita. Dermatol Clin. 2011;29:643-7
-
104Delgado L, Aoki V, Santi C, Gabbi T, Sotto M, Maruta C. Clinical and immunopathological evaluation of epidermolysis bullosa acquisita. Clin Exp Dermatol. 2011;36:12-8
-
105Gürcan HM, Ahmed AR. Current concepts in the treatment of epidermolysis bullosa acquisita. Expert Opin Pharmacother. 2011 ;12:1259-68.
-
106Goyal N, Rao R, Balachandran C, Pai S, Bhogal BS, Schmidt E, et al. Childhood Epidermolysis Bullosa Acquisita: Confirmation of Diagnosis by Skin Deficient in Type VII Collagen, Enzyme-linked Immunosorbent Assay, and Immunoblotting. Indian J Dermatol. 2016;61:329-32.
-
107Yang B, Wang C, Wang N, Pan F, Chen S, Zhou G, et al. Childhood epidermolysis bullosa acquisita: report of a Chinese case. Pediatr Dermatol. 2012;29:614-7.
-
108Mutasim DF. Autoimmune bullous dermatoses in the elderly: an update on pathophysiology, diagnosis and management. Drugs Aging. 2010;27:1-19.
-
109D Dasgeb B, Kornreich D, McGuinn K, Okon L, Brownell I, Sackett DL. Colchicine: an ancient drug with novel applications. Br J Dermatol. 2018;178:350-6
-
110Adachi A, Komine M, Suzuki M, Murata S, Hirano T, Ishii N, et al. Oral colchicine monotherapy for epidermolysis bullosa acquisita: Mechanism of action and efficacy. J Dermatol. 2016;43:1389-91.
-
111Culton DA, Diaz LA. Treatment of subepidermal immunobullous diseases. Clin Dermatol. 2012;30:95-102.
-
112Kawase K, Oshitani Y, Mizutani Y, Shu E, Fujine E, Seishima M. Inflammatory epidermolysis bullosa acquisita effectively treated with minocycline. Acta Derm Venereol. 2014;94:615-6.
-
113Tu J, Kumarasinghe PW. Epidermolysis bullosa acquisita with moderately severe Dysphagia due to esophageal strictures. Indian J Dermatol. 2011;56:224-7.
-
114Meurer M. Immunosuppressive therapy for autoimmune bullous diseases. Clin Dermatol. 2012;30:78-83.
-
115Sami N. Mycophenolate mofetil (MMF) in the treatment of epidermolysis bullosa acquisita (EBA) long-term follow-up. JAAD Case Rep. 2015;1:321-3.
-
116Yan TM, He CX, Hua BL, Li L, Jin HZ, Liu YH, et al. Coexistence of acquired hemophilia A and epidermolysis bullosa acquisita: Two case reports and published work review. J Dermatol. 2017;44:76-9.
-
117Mosqueira CB, Furlani Lde A, Xavier AF, Cunha PR, Galvão AM. Intravenous immunoglobulin for treatment of severe acquired bullous epidermolysis refractory to conventional immunosuppressive therapy. An Bras Dermatol. 2010;85:521-4.
-
118Ahmed AR, Gürcan HM. Treatment of epidermolysis bullosa acquisita with intravenous immunoglobulin in patients non-responsive to conventional therapy: clinical outcome and post-treatment long-term follow-up. J Eur Acad Dermatol Venereol. 2012;26:1074-83.
-
119Tayal U, Burton J, Dash C, Wojnarowska F, Chapel H. Subcutaneous immunoglobulin therapy for immunomodulation in a patient with severe epidermolysis bullosa acquisita. Clin Immunol. 2008;129:518-9.
-
120Sanli H, Akay BN, Ayyildiz E, Anadolu R, Ilhan O. Remission of severe autoimmune bullous disorders induced by long-term extracorporeal photochemotherapy. Transfus Apher Sci. 2010;43:353-9.
-
121Baroudjian B, Le Roux-Villet C, Bréchignac S, Alexandre M, Caux F, Prost-Squarcioni C, et al. Long-term efficacy of extracorporeal photochemotherapy in a patient with refractory epidermolysis bullosa acquisita. Eur J Dermatol. 2012;22:795-7.
-
122Liszewski W, Omland SH, Gniadecki R. The successful use of extracorporeal photopheresis in a 12-year-old patient with refractory epidermolysis bullosa acquisita. Pediatr Dermatol. 2015;32:e60-1.
-
123Adamski J, Kinard T, Ipe T, Cooling L. Extracorporeal photopheresis for the treatment of autoimmune diseases. Transfus Apher Sci. 2015;52:171-82.
