Resumos
A alcaptonúria (ocronose) é um erro inato do metabolismo da fenilalanina e tirosina, transmitido de forma autossômica recessiva. Resulta da deficiência completa da enzima ácido homogentísico oxidase (HGO), causada por mutação no gene 3q (3q21 - q23), levando ao acúmulo do ácido em diversos órgãos e tecidos, com aumento de sua excreção urinária. Os achados clínicos característicos incluem artropatia ocronótica, pigmentação anormal da cartilagem de outros tecidos conjuntivos e urina enegrecida. A incidência é rara, estimada em 1-4: 1.000.000 indivíduos, tendo maior prevalência em populações com alto grau de consangüinidade. A seguir, relataremos o caso de dois irmãos (um homem e uma mulher) com ocronose, discutindo sua patogênese, manifestações clínicas, diagnóstico e tratamento(1, 2).
ocronose; alcaptonúria; artropatia ocronótica
Alkaptonuria (ochronosis) is an autossomal recessive disorder which results from an innate error of phenylalanine and tyrosine’s metabolism. This defect causes accumulation of homogentisic acid on the tissue and usually is excreted in the urine. This disorder results from a complete deficiency of the homogentisate 1,2 - dioxygenase (HGO) caused by chromosome 3q(3q21-q23) mutation. The clinical features include: ochronotic artropathy, black-gray pigmentation of the cartilage and the urine turns black. This is a rare disorder and incidence is about 1-4 in 1.000.000 of people and the prevalence is higher in consanguineous groups. The author will describe two affected brothers and discuss the pathophysiology, clinical features, diagnosis and treatment of ochronosis.
ochronosis; alkaptonuria; ochronotic artropathy
RELATO DE CASO CASE REPORT
Alcaptonúria (ocronose): relato de dois casos
Alkaptonuria (ochronosis): report of two cases
Letícia R. BrandãoI; Brunela P. BorjailleI; Tatiana M. HasegawaII; Renata F. RosaIII; Elaine AzevedoIV; Wiliam H. ChahadeV
IMédica estagiária (E2) do Serviço de Reumatologia do HSPE-FMO
IIMédica residente (R2) em dedicação plena do Serviço de Reumatologia do HSPE-FMO
IIIMédica assistente do Serviço de Reumatologia do HSPE-FMO
IVMédica encarregada do Ambulatório de Doenças Osteometabólicas do Serviço de Reumatologia do HSPE-FMO
VDiretor do Serviço de Reumatologia do HSPE-FMO
Endereço para correspondência Endereço para Correspondência: Hospital Servidor Público Estadual (HSPE) Rua Pedro de Toledo, 1800, Vila Clementino Caixa Postal 8570, São Paulo, SP, Brasil telefone: (11) 5088-8099
RESUMO
A alcaptonúria (ocronose) é um erro inato do metabolismo da fenilalanina e tirosina, transmitido de forma autossômica recessiva. Resulta da deficiência completa da enzima ácido homogentísico oxidase (HGO), causada por mutação no gene 3q (3q21 q23), levando ao acúmulo do ácido em diversos órgãos e tecidos, com aumento de sua excreção urinária. Os achados clínicos característicos incluem artropatia ocronótica, pigmentação anormal da cartilagem de outros tecidos conjuntivos e urina enegrecida. A incidência é rara, estimada em 1-4: 1.000.000 indivíduos, tendo maior prevalência em populações com alto grau de consangüinidade. A seguir, relataremos o caso de dois irmãos (um homem e uma mulher) com ocronose, discutindo sua patogênese, manifestações clínicas, diagnóstico e tratamento(1, 2).
Palavras-chave: ocronose, alcaptonúria, artropatia ocronótica.
ABSTRACT
Alkaptonuria (ochronosis) is an autossomal recessive disorder which results from an innate error of phenylalanine and tyrosines metabolism. This defect causes accumulation of homogentisic acid on the tissue and usually is excreted in the urine. This disorder results from a complete deficiency of the homogentisate 1,2 - dioxygenase (HGO) caused by chromosome 3q(3q21-q23) mutation. The clinical features include: ochronotic artropathy, black-gray pigmentation of the cartilage and the urine turns black. This is a rare disorder and incidence is about 1-4 in 1.000.000 of people and the prevalence is higher in consanguineous groups. The author will describe two affected brothers and discuss the pathophysiology, clinical features, diagnosis and treatment of ochronosis.
