Resumos
A esclerose sistêmica (ES) é uma doença sistêmica caracterizada por microangiopatia, exuberante atividade do fibroblasto e anormalidades imunológicas. A faceta microangiopática é responsável pela maioria das complicações com potencial mortalidade. A interação entre o endotélio e as plaquetas desempenha um papel importante na fisiopatologia da ES. Evidências de ativação plaquetária na ES incluem níveis séricos elevados de substâncias derivadas das plaquetas (fator 4 plaquetário, fator de Von Willenbrand, tromboxane A2 e ß-tromboglobulina), agregados plaquetários circulantes, anormalidades ultraestruturais nas plaquetas e a presença de plaquetas aderidas ao endotélio. A presente revisão aborda de forma crítica os princípios e problemas potenciais dos principais métodos de avaliação da função e ativação plaquetárias. A avaliação da capacidade de agregação das plaquetas é feita com e sem a adição de agonistas (adenosina difosfato, colágeno e adrenalina). A ativação plaquetária pode ser avaliada de duas formas: pela dosagem da concentração plasmática de substâncias que são liberadas pela ativação plaquetária (ex., tromboxane, fator 4 plaquetário) e pela mensuração da expressão em membrana plaquetária de moléculas que são transportadas para a membrana plaquetária durante o processo de ativação das plaquetas (ex., GMP-140 e glicoproteína IIb/IIIa). Uma contribuição recente para este campo foi dada pela demonstração de que a enolase específica de neurônios (NSE) é liberada das plaquetas para o plasma em pacientes com ES em atividade. Assim, a dosagem da NSE, que é um exame disponível nos principais laboratórios com ênfase na dosagem de marcadores tumorais, pode se tornar um marcador de atividade plaquetária de grande utilidade em diversas condições clínicas.
esclerose sistêmica; agregação plaquetária; ativação plaquetária
Systemic sclerosis (SSc) is a multisystemic disease characterized by microangiopathy, exuberant fibroblast activity and immunologic abnormalities. The microangiopathic aspect is responsible for the most severe and life-threatening features of the disease. The interaction between endothelium and platelets plays an important role in the pathophysiology of SSc. Evidence of platelet activation in SSc includes high plasma levels of platelet-derived substances (Von Willenbrand factor, platelet factor 4, thromboxane A2, and ß-thromboglobulin), circulating platelet aggregates, ultra structural abnormalities in platelets, and the presence of platelets adhered to the endothelium. This review addresses the principles and possible pitfalls of the main methods for evaluation of platelet activation and function. The assessment of the ability of platelet aggregation is performed with and without the addition of agonists (adenosine diphosphate, collagen, adrenaline). Platelet activation may be assessed by two ways: by measurement of plasma concentration of substances that are released as the platelets are activated (e.g., thromboxane, platelet factor 4) and by the measurement of membrane expression of molecules that are transported to the platelet membrane during the activation process (e.g., GMP-140, glycoprotein IIb/IIIa). A recent contribution to this field was the demonstration that neuron-specific enolase (NSE) is released from the platelets into the blood in patients with active SSc. NSE, which is readily available in clinical laboratories with emphasis in tumor markers, may become a useful platelet activation marker in a series of clinical conditions.
systemic sclerosis; platelet aggregation; platelet activation
ARTIGO DE REVISÃO REVIEW ARTICLE
Ativação plaquetária na esclerose sistêmica e alternativas metodológicas para sua avaliação(* * Disciplina de Reumatologia, Escola Paulista de Medicina, Universidade Federal de São Paulo (EPM-UNIFESP). )
Platelet activation in systemic sclerosis and methodological options to its assessment
Paulo Sergio MassabkiI; Dayse Maria LourençoII; Luís Eduardo Coelho AndradeI
IDisciplina de Reumatologia. EPM-UNIFESP
IIDisciplina de Hematologia. EPM-UNIFESP
Endereço para correspondência Endereço para correspondência: Luís Eduardo C. Andrade Rua Botucatu 740 CEP 04029-062, Vila Clementino, São Paulo, SP E-mail: luis@reumato.epm.br
RESUMO
A esclerose sistêmica (ES) é uma doença sistêmica caracterizada por microangiopatia, exuberante atividade do fibroblasto e anormalidades imunológicas. A faceta microangiopática é responsável pela maioria das complicações com potencial mortalidade. A interação entre o endotélio e as plaquetas desempenha um papel importante na fisiopatologia da ES. Evidências de ativação plaquetária na ES incluem níveis séricos elevados de substâncias derivadas das plaquetas (fator 4 plaquetário, fator de Von Willenbrand, tromboxane A2 e ß-tromboglobulina), agregados plaquetários circulantes, anormalidades ultraestruturais nas plaquetas e a presença de plaquetas aderidas ao endotélio. A presente revisão aborda de forma crítica os princípios e problemas potenciais dos principais métodos de avaliação da função e ativação plaquetárias. A avaliação da capacidade de agregação das plaquetas é feita com e sem a adição de agonistas (adenosina difosfato, colágeno e adrenalina). A ativação plaquetária pode ser avaliada de duas formas: pela dosagem da concentração plasmática de substâncias que são liberadas pela ativação plaquetária (ex., tromboxane, fator 4 plaquetário) e pela mensuração da expressão em membrana plaquetária de moléculas que são transportadas para a membrana plaquetária durante o processo de ativação das plaquetas (ex., GMP-140 e glicoproteína IIb/IIIa). Uma contribuição recente para este campo foi dada pela demonstração de que a enolase específica de neurônios (NSE) é liberada das plaquetas para o plasma em pacientes com ES em atividade. Assim, a dosagem da NSE, que é um exame disponível nos principais laboratórios com ênfase na dosagem de marcadores tumorais, pode se tornar um marcador de atividade plaquetária de grande utilidade em diversas condições clínicas.
Palavras-chave: esclerose sistêmica, agregação plaquetária, ativação plaquetária.