-
124Knobler R, Berlin G, Calzavara-Pinton P, Greinix H, Jaksch P, Laroche L, et al. Guidelines on the use of extracorporeal photopheresis. J Eur Acad Dermatol Venereol. 2014;28(Suppl 1):1-37
-
125Lamberts A, Euverman HI, Terra JB, Jonkman MF, Horváth B. Effectiveness and Safety of Rituximab in Recalcitrant Pemphigoid Diseases. Front Immunol. 2018;9:248.
-
126Joly P, Maho-Vaillant M, Prost-Squarcioni C, Hebert V, Houivet E, Calbo S, et al. First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): a prospective, multicentre, parallel-group, open-label randomised trial. Lancet. 2017;389:2031-2040
-
127Hebert V, Joly P. Rituximab in pemphigus. Immunotherapy. 2018;10:27-37.
-
128Saha M, Cutler T, Bhogal B, Black MM, Groves RW. Refractory epidermolysis bullosa acquisita: successful treatment with rituximab. Clin Exp Dermatol. 2009;34:e979-80
-
129Kim JH, Lee SE, Kim SC. Successful treatment of epidermolysis bullosa acquisita with rituximab therapy. J Dermatol. 2012;39:477-9.
-
130McKinley SK, Huang JT, Tan J, Kroshinsky D, Gellis S. A case of recalcitrant epidermolysis bullosa acquisita responsive to rituximab therapy. Pediatr Dermatol. 2014;31:241-4
-
131Cavailhes A, Balme B, Gilbert D, Skowron F. Successful use of combined corticosteroids and rituximab in the treatment of recalcitrant epidermolysis bullosa acquisita. Ann Dermatol Venereol. 2009;136:795-9
-
132Oktem A, Akay BN, Boyvat A, Kundakci N, Erdem C, Bostanci S, et al. Long-term results of rituximab-intravenous immunoglobulin combination therapy in patients with epidermolysis bullosa acquisita resistant to conventional therapy. J Dermatolog Treat. 2017;28:50-4.
-
133Kubisch I, Diessenbacher P, Schmidt E, Gollnick H, Leverkus M. Premonitory epidermolysis bullosa acquisita mimicking eyelid dermatitis: successful treatment with rituximab and protein A immunoapheresis. Am J Clin Dermatol. 2010;11:289-93.
-
134Kolesnik M, Becker E, Reinhold D, Ambach A, Heim MU, Gollnick H, et al. Treatment of severe autoimmune blistering skin diseases with combination of protein A immunoadsorption and rituximab: a protocol without initial high dose or pulse steroid medication. J Eur Acad Dermatol Venereol. 2014;28:771-80.
-
135Prost-Squarcioni C, Ingen-Housz-Oro S, Joly P, Bernard P, Bedane C; Centres de référence des maladies bulleuses auto-immunes. Société Française deDermatologie. Epidermolysis bullosa acquisita. Guidelines for the diagnosis and treatment. Centres deréférence des maladies bulleuses auto-immunes. Société Française de Dermatologie. Ann Dermatol Venereol. 2011;138:274-9.
-
136Bauer JW, Schaeppi H, Metze D, Muss W, Pohla-Gubo G, Hametner R, et al. Ocular involvement in IgA-epidermolysis bullosa acquisita. B Br J Dermatol. 1999;141:887-92.
-
137Caux F, Kirtschig G, Lemarchand-Venencie F, Venencie PY, Hoang-Xuan T, Robin H, et al. IgA-epidermolysis bullosa acquisita in a child resulting in blindness. Br J Dermatol. 1997;137:270-5.
-
138Lang PG Jr, Tapert MJ. Severe ocular involvement in a patient with epidermolysis bullosa acquisita. JJ Am Acad Dermatol. 1987;16:439-43.
-
139Benton EC, Bhogal B, Oakley R, Groves RW. Beware the blistering patient with dysphonia. Clin Exp Dermatol. 2013;38:691-2.
-
140Clement M, Ratnesar P, Thirumoorthy T, McGrath J, Black MM. Epidermolysis bullosa acquisita--a case with upper airways obstruction requiring tracheostomy and responding to cyclosporin. Clin Exp Dermatol. 1993;18:548-51.
-
141Ishii N, Furumura M, Hamada T, Mori O, Ohzono A, Ueda A, et al. Oesophageal involvement in epidermolysis bullosa acquisita. Br J Dermatol. 2015;172:288-90.
-
142Luke MC, Darling TN, Hsu R, Summers RM, Smith JA, Solomon BI, et al. Mucosal morbidity in patients with epidermolysis bullosa acquisita. Arch Dermatol. 1999;135:954-9.
Publication Dates
-
Publication in this collection
30 June 2019 -
Date of issue
Mar-Apr 2019
History
-
Received
14 Sept 2018 -
Accepted
30 Jan 2019