Keywords: ochronosis, alkaptonuria, ochronotic artropathy.
INTRODUÇÃO
A alcaptonúria é uma doença hereditária autossômica recessiva, caracterizada pela deposição do ácido homogentísico em vários órgãos e tecidos(1).
O termo ocronose introduzido por Virchow refere-se à coloração ocre observada à microscopia dos tecidos acometidos. Os pacientes excretam na urina grandes quantidades de ácido homogentísico, o qual pode ser espontaneamente oxidado em pigmento enegrecido. Esta reação pode ser acelerada pela adição de solução alcalina, dando origem ao termo alcaptonúria por Boedecker em 1866(1, 3).
As primeiras manifestações da doença ocorrem na infância como urina e cerume enegrecidos, porém os pacientes tornam-se sintomáticos somente em torno da quarta década de vida(1, 4).
As complicações mais freqüentes são artropatia ocronótica, ocronose cardiovascular, cutânea e ocular, além de obstrução do trato genitourinário por cálculos ocronóticos. A artropatia ocronótica, manifestação mais incapacitante da doença, acomete, primariamente, grandes articulações e a coluna vertebral(4, 5).
DESCRIÇÃO DO CASO
CASO 1
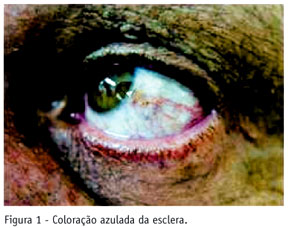
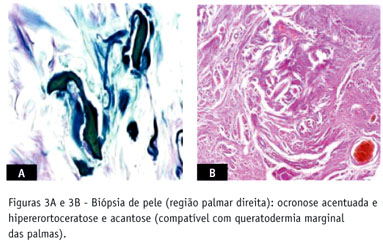
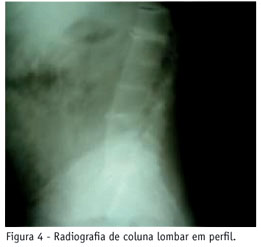
V.V., 61 anos, sexo masculino, branco, natural de São Paulo. O paciente procurou o Serviço de Reumatologia do Hospital do Servidor Público Estadual de São Paulo (HSPE), em 2001, com história de lombalgia mecânica, há 16 anos, com diminuição da mobilidade. Há sete anos, apresentou gonalgia bilateral de início insidioso, protocinética, com períodos de artrite e limitação funcional progressiva e artralgia em ombros com limitação da mobilização. Relatava urina escura desde a infância. Ao exame físico: escleras de coloração azulada (Figura 1), pigmentação azulada em região palmar (Figura 2) e ouvido externo, cerume de coloração enegrecida; marcha antálgica; hiperextensão cervical com limitação a lateralização (20ºD/20ºE) e rotação (40ºD/40ºE); limitação a abdução em ombros (50ºD/60ºE); derrame articular em joelho esquerdo; teste de Schöber < 5 cm. Biópsia de pele da região palmar direita: ocronose acentuada (Figura 3). Exames laboratoriais: sinoviograma com líquido inflamatório sem cristais. O hematológico e bioquímica sérica foram normais. Exames ultra-sonográficos: vias urinárias e próstata normais; joelho esquerdo com bursite subquadricipital e calcificações livres em seu interior, sinais de sinovite, tendinopatia crônica quadriciptal e patelar calcificada. Ecocardiograma com calcificações em valvas mitral e aórtica. Exames de raio X simples: a) coluna dorsolombar - calcificações e estreitamento dos discos intervertebrais, presença de sinal do vácuo e esclerose de platôs vertebrais (Figura 4); b) joelhos - diminuição do espaço articular e hipertrofia das espinhas tibiais (Figura 5); c) ombro direito - osteoartrite acromioclavicular e diminuição do espaço glenoumeral (Figura 6); d) sacroilíacas - esclerose bilateral e simétrica. Ressonância magnética de ombro esquerdo - osteoartrite acromioclavicular, glenoumeral e ruptura do manguito rotador (Figura 7). Sua urina, quando exposta ao sol, tornou-se enegrecida. A pesquisa do ácido homogentísico nesta foi positiva.