ABSTRACT
Systemic sclerosis (SSc) is a multisystemic disease characterized by microangiopathy, exuberant fibroblast activity and immunologic abnormalities. The microangiopathic aspect is responsible for the most severe and life-threatening features of the disease. The interaction between endothelium and platelets plays an important role in the pathophysiology of SSc. Evidence of platelet activation in SSc includes high plasma levels of platelet-derived substances (Von Willenbrand factor, platelet factor 4, thromboxane A2, and ß-thromboglobulin), circulating platelet aggregates, ultra structural abnormalities in platelets, and the presence of platelets adhered to the endothelium. This review addresses the principles and possible pitfalls of the main methods for evaluation of platelet activation and function. The assessment of the ability of platelet aggregation is performed with and without the addition of agonists (adenosine diphosphate, collagen, adrenaline). Platelet activation may be assessed by two ways: by measurement of plasma concentration of substances that are released as the platelets are activated (e.g., thromboxane, platelet factor 4) and by the measurement of membrane expression of molecules that are transported to the platelet membrane during the activation process (e.g., GMP-140, glycoprotein IIb/IIIa). A recent contribution to this field was the demonstration that neuron-specific enolase (NSE) is released from the platelets into the blood in patients with active SSc. NSE, which is readily available in clinical laboratories with emphasis in tumor markers, may become a useful platelet activation marker in a series of clinical conditions.
Palavras chaves: systemic sclerosis, platelet aggregation, platelet activation.
INTRODUÇÃO
A esclerose sistêmica é um distúrbio sistêmico de etiologia desconhecida, caracterizada por microangiopatia e fibrose da pele e de vísceras, incluindo o trato gastrintestinal, os pulmões, o coração e os rins(1-18). O grau e o ritmo de acometimento da pele e dos órgãos internos variam entre os pacientes. Entretanto, podem ser identificados dois subgrupos, apesar de existir alguma sobreposição. Um subgrupo recebe a designação de esclerose sistêmica difusa e caracteriza-se pelo surgimento rápido de espessamento simétrico da pele nas partes proximal e distal dos membros, na face e no tronco. Nesses pacientes ocorre um risco maior de manifestações viscerais, especialmente nos primeiros anos da enfermidade. O outro subgrupo é o da esclerose sistêmica limitada, definida pelo espessamento cutâneo simétrico limitado aos dedos ou à extremidade distal dos membros e à face. Com freqüência esse subgrupo possui características que configuram a chamada síndrome CREST (calcinose, fenômeno de Raynaud, dismotilidade esofagiana, esclerodactilia e teleangiectasia)(19,24).
A esclerose sistêmica apresenta distribuição mundial e afeta todas as raças(25,26). O início da doença ocorre em geral entre a terceira e quinta décadas e as mulheres são afetadas cerca de três vezes mais do que os homens e ainda de modo mais freqüente durante os anos férteis. Vários fatores ambientais têm sido associados com o surgimento de esclerose sistêmica e de outras doenças semelhantes à esclerodermia como, por exemplo, a exposição ao pó de sílica, cloreto de polivinil, bleomicina e silicone, entre outros(27-30).
Sabe-se que a esclerose sistêmica tem como característica mais proeminente a hiperprodução de colágeno cuja qualidade é normal. Entretanto, a etiologia e a patogênese dessa doença permanecem desconhecidas. Atualmente, os estudos apontam alguns pólos fisiopatológicos para a esclerose sistêmica(31) tais como: anormalidades da microcirculação; fatores citotóxicos que podem causar alterações da microcirculação; interações entre os fibroblastos e a matriz celular; anormalidades do sistema imunológico, em particular o papel dos auto-anticorpos e o papel dos mastócitos.
A fibrose generalizada que ocorre na pele e em outros órgãos que contêm tecido conjuntivo tem atraído a atenção, não só dos estudiosos afeitos à Reumatologia, mas também de diversas outras disciplinas, pois serve como modelo para estudos dos eventos fisiopatológicos que levam à formação de fibrose.
Nos últimos anos têm surgido várias evidências de conexão entre os produtos derivados de plaquetas ativadas e dois elementos fundamentais da esclerose sistêmica - hiperatividade dos fibroblastos e microangiopatia. E, de fato, tem se demonstrado de forma consistente um acentuado grau de ativação plaquetária na esclerose sistêmica.
ATIVAÇÃO PLAQUETÁRIA
Há diversas evidências de ativação generalizada das plaquetas na esclerose sistêmica. Algumas substâncias plaquetárias estão em concentração elevada no soro de pacientes com esclerose sistêmica, tais como o fator de Von Willenbrand, fator plaquetário 4, tromboxane A2 e beta-tromboglobulina(32). Ademais, há demonstração de agregados plaquetários circulantes bem como documentação ultramicroscópica da existência de alterações morfológicas plaquetárias, da presença de plaquetas aderidas às células endoteliais e, ocasionalmente neutrófilos carregando em sua superfície plaquetas ativadas. Todas essas alterações são compatíveis com intensa ativação plaquetária na esclerose sistêmica(33,34).
Uma endoarterite proliferativa de pequenas artérias é a base fisiopatológica para grande parte dos sinais e sintomas observados na esclerose sistêmica. As plaquetas parecem ter papel importante na evolução da microangiopatia da esclerose sistêmica(35). Elas interagem com o subendotélio exposto após a lesão endotelial primária causando agregação e descarga do seu conteúdo granular, principalmente adenosina difosfato (ADP), serotonina e cálcio. ADP é um importante ativador secundário das plaquetas, tendo grande atividade no recrutamento de plaquetas íntegras para o local do dano vascular. Além disso, a degranulação dos grânulos alfa provoca a liberação de compostos ativos tais como fatores de crescimento, moléculas de adesão (P-selectina) e fatores de coagulação. A aderência ao subendotélio exposto e o grau de proliferação luminal são conseqüências diretas da extensão da perda endotelial da parede do vaso(36-38).
As proteínas secretadas pelas plaquetas têm várias funções biológicas, incluindo pró-coagulação, anti-heparínica e promoção de crescimento celular. Algumas dessas proteínas, tais como fator 4 plaquetário e a beta-tromboglobulina, são existentes apenas nas plaquetas e assim seus níveis séricos servem como marcadores de ativação das mesmas(39-41). Além desses, outro fator importante na interação das plaquetas com a parede vascular é o fator de Von Willebrand (vWF). É provável que o aumento na concentração plasmática do vWF seja derivado das células endoteliais. Nesse caso, ele perpetuaria a lesão endotelial, tendo participação na agregação plaquetária e adesão ao subendotélio da parede do vaso lesado. Portanto, a medida do vWF no soro pode ser útil como marcador de dano endotelial(37).