CASO 2
M.V., 52 anos, sexo feminino. Encaminhada para investigação, após realização de densitometria óssea, a qual sugeria calcificação de discos intervertebrais. Queixa de lombalgia de caráter mecânico de longa data e gonalgia progressiva bilateral. Apresentava quadro clínico e radiológico semelhante ao do irmão (descritos anteriormente).
A seguir, ilustraremos os casos relatados:
DISCUSSÃO
O ácido homogentísico se fixa no colágeno, sendo, portanto, depositado nos tecidos conjuntivos, resultando em uma coloração azul escura na pele, orelhas, escleras e cartilagem(1, 6).
O diagnóstico de alcaptonúria foi confirmado nos pacientes acima, baseado nos achados clínicos de comprometimento cutâneo, ocular, articular e cardiovascular, alterações radiográficas e pesquisa do ácido homogentísico na urina(1, 7).
A seguir, discorreremos sobre as principais manifestações clínicas da doença.
ARTROPATIA OCRONÓTICA
A patogênese da alcaptonúria envolve depósito de pigmento na cartilagem, cápsula articular, tendões e ligamentos. A cartilagem articular apresenta coloração marrom enegrecida mais evidente nas camadas profundas, devido a alta afinidade das fibrilas de colágeno pelo pigmento ocronótico. Erosões podem alcançar o osso subcondral em casos avançados. Joelhos, ombros e quadris são as articulações mais comumente acometidas. O envolvimento axial inclui hérnia discal, osteofitose e espondilose. Pacientes queixam-se de lombalgia, diminuição da mobilidade e da estatura(1, 4, 8, 9).
CUTÂNEAS
A cartilagem do ouvido externo é tipicamente acometida, sendo freqüentemente o primeiro sítio cutâneo envolvido. A membrana timpânica e ossículos podem demonstrar alterações ocronóticas, caracterizando-se com hipoacusia ou surdez.
A pele exibe coloração azul enegrecida em regiões de alta densidade de glândulas sudoríparas e de tecido cartilaginoso(1, 8).
OCULAR
O pigmento ocronótico deposita-se, primariamente, na esclera, conjuntiva, fissura interpalpebral, placa tarsal, córnea e pálpebras. A pigmentação da esclera está confinada a áreas expostas ao sol e, geralmente, precede o quadro articular. Geralmente, não ocorre alteração visual(1, 10).
CARDIOVASCULARES
Depósitos de pigmento ocronótico no sistema cardiovascular estão presentes no endocárdio, valvas cardíacas (aórtica e mitral) e na parede vascular. Sugere-se que estes sirvam de estímulo para calcificações distróficas, que eventualmente resultarão em estenose valvar(1, 11).
SISTEMA RESPIRATÓRIO
Presença de pigmento ocronótico na cartilagem hialina torna-se evidente na laringe, traquéia e cartilagem brônquica, sendo as repercussões clínicas raras como disfagia e rouquidão. A rigidez da cartilagem costal pode comprometer a função respiratória(1, 6).
TRATO GENITOURINÁRIO
O pigmento ocronótico pode formar cálculos na bexiga, rins, ureteres e uretra. O local mais acometido é a próstata, cujo pH alcalino resulta na rápida polimerização do ácido homogentísico. Os cálculos resultam tanto em obstrução vesical como prostática, ocorrendo infecções repetidas do trato urinário e uremia, respectivamente(1, 4, 6).
OUTROS SÍTIOS
Outros locais menos freqüentes de depósito incluem os dentes, sistema nervoso central (SNC) e órgãos endócrinos(1, 4).
A diversidade de manifestações clínicas são suficientes para se chegar ao diagnóstico(1, 6).
A história familiar presente, dosagem do ácido homogentísico urinário e histopatologia ajudam a confirmar seu diagnóstico(1).
Na histopatologia, observamos o pigmento amarelo-acastanhado ou ocre na derme através da coloração de HE. Grânulos finos de pigmento localizam-se nos macrófagos da derme. O pigmento também deposita-se nas células endoteliais, membrana basal e células secretoras das glândulas sudoríparas(1, 6).