Vários mediadores derivados das células endoteliais inibem a agregação plaquetária, principalmente a prostaciclina (PGI2). Além do mais, heparina e a trombomodulina na superfície endotelial inibem a ativação da trombina plaquetária(42). Portanto, o comprometimento da função endotelial pode favorecer diretamente a ativação plaquetária e essas, uma vez ativadas, desempenham papel importante na microangiopatia da esclerose sistêmica(35-37).
A molécula de adesão P-selectina expressa nas células endoteliais serve como mediador da aderência e agregação plaquetária, uma vez que a deficiência desse mediador nas células endoteliais provoca falha na adesividade plaquetária(43). Esses dados são corroborados por estudos mostrando que a inibição da expressão da P-Selectina previne lesão de reperfusão após isquemia miocárdica(44). A prostaciclina tem ação oposta à da P-selectina e reduz o tamanho do infarto em associação com diminuição do acúmulo de plaquetas nos vasos isquêmicos(45).
Vale ressaltar que a ativação plaquetária na esclerose sistêmica pode contribuir para a ocorrência de doença arterial oclusiva pela liberação de fatores de crescimento; pode contribuir também para a fibrose tecidual uma vez que produtos plaquetários estimulam a síntese de glicosaminoglicanos, particularmente pelos fibroblastos(46,47). Os fibroblastos dos pacientes com esclerose sistêmica apresentam anormalidades na membrana plasmática e ligam-se ao colágeno via GPIa/IIa, eletroforeticamente e imunologicamente idêntica à integrina α2/β1, que também regula a ligação do colágeno e laminina com as células endoteliais, queratinócitos e células epiteliais. Portanto, aumento na resposta plaquetária ao colágeno na esclerose sistêmica pode refletir uma alteração generalizada na membrana celular, mas se faz necessário uma melhor investigação da expressão da integrina nas membranas celulares, em particular, naquelas observadas na esclerose sistêmica(46,47).
A adesão plaquetária ao subendotélio tem se mostrado fundamental na evolução da proliferação vascular da esclerose sistêmica. As plaquetas interagem com o subendotélio exposto após lesão levando à agregação plaquetária e descarga de seu conteúdo granular. Vários fatores governam essa interação plaqueta-parede vascular: extensão da lesão endotelial, velocidade e característica do fluxo sangüíneo, concentração sérica do fator de Von Willebrand e relação tromboxane/prostaciclina no microambiente vascular. Há evidências in vitro da adesão plaquetária ao subendotélio na esclerose sistêmica que parecem preceder o desenvolvimento das lesões vasculares proliferativas. Além disso, a doença vascular na esclerose sistêmica atinge preferencialmente pequenas arteríolas e microcirculação onde o fluxo é mais constante e menos pulsátil e justamente onde a aderência plaquetária ao subendotélio é maior(46).
O mecanismo exato pelo qual ocorre aumento da adesividade das plaquetas esclerodérmicas ao colágeno permanece desconhecido. As diferenças funcionais de populações plaquetárias em controles e pacientes com esclerose sistêmica podem ser a base para as diferenças na adesão. Por exemplo, plaquetas jovens têm sido relatadas como tendo maior afinidade pelo colágeno do que as plaquetas mais velhas(48). É possível que a população de plaquetas esclerodérmicas seja mais jovem que o normal em vista do elevado nível plasmático de beta-tromboglobulina e de agregados plaquetários circulantes na esclerose sistêmica. Essas alterações indicam aumento da utilização plaquetária, uma vez que os níveis plasmáticos de beta-tromboglobulina e o número de agregados plaquetários circulantes correlacionam-se inversamente com o tempo de sobrevida das plaquetas(49,50). Entretanto, não há estudos específicos sobre o tempo de sobrevida das plaquetas na esclerose sistêmica.
MARCADORES DE ATIVAÇÃO PLAQUETÁRIA
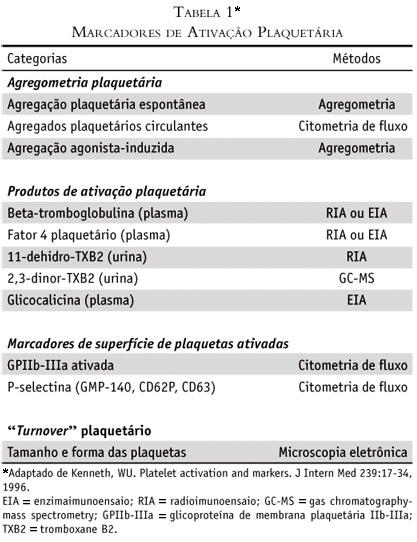
Diversos métodos têm sido utilizados, principalmente nas duas últimas décadas, para avaliar a função e o grau de ativação plaquetária (Tabela 1). Testes que estudam a função plaquetária medem a sua capacidade de adesão, agregação e/ou ativação. Portanto, é necessário compreendermos um pouco da fisiologia das plaquetas. As plaquetas são fragmentos celulares anucleados e contêm três tipos de organelas maiores: grânulos alfa, lisossomos e corpos densos. Os grânulos alfa contêm fibrinogênio, fator 4 plaquetário, proteína 140 da membrana do grânulo (GMP-140), P-selectina, CD62p, trombospondina, beta-tromboglobulina e fatores de crescimento. Os lisossomos contêm hidrolases ácidas e o antígeno CD63. Os corpos densos contêm adenosina trifosfato, adenosina difosfato e serotonina. Deve-se ressaltar que secreção e agregação não estão sempre ligadas, ou seja, é possível que plaquetas sem estímulo à secreção possam estar agregadas(51,52).
Após ativação as plaquetas sofrem diversas alterações bioquímicas e morfológicas(53) com liberação do conteúdo granular e exposição de epítopos de membrana dependentes da ativação. Alguns métodos bioquímicos são capazes de detectar essas alterações mesmo que não haja alteração na agregação. Entre eles estão o radioimunoensaio (RIA), o enzimaimunoensaio (ELISA) e a citometria de fluxo(52,53), capazes de detectar neoantígenos de superfície bem como dosar os níveis séricos de produtos de ativação plaquetária.
O interesse na área de agregação plaquetária foi estimulado pela disponibilidade do agregômetro plaquetário desenvolvido por Born(54) em 1960. A medida da agregação plaquetária é simples. Sangue venoso citratado é centrifugado a 2.000 rpm por 10 minutos para obtenção do plasma rico em plaquetas (PRP). Em seguida alíquotas do PRP ajustadas para 300 x 109/L são colocadas em cubetas sob agitação contínua e a extensão da agregação é registrada no agregômetro pelo grau de intensidade de transmissão da luz. O teste de agregação plaquetária deve ser conduzido com ou sem a adição de agonistas fisiológicos. Na ausência desses, a ocorrência de aumento de 20% na transmissão da luz dentro de 10 minutos após a adição do PRP no agregômetro reflete um teste positivo para agregação plaquetária espontânea. Além da agregação plaquetária espontânea é interessante medir a agregação induzida por agonistas: adiciona-se ADP, colágeno ou adrenalina e a agregabilidade é definida como a concentração mínima de um agonista em induzir agregação máxima. Como é difícil quantificar precisamente a agregação, cuidados devem ser tomados visando reduzir os fatores extrínsecos que a influenciam. Por isso, o PRP deve ser preparado a fresco e usado no teste de agregação até três horas após a preparação.
A liberação do conteúdo das organelas plaquetárias pode ser usada como um indicador de ativação plaquetária. O conteúdo dessas organelas é de dois tipos: substâncias que são liberadas no plasma tais como a serotonina e aquelas que permanecem ligadas à membrana tais como GMP-140. No primeiro caso amostras simples de plasma são necessárias para análise enquanto que aquelas que permanecem ligados à membrana requerem amostras de plaquetas para análise(55). A beta-tromboglobulina e o fator 4 plaquetário são proteínas específicas liberadas dos grânulos alfa das plaquetas ativadas(56). Níveis séricos alterados de ambas as proteínas têm sido descritos em várias doenças, incluindo câncer, hipertensão arterial grave, doença arterial coronariana e diabetes melito(57-60). Entretanto, como elas são de excreção renal, a função renal deve ser avaliada cuidadosamente na medida de seus níveis plasmáticos. Outros pesquisadores têm encontrado flutuações nos níveis plasmáticos de voluntários sadios dependendo da hora do dia, idade, técnica de flebotomia, tipo de anticoagulação e preparação da amostra(61-65) e ainda pelo uso de certas drogas, como por exemplo, os beta bloqueadores(66). A beta-tromboglobulina, composta de 81 aminoácidos dispostos em quatro subunidades idênticas, foi primeiramente descrita por Moore et al., em 1975(67). Ocupa cerca de 10% do conteúdo dos grânulos alfa, sendo liberada sob influência de conhecidos ativadores plaquetários, tais como ADP, colágeno, imunocomplexos e trombina. Sua meia-vida plasmática é de aproximadamente 100 minutos e é excretada na urina(55). O fator 4 plaquetário foi descrito em 1969 por Niewiaroswski e Thomas. É uma proteína de 70 aminoácidos secretada como um complexo de 2 tetrâmeros(68-70). Age por interferência na ação da heparina e, portanto, promove a coagulação. Em razão de o fator 4 plaquetário ser uma proteína ligada à heparina, a presença de anticoagulante no sangue tem influência na determinação de seus níveis. Além disso, o ácido acetil-salicílico causa uma redução na concentração plasmática de fator 4 plaquetário(71). O fator 4 plaquetário e a beta-tromboglobulina diferem apenas em sua porção amino-terminal 4, o que lhes confere similaridade química e imunológica(72), embora tenham funções inteiramente distintas. Ambas as proteínas podem ser medidas de maneira eficaz tanto por radioimunoensaio quanto por enzimaimunoensaio(39-41,56,73-76).
A preparação inicial da amostra biológica é extremamente importante para a dosagem sérica de qualquer substância derivada das plaquetas, incluindo a beta-tromboglobulina e o fator 4 plaquetário. Sangue venoso é coletado cuidadosamente, sem garroteamento excessivo, com agulha de médio calibre, em seringa de plástico polipropileno previamente resfriada. Imediatamente a amostra deve ser transferida para tubo de plástico com solução anticoagulante (ácido cítrico 38 mM, citrato de sódio 60 mM e dextrose 14 mM) na proporção 9:1 v/v. Em aproximadamente 15 minutos deve-se centrifugar a amostra a 2.000 rpm por 30 minutos a 4ºC, para obtenção do plasma pobre em plaquetas (PPP). O terço médio do PPP é cuidadosamente aspirado e armazenado a -20ºC até a dosagem do marcador. Esta será efetuada por ELISA ou RIA através de kits comercialmente disponíveis.
A ativação plaquetária é acompanhada também pela síntese de tromboxane A2 (TXA2). O TXA2 é liberado na circulação sangüínea e é rapidamente convertido em TXB2, um produto estável, porém inativo. Este por sua vez, é convertido em uma série de compostos, incluindo o 2,3-dinor-TXB2 e o 11-dehidro-TXB2, que são excretados na urina. A combinação de dosagem desses dois elementos, por ELISA, pode fornecer informações úteis a respeito da síntese de TXA2(55,77). A capacidade de síntese de TXA2 pelas plaquetas ativadas também pode ser estimada pela medida de TXB2 sérico, entretanto o uso do TXB2 com marcador de ativação plaquetária não tem sido avaliado em detalhes(39). A glicocalicina(55), fragmento proteolítico da gpIb, pode ser facilmente medida por ELISA(78). Níveis séricos elevados desse marcador estão presentes na púrpura trombocitopênica idiopática(79) e na trombocitopenia da infecção por HIV(78). Níveis plasmáticos normais têm sido relatados em pacientes com hipoplasia de medula óssea e baixos níveis são encontrados no sangue venoso umbilical(80). Interessantemente, em um estudo ex vivo, níveis séricos da glicocalicina não se correlacionaram com a concentração de P-selectina de membrana e beta-tromboglobulina, considerando-se a possibilidade de que níveis elevados de glicocalicina podem não refletir realmente ativação plaquetária e sim, simplesmente, algum outro aspecto da fisiologia plaquetária(81).