Ocorre deposição de pigmento no citoplasma dos condrócitos, resultando em degeneração das fibras de colágeno. Há fragmentação da cartilagem articular e fagocitose das fibrilas de colágeno pelos macrófagos sinoviais. Os fragmentos levam a um quadro de sinovite inespecífica(1, 6).
O diagnóstico diferencial deve incluir artropatia hemofílica, sinovite vilonodular pigmentada, artropatia associada a hemocromatose e espondilite anquilosante(1).
Artropatia ocronótica não apresenta cura até o presente momento, pois a deficiência da enzima não pode ser tratada. Deve ser feita restrição de tirosina e fenilalanina da dieta, o que reduziria a excreção do ácido homogentísico na urina(1, 12). A artropatia é irreversível, embora a dieta possa prevenir a progressão da doença. Alguns estudos sugerem que altas doses do ácido ascórbico previnam a interação do pigmento ocronótico com os tecidos. Ensaios clínicos estão avaliando a eficácia do nitisinose (inibidor da enzima precursora do ácido homogentísico)(1, 12). A terapêutica baseia-se em medidas sintomáticas como o uso de analgésicos, antiinflamatórios não-hormonais (AINHs), fisioterapia, infiltração intra-articular com corticóide. Finalmente, artroplastia de ombros, quadris e joelhos se faz necessária naqueles casos graves de artropatia ocronótica(1). O paciente do caso 1, com artropatia ocronótica, evoluiu para prótese de ombro esquerdo aos 59 anos de idade e está aguardando prótese em joelhos.
Declaramos a inexistência de conflitos de interesse.
Recebido em 22/01/06.
Aprovado, após revisão, em 16/07/06.
Serviço de Reumatologia do Hospital do Servidor Público Estadual de São Paulo "Francisco Morato de Oliveira" (HSPE-FMO).
-
1Daniel K, Thomas B: Ochronosis. In Klippel's textbook of Rheumatology. Phyladelphia, Mosby 2: 28.1-28.4, 1998.
-
2Janocha S, Wolz W, Srsen S, et al: The human gene for alkaptonuria (AKU) maps to chromosome 3q. Genomics 19: 5-8, 1994.
-
3Phornphutkul C, Introne WJ, Perry MB, et al: Natural history of alkaptonuria. N Engl J 374: 2111-21, 2002.
-
4Mulier M, Willems K: Ochronosis: Clinical manifestations. Belgian orthweb project, case of month, august 1997. http\\www.orthoweb.be/cases/
-
5Lima ACR, Navarro ML, Provenza JR, Bonfiglioli Rl: artropatia Ocronótica: Relato de Caso. Rev Bras Reumatol 40: 213-6, 2000.
-
6Knubone TS, Selner AJ: Ochronosis and alkaptonuria. Case Report and Literature Review. J am Pediatr Med Assoc 85: 554-5, 1995.
-
7Alparslan L, Weissman BN: Imagin. In: Harris Jr, RC Budd, GS Firestein et al(eds). In:Kelley's Text book of Rheumatology. Philadelphia, W.B. Saunders 1: 763-4, 2005.
-
8Gutzmer R, Herbst RA, Kiehl P, Kapp A, Weiss J: Alkaptonuric ochronosis: report of two affected brothers. J Am Acad Dermatol 37: 305-7, 1997.
-
9Cortina R, Moris C, Astudillo A, Gosálbez F, Cortina A: Familial ochronosis. Eur Heart J 16: 285-6, 1995.
-
10Zannoni VG, Lomtevas N, Goldfinger S: Oxidation of homogentisic acid to ochronotic pigment in connective tissue. Biochim Biophys Acta 177: 94-105, 1969.
-
11Kocyigit H, Gurgan A, Terzioglu R, Gurgan U: Clinical, radiographic and echocardiographic findings in a patient with ochronosis. Clin Rheumatol 17: 403-6, 1999.
-
12Forslind K, Wollhein FA, Akesson B, Rydholm U: Alkaptonuria and ochronosis in three siblings: ascorbic acid treatment monitored by urinary HGA excretion. Clin Exp Rheumatol 6: 289-92, 1988.
Endereço para Correspondência:
Datas de Publicação
-
Publicação nesta coleção
05 Dez 2006 -
Data do Fascículo
Out 2006
Histórico
-
Recebido
22 Jan 2006 -
Aceito
16 Jul 2006