Entretanto, a dosagem de outros produtos de ativação plaquetária, tais como metabólitos do tromboxane e a glicocalicina, tem pouca aplicabilidade no momento.
Quando falamos em avaliação de plaquetas ativadas propriamente dita e não em produtos de ativação plaquetária, o método empregado é o da citometria de fluxo. Como mostrado na Tabela 1 a citometria de fluxo é útil na detecção de glicoproteínas de membrana tais como a GMP-140 e a glicoproteína IIb/IIIa. Para analisar a expressão dessas glicoproteínas na superfície plaquetária coleta-se sangue venoso em tubo contendo 100 µL de EDTA. O plasma rico em plaquetas é incubado por duas horas a 4ºC em 1mL de paraformaldeído 1%. As células fixadas são então centrifugadas a 2.000 rpm por cinco minutos em temperatura ambiente. Em seguida, o botão de células é lavado duas vezes com 1 mL de PBS. Após, 100 µL da suspensão fixada é incubada com 10 µL de monoclonal anti-GPIb conjugado a fluoresceína e 10 µL de monoclonal antiCD62P conjugado a ficoeritrina ou monoclonal anti-CD63 conjugado a ficoeritrina. Depois de 20 minutos de incubação em temperatura ambiente, as amostras são lavadas uma vez com PBS e novamente fixadas adicionando-se 1 mL de paraformaldeído a 1% e armazenadas no escuro a 4ºC. Posteriormente, são levadas ao aparelho para análise citométrica(82-84).
Algumas considerações metodológicas se fazem necessárias. Todos os métodos descritos acima estão sujeitos a interpretações errôneas principalmente em razão da possibilidade de ativação plaquetária in vitro que pode ocorrer durante a coleta e processamento da amostra. É extremamente importante que se observem alguns detalhes tais como calibre da agulha utilizada na punção venosa, tempo de coleta, qualidade do material utilizado bem como dos reagentes empregados no processamento da amostra. Deve-se evitar uso de coquetéis de anticoagulantes e materiais de vidro e, se possível, realizar a dosagem dos marcadores em laboratórios especializados. O plasma deve ser o mais pobre possível em plaquetas e separado imediatamente após a coleta.
Como descrito acima, há diversos métodos para avaliação da função plaquetária, especificamente com relação à sua agregabilidade e ao seu grau de ativação. Então, surge a pergunta: quais são os melhores métodos para monitorar a função plaquetária?
Algumas pessoas consideram o método fisiológico, ou seja, o teste de agregação plaquetária, o melhor teste. Porém o maior problema com esse método é que ele é apenas um indicador grosseiro do que acontece com as plaquetas. Ele pode, certamente, mostrar que a função plaquetária foi inibida, mas não pode, efetivamente, mostrar que as plaquetas tornaram-se ativadas e nem detectar mudanças sutis na função plaquetária. Outro problema é que esse método requer uma quantidade muito grande de plaquetas para sua execução e, por exemplo, no caso de pacientes com trombocitopenia é quase impossível obter o número de plaquetas suficiente para realizar o teste de agregação.
Por outro lado, a medida dos principais marcadores solúveis e de membrana pode mostrar mudanças no estado de ativação plaquetária antes de ser detectada pelo método fisiológico de agregação plaquetária. A dosagem plasmática de produtos de degranulação plaquetária por ELISA tem a vantagem de ser de fácil interpretação, de ser quantitativa e não requerer equipamento caro. Por outro lado, a citometria de fluxo para detecção de marcadores de membrana nos permite enxergar alterações individuais das plaquetas. O ELISA pode dosar um determinado constituinte plaquetário em uma suspensão de plaquetas, ao passo que a citometria de fluxo indicará a percentagem de plaquetas contendo diferentes quantidades desse constituinte. A citometria de fluxo tem a desvantagem de não ser quantitativa, porém uma contagem com calibração rigorosa tem tornado o método mais eficaz, dando uma melhor aproximação à quantificação do antígeno em questão. Apesar da citometria de fluxo ser o método mais acurado, a facilidade do ELISA torna-o o método mais utilizado. Contudo, pode haver dificuldades técnicas com alguns marcadores, em especial, a GMP-140 que não se conserva bem, principalmente após fixação. Já o antígeno CD63 é estável mesmo se as plaquetas forem armazenadas e fixadas por algum tempo. Outro fator a ser considerado é que o comportamento desses marcadores pode ter diferentes significados. Por exemplo, o antígeno CD63 requer intensa estimulação para ser liberado, enquanto a GMP-140 pode ser liberada sob moderada estimulação.
Recentemente, demonstramos uma alta freqüência de níveis séricos elevados de enolase específica de neurônio (NSE) em pacientes com esclerose sistêmica(85). A NSE foi descrita originalmente por Moore e McGregor, em 1965(86), utilizando fracionamento cromatográfico e eletroforético de proteínas solúveis do cérebro e do fígado. Ela está presente em neurônios e tecidos neuroendócrinos periféricos. Segundo Day e Thompsom(87), a NSE ocorre também em plaquetas em níveis equivalentes à sua concentração no cérebro, e em eritrócitos em níveis aproximadamente 10% dessa. É considerada marcadora de câncer de pequenas células de pulmão(88-90) e neuroblastoma(91-93).
Em nosso recente estudo(85), demonstramos níveis séricos anormais de NSE em pacientes com esclerose sistêmica em uma freqüência inusitadamente aumentada. Este achado revestiu-se de maior relevância diante do fato de que pacientes com outras enfermidades reumáticas auto-imunes não apresentaram níveis séricos de NSE elevados. O encontro de NSE sérica elevada na esclerose sistêmica associou-se à doença mais precoce e a comprometimento mais extenso e intenso da pele, o que sugere fortemente uma associação com atividade de doença. Por outro lado não foi encontrada associação com dismotilidade esofágica e com doença pulmonar intersticial. Demonstrou-se também uma relação inversamente proporcional entre a concentração de NSE no soro e nas plaquetas, o que aponta fortemente para a hipótese de que os níveis séricos elevados de NSE na esclerose sistêmica sejam ocasionados por liberação desta enzima a partir de seus estoques plaquetários em função do processo de ativação plaquetária observado nessa enfermidade. Se a NSE pode ser um útil marcador de ativação plaquetária na esclerose sistêmica bem como em outras condições clínicas, mais estudos serão necessários para tal discernimento.
Atualmente muito tem se estudado sobre ativação plaquetária na esclerose sistêmica e não sabemos se a ativação plaquetária constitui um evento primário ou se é meramente uma resposta secundária à agressão endotelial. Entretanto, a hipótese de que substâncias com capacidade quimiotática e mitogênica secretadas pelos grânulos plaquetários possam aumentar a lesão endotelial e contribuir para alterações vasculares e perivasculares da esclerose sistêmica garante um excelente campo para novos estudos. Ademais, é possível que o grau de ativação plaquetária guarde correlação com o grau de gravidade e/ou atividade da doença. Essa possibilidade é saudada com entusiasmo visto não haver hoje em dia marcadores de atividade da esclerose sistêmica disponíveis. Nesse sentido, ensaios laboratoriais de execução simples e disponibilidade em laboratórios clínicos teriam potencial aplicação no monitoramento de pacientes com esclerose sistêmica.
Trabalho recebido em 01/12/03.
Aprovado, após revisão, em 26/01/04.
-
1Medsger TA Jr, Masi AT, Rodnan JP. Survival with systemic sclerosis (scleroderma): A life-table analysis of clinical and demographic factors in 309 patients. Ann Int Med 75:369-76, 1971.
-
2Medsger TA Jr. Progressive systemic sclerosis. Clin Rheum Dis 9: 9-16, 1983.
-
3Medsger TA Jr. Systemic sclerosis (scleroderma), localized scleroderma, eosinophilic fasciitis and calcinosis. In McCarty DJ, Koopman WJ, eds. Arthritis and allied conditions, 12th ed. Philadelphia: Lea and Febiger; p.1113-43, 1993.
-
4LeRoy EC, Medsger TA Jr. Criteria for the classification of early sistemica sclerosis. J Rheumatol 28:1573-6, 2001.
-
5LeRoy EC, Black CM, Fleischmajer R, Jablonska S, Krieg T, Medsger TA, et al. Scleroderma (systemic sclerosis): classifications, subsets and pathogenesis. J Rheum 15:202-5, 1988.
-
6LeRoy EC. A brief overview of the pathogenesis of scleroderma (systemic sclerosis). Ann Rheum Dis 51:286-9, 1992.
-
7LeRoy EC. Systemic sclerosis (scleroderma). In: Bennet JC, Plumm F, Glass DN, eds. Cecil's textbook of medicine, 20th ed. Philadelphia: Saunders; p.1483-8, 1996.
-
8Seibold JR. Scleroderma. In: Kelley WN, Harris Ed, Rudy S, Sledge CB, eds. Textbook of rheumatology, 5th ed. Philadelphia: Saunders; p.1133-63, 1997.
-
9Seibold JR. Clinical trials: types, design, and end points. Curr Opin Rheumatol 13:512-5, 2001.
-
10Steen VD, Medsger TA. Epidemiology and natural history of systemic sclerosis. Rheum Dis Clin North Am 16:1-10, 1990.
-
11Steen VD. Improvement in skin thickening in systemic sclerosis associated with improved survival. Arthritis Rheum 44:2828-35, 2001.
-
12Black CM, Dieppe P, Huskisson T. Regressive systemic sclerosis. Ann Rheum Dis 45:384-7, 1986.
-
13Black CM. The aetiopathogenesis of systemic sclerosis. J Intern Med 234:3-9, 1993.
-
14Black CM. Systemic sclerosis "State of the art" 1995. Scand J Rheumatol 24:194:6, 1995.
-
15Rodnan GP, Benedek TG. An historical account of the study of progressive systemic sclerosis. Ann Int Med 57:305-19, 1962.
-
16Rodnan GP, Jablonska S, Medsger TA Jr. Classification and nomenclature of progressive systemic sclerosis (scleroderma). Clin Rheum Dis 5:5-13, 1979.
-
17Marques Neto JF, Sampaio-Barros PD. Conferência Internacional sobre Esclerose Sistêmica – Termas de Montecatini, março/1998. Rev Bras Reumatol 38:264-8, 1998.
-
18Andrade LEC, Pucinelli MLC. Esclerose sistêmica. In: Prado FC, Ramos J, Valle JR. Atualização terapéutica, 20ª ed. São Paulo: Artes Médicas; p.1387-90, 2001.
-
19Tan EM, Rodnan GP, Garcia I. Diversity of antinuclear antibodies in progressive systemic sclerosis: Anti-centromere antibody and its relationship to CREST syndrome. Arthritis Rheum 23:617, 1980.
-
20Steen VD, Ziegler GL, Rodnan GP. Clinical and laboratory association of anticentromere antibody in patients with progressive systemic sclerosis. Arthritis Rheum 27:121-30, 1984.
-
21LeRoy EC. Systemic sclerosis: A vascular perspective. Rheum Dis Clin North Am 22:675-94, 1996.
-
22Maricq HR. Microvascular abnormalities as possible predictors of diseases subsets in Raynaud phenomenon and early connective tissue diseases Clin Exp Rheumatol 1:195, 1983.
-
23White B, Bauer EA, Goldsmith LA. Guidelines for clinical trials in systemic sclerosis. Arthritis Rheum 38:351-60, 1995.
-
24Krieg T, Meurer M. Systemic scleroderma. J Am Acad Dermatol 18:788-91, 1988.
-
25Medsger TA Jr, Masi AT. Epidemiology of systemic sclerosis (scleroderma). Ann Intern Med 74:14-21, 1971.
-
26Medsger TA Jr. Epidemiology of systemic sclerosis. Clin Dermatol 12:207-16, 1994.
-
27Vallyatan V. Generation of free radicals from fresh fractured silica dust. Am Rev Respir Dis 138:1213-9, 1988.
-
28Nietert PJ, Silver RM. Systemic sclerosis: environmental and occupational risk factors. Curr Opin Rheumatol 12:520-6, 2000.
-
29Finch WR, Rodnan GP, Buckingham RB. Bleomycin-induced scleroderma. J Rheumatol 7:651-4, 1980.
-
30Endo LP, Edwards NL, Longley S. Silicone and rheumatic diseases. Semin Arthritis Rheum 17:112-8, 1987.
-
31Murell DF. A radical proposal for the pathogenesis of scleroderma. Acad Dermatol 28:78-85, 1993.
-
32Herrick AL, Illingworth K, Blann A, Hay CRM, Hollis S, Jayson MV. Von Willebrand factor, thrombomodulina, thromboxane Betathromboglobulina, and markers of fibrinolysis in primary Raynaud's phenomenon and systemic sclerosis. Ann Rheum Dis 55122-7, 1996.
-
33Kahaleh MB, Osborn I, LeRoy EC. Elevated levels of circulating platelet aggregates and Beta-thromboglobulina in scleroderma. Ann Intern Med 96:610-3, 1982.
-
34Blann AD, Illingworth K, Jayson MIV. Mechanisms of endothelial cell damage in systemic sclerosis and Raynaud's phenomenon. J Rheumatol 20:1325-30, 1993.
-
35Kahaleh MB, Le Roy EC. Endothelial injury in scleroderma. J Lab Clin Med 101:553-60, 1983.
-
36Freedman Rf, Girgis R, Mayes MD. Endothelial and adrenergic disfunction in Raynaud's phenomenon and scleroderma. J Rheumatol 26:2386-8, 1999.
-
37Kahaleh MB, Leroy EC. Autoimmunity and vascular involvement in systemic sclerosis. Autoimmunity 31:195-214, 1999.
-
38Herrick AL. Vascular function in systemic sclerosis Curr Opin Rheumatol 12:527-33, 2000.
-
39Kenneth WU. Review: Platelet activation and markers. J Intern Med 239:17-34, 1996.
-
40Matzdorf AC, Kemkes-Matthes B, Voss R, Pralle H. Comparison of Beta-Thromboglobulina, flow cytometry, and platelet aggregometry to study platelet activation. Haemostasis 26:98-106, 1996.
-
41Cox D. Methods for monitoring platelet function. Am Heart J 135:s160-9, 1998.
-
42Blann AD, Taberner DA. A reliable marker of endothelial cell dysfunction: Does it exist ? Br J Haematol 90:244-48, 1995.
-
43Massberg S, Enders G, Leiderer R, Eisenmenger S, Vestweber d, Krombach F, et al. Platelet-endothelial cell interactions durin ischemia/reperfusion: the role of P-selectin. Blood 92:507-15, 1998.
-
44Scalia R, Murohara T, Delyani JA, Nossuli TO, Lefer AM. Myocaridial protection by N,N,N-trimethylsphingosine in ischemia reperfusion injury is mediated by inhibition of P-selectin. J Leukoc Biol 59:317-24, 1996.
-
45Wigley FM, Seibold JR, Wise RA, McCloskey DA, Dole WP. Intravenous Iloprost treatment of Raynaud's phenomenon and ulcers secondary to systemic sclerosis. J Rheumatol 19:407-14, 1992.
-
46Kahaleh MB, Scharstein KK, LeRoy EC. Enhanced platelet adhesion to collagen in scleroderma. Effect of scleroderma plasma and scleroderma platelets. J Rheumatol 12:468-71, 1985.
-
47Goodfield MJD, Orchard MA, Rowell NR. Woole blood platelet aggregation and coagulation factors in patients with systemic sclerosis. Br J Haematol 84:675-80, 1993.
-
48Hirsh J, Glynn MF, Mustard JF. The effect of platelet age on platelet adherence to collagen. J Clin Invest 47:463-6, 1968.
-
49Salem HH, Koutts J, Firkin BG. Circulating platelet aggregates in ischaemic heart disease and their correlation to platelet-life span. Thrombos Res 17:707-11, 1980.
-
50Doyle DJ. Plasma concentrations of platelet-specific proteins correlated with platelet survival. Blood 55:82-4, 1980.
-
51Kaplan KL, Broeckman MJ, Chernoff A, Lesznik GR, Drillings M. Platelet a granule proteins: studies on release and subcellular localization. Blood 53:604, 1979.
-
52Holmsen H. Platelet secretion and energy metabolism. In: Colman R, Hirsh J, Marder V, Salzman E: Hemostasis and thrombosis: basic principles and clinical practice, 3rd ed. Philadelphia: JB Lipincott, 1994.
-
53Siess W. Molecular mechanisms of platelet activation. Physiol Rev 69:58-178, 1989.
-
54Born GVR. Aggregation of blood platelet by adenosine diphosphate and its reversal. Nature 194:927-9, 1962.
-
55Gourney D, Lip GYH, Blann AD. A reliable plasma marker of platelet activation: Does it exist? Am J Haematol 70:139-44, 2002.
-
56Kaplan KL, Owen J. Plasma levels of Beta- thromboglobulin and platelet factor 4 as indices of platelet activation in vivo. Blood 57: 199-202, 1981.
-
57Al Mondhiry H. Beta-thromboglobulin and platelet factor 4 in patients with cancer: correlation with the stage of the disease and the effect of chemotherapy. Am J Hematol 14:105-11, 1983.
-
58White GC, Marouf AA. Platelet factor 4 in patients with coronary artery disease. J Clin Med 97:369-78, 1981.
-
59Lane DA, Ireland H, Wolff S, Ranasinghe E, Dawes J. Detection of enhanced in-vivo platelet alpha granule release in different patients groups. Comparison of beta-thromboglobulin, platelet factor 4 and thrombospondim assays. Thromb Haemost 52:183-7, 1984.
-
60Voisin PJ, Rouselle D, Streiff F, Debry G, Stotlz JF, Drouin P. Reduction of Beta-thromboglobulin levels in diabetics controlled by an artificial pancreas. Metabolism 32:138-41, 1983.
-
61Walz DA. Platelet released proteins as molecular markers for the activation process. Semin Thromb Haemost 10:270-9, 1984.
-
62Pechan J, Mikulecky M, Okrucka A. circadian rhythm of plasma Beta-thromboglobulin in healthy human subjects. Blood Coagul Fibrinolisys 3:105-7, 1992.
-
63Zahavi J, Cella G, Dubiel M, Kakkar VV. The variability of plasma beta-thromboglobulin in healthy individuals. Thromb Haemost 40:565-7, 1979.
-
64Chong BH, Murray B, Berndt MC, Dunlop LC. Plasma P-Selectin is increased in thrombotic consumptive disorders. Blood 83:1535-41, 1994.
-
65Ludlam CA, Cash JD. Studies of liberation of Beta-thromboglobulin from human platelets in vitro. Br J Haematol 33:239-47, 1976.
-
66Anfossi G. Influence of propranolol on platelet aggregation and thromboxane B2 production from platelet rich plasma and whole blood. Prostanglandins Leukot Essent Fatty Acids 36:1-7, 1981.
-
67Moore S, Pepper DS. The isolation of a platelet specific betathromboglobulin and the detection of antiurokinase antiplasmin released from thrombin aggregated washed human platelets. Biochim Biophys Acta 379:360-9, 2002.
-
68Niewiarowski S, Thomas DP. Platelet factor 4 and ADP release during human platelet aggregation. Nature 222:1269-70, 1969.
-
69Kaser-Glanzmann R, Jakabova M, Luscher EF. Isolation and some properties of the heparin neutralising factor (PF4) released from human blood platelets. Experimentia 28:1221-2, 1972.
-
70Dawes J, Smith RC, Pepper DC. The release, distribution and clearance of human Beta-thromboglobulin and platelet factor 4. Thromb Res 12:569-82, 1978.
-
71Minar E, Ehringer H. Influence of acetylsalicylic acid (1.0 g/day) on platelet survive time, Beta-thromboglobulin and platelet factor 4 in patients with peripheral arterial acclusive disease. Thromb Res 45:791-802, 1987.
-
72Rucinski B, Niewiaroswski S, James P. Antiheparin proteins secreted by human platelets: Purification, characterization, and radioimmunoassay. Blood 53:47-62, 1979.
-
73Ludlam CA, Moore S, Bolton AE. The release of a human plateletspecific protein measured by radioimmunoassay. Thromb Res 6: 543-8, 1975.
-
74Amrani DL, Stojanovic L, Mosesson MN, Shalev Y, mosesson MW. Development of a whole platelet ELISA to detect circulating activated platelets. J Lab Clin Med 126:603-11, 1995.
-
75Woodhams BJ, Kernoff PBA. The application of polyethylene glycol to radioimmunoassays used in haemostasis. Thromb Res 29:333-41, 1983.
-
76Abrams C, Shattil SJ. Immunological detectionof activated platelets in clinical disorders. Thromb Haemost 65:467-73, 1991.
-
77FitzGerald GA, Pedersen AK, Patrono C. Analysis of prostacyclin and thromboxane biosynthesis in cardiovascular disease. Circulation 67:1174-7, 1983.
-
78Beer JH, Steiner B. Glycocalicin: a new assay: The normal plasma levels and its potential usefulness in selected diseases. Blood 83: 691-702, 1994.
-
79Kurata Y, Hayashi S, Kiyoi T, Kosugi S, Honda S, Tomiyama Y. Diagnostic value of tests for reticulated platelets, plasma glycocalicin and thrombopoietin levels for discriminating between hyperdestructive and hypoplastic thrombocytopenia. Am J Clin Pathol 115:656-64, 2001.
-
80Jilma-Stohlawetz P, Homoncik M, Jilma B, Folman B, Folman CC, von dem Borne AE, Bernaschek G, et al. High levels of reticulated platelets and thrombopoietin characterize fetal platelets. Br J Haematol 112:466-8, 2001.
-
81Kostelijk EH, Folman CC, Gouwerok CW, Kramer CM, Verhoeven AJ, de Korte D. Increase in glycocalicin levels in platelet concentrates stored in plasma or synthetic medium for 8 days: comparison with other platelet activation markers. Vox Sang 79:21-6, 2000.
-
82Shattil SJ, Cunningham M, Hoxie JA. Detction of activated platelets in whole blood using activation-dependent monoclonal antibodies and flow cytometry. Blood 70:307-15, 1987.
-
83Michelson AD. Flow citometry: a clinical test of platelet function. Blood 87:4925-36, 1996.
-
84Ginsberg MH, Frelinger AL, Lam SC, Forsyth J, McMillan R, Plow EF, Shattil SJ. Analysis of platelet aggregation disorders based on flow cytometric analysis of membrane glycoprotein IIb/IIIa with conformation-specific monoclonal antibodies. Blood 76:2017, 1990.
-
85Massabki PS, Silva NP, Lourenço DM, Andrade LEC. Neuron specific enolase concentration is increased in serum and decreased in platelets of patients with systemic sclerosis. J Rheumatol 30: 2606-12, 2003.
-
86Moore BW, McGregor T. Chromatoghraphic and electrophoretic fractionation of soluble proteins of brain and liver. J Biol Chem 240:1647-53, 1965.
-
87Day INM, Thompson RJ. Levels of immunorective aldolase C, creatine kinase-BB, neuronal and non-neuronal enolase, and 14-3-3 protein in circulating human blood cells. Clin Chim Acta 136: 219-28, 1984.
-
88Carney DN. Serum neuron-specific enolase: a marker for disease extent and response to therapy of small cell lung cancer. Lancet 1:583-5, 1982.
-
89Cooper EH, Splinter TAW. Neuron-specific enolase (NSE): a useful marker in small-cell lung cancer. Lung Cancer 3:61-6, 1987.
-
90Jörgensen L, Österlin K, Cooper EH, Hansen HH. The prognostic influence of neuron-specific enolase in small-cell lung cancer. Br J Cancer 58:805-7, 1988.
-
91Zeltzer PM, Marangos PJ, Evans AE, Schneider PS. Serum neuronspecific enolase in children with neiroblastoma. Relationship to stage and disease course. Cancer 57:1230-4, 1986.
-
92Cooper EH, Prichard J, Bailey CC, Ninane J. Serum neuron-specific enolase in children's cancer. Br J Cancer 56:65-7, 1987.
-
93Villard JL, Tiget F, Hartmann O. Serum neuron-specific enolase / nonneuronal enolase ratio in diagnosis of neuroblastomas. Cancer 62:2546-53, 1988.
Datas de Publicação
-
Publicação nesta coleção
09 Maio 2011 -
Data do Fascículo
Fev 2004
Histórico
-
Aceito
26 Jan 2004 -
Recebido
01 Dez 2003