Resumos
Arterite de Takayasu (AT) é uma doença rara caracterizada por vasculite dos grandes vasos, principalmente aorta e seus ramos. A inflamação vascular leva a irregularidades na parede do vaso, causando estenoses e aneurismas. O envolvimento da artéria pulmonar (AP) é freqüente na AT. Apesar disso, o desenvolvimento de hipertensão arterial pulmonar (HAP) é menos comum, alterando o tratamento e o prognóstico dos pacientes. Descrevemos um caso de uma paciente com AT com estenoses múltiplas em artérias pulmonares, HAP e doença cerebrovascular, além de revisão de literatura sobre o assunto.
vasculite; arterite de Takayasu; hipertensão pulmonar
Takayasu arteritis (TA) is a rare illness characterized by vasculitis of great vessels, mainly of aorta and its branches. The vascular inflammation leads to irregularities of the vessel wall causing stenosis and aneurysms. The pulmonary artery (PA) involvement is frequent in TA. Despite this, the development of pulmonary arterial hypertension is less common, modifying the treatment and the prognosis of the patients. We describe a case of a patient with TA with multiple stenosis in pulmonary arteries, pulmonary arterial hypertension and cerebrovascular disease, as well as review of the literature on the subject.
vasculitis; Takayasu arteritis; pulmonary hypertension
RELATO DE CASO CASE REPORT
Hipertensão arterial pulmonar e acidente vascular encefálico em paciente com Arterite de Takayasu
Pulmonary arterial hypertension and cerebrovascular disease in patient with Takayasu Arteritis
Carlos Frederico Rodrigues ParchenI; Carolina de Souza MüllerII; Salun AragãoIII; Eduardo Santos PaivaIV; Sebastião RadominskiIV
IMédico do Curso de Especialização em Reumatologia do HC-UFPR
IIMédica Residente em Reumatologia do HC-UFPR
IIIMédico Residente em Clínica Médica do HC-UFPR
IVMédico do Serviço de Reumatologia do HC-UFPR
Endereço para correspondência Endereço para correspondência: Rua Frei Fabiano de Cristo, 603 CEP 81530-110, Curitiba, PR, Brasil telefone: (41) 3266-2467 e-mail: carlosparchen@yahoo.com.br
RESUMO
Arterite de Takayasu (AT) é uma doença rara caracterizada por vasculite dos grandes vasos, principalmente aorta e seus ramos. A inflamação vascular leva a irregularidades na parede do vaso, causando estenoses e aneurismas. O envolvimento da artéria pulmonar (AP) é freqüente na AT. Apesar disso, o desenvolvimento de hipertensão arterial pulmonar (HAP) é menos comum, alterando o tratamento e o prognóstico dos pacientes. Descrevemos um caso de uma paciente com AT com estenoses múltiplas em artérias pulmonares, HAP e doença cerebrovascular, além de revisão de literatura sobre o assunto.
Palavras-chave: vasculite, arterite de Takayasu, hipertensão pulmonar.
ABSTRACT
Takayasu arteritis (TA) is a rare illness characterized by vasculitis of great vessels, mainly of aorta and its branches. The vascular inflammation leads to irregularities of the vessel wall causing stenosis and aneurysms. The pulmonary artery (PA) involvement is frequent in TA. Despite this, the development of pulmonary arterial hypertension is less common, modifying the treatment and the prognosis of the patients. We describe a case of a patient with TA with multiple stenosis in pulmonary arteries, pulmonary arterial hypertension and cerebrovascular disease, as well as review of the literature on the subject.
Keywords: vasculitis, Takayasu arteritis, pulmonary hypertension.
INTRODUÇÃO
Classicamente, define-se a arterite de Takayasu (AT) como uma doença inflamatória rara, crônica, progressiva e oclusiva da aorta e seus ramos. É uma doença sistêmica que pode apresentar manifestações isoladas, atípicas e catastróficas. Historicamente, Takayasu, em 1908, descreveu as alterações retinianas da doença, mesma época na qual foram relatadas as alterações vasculares, como a ausência de pulso. Entretanto, somente após 1954 a doença recebeu o nome que leva até hoje(1, 11).
Nos Estados Unidos, a incidência anual estimada é de 2,6 casos por 1 milhão de habitantes, aproximadamente cem vezes menos que no Japão. Embora a AT apresente distribuição mundial, é mais freqüentemente vista na Ásia que na Europa e Américas, sendo que 80% dos pacientes são do sexo feminino. A maioria dos pacientes encontra-se entre 5 e 60 anos de idade, e a idade média de aparecimento está na quarta década de vida. A auto-imunidade celular parece ter relação direta com a injúria vascular. Encontra-se inflamação granulomatosa envolvendo macrófagos, linfócitos e células gigantes multinucleadas. Alternando períodos de atividade e remissão, a taxa de sobrevida de 15 anos é descrita como de 90 a 95%(1).
Diante do exposto e da possibilidade de acompanhamento desses pacientes em nosso Serviço de Reumatologia, relatamos o caso de uma paciente com AT com estenoses múltiplas em artérias pulmonares e doença cerebrovascular.
RELATO DE CASO
Paciente feminina, 30 anos, com diagnóstico de AT cinco meses antes da admissão, baseado nos seguintes sintomas: carotidínea persistente, claudicação de membros superiores, ausência de pulso palpável em membros superiores e incapacidade de se mensurar a pressão arterial nos membros superiores. O diagnóstico foi confirmado por arteriografia cérvico-torácica, apresentando estenose crítica em artéria carótida comum em subclávias direita e esquerda, sem acometimento de circulação vertebral. A paciente estava usando prednisona diariamente e metotrexato semanal, com controle adequado da doença após o diagnóstico.
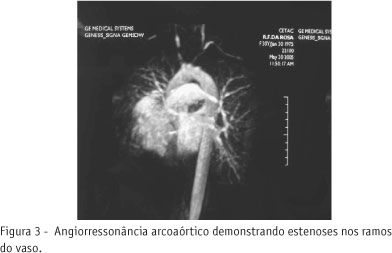
Foi internada por quadro súbito de afasia de expressão, sem outros déficits focais. Apresentava ainda queixa de dispnéia aos esforços há dois meses. Ao exame físico, apresentava ausência de pulsos assim como pressão arterial inaferível em membros superiores, sopro em região de carótidas e de subclávias, hiperfonese de B2 no foco pulmonar e afasia. Tomografia computadorizada (TC) de crânio na admissão era normal. A radiografia de tórax era normal. Exames laboratoriais de entrada mostravam: hemograma com leucocitose, plaquetas normais, coagulograma, provas de função renal e hepática, velocidade de hemossedimentação (VHS) e proteína C reativa (PCR) normais. Optou-se por realizar pulsoterapia com metilprednisolona 1 g por três dias. No terceiro dia da pulsoterapia, a paciente ainda apresentava afasia e começou a desenvolver hemiparesia à direita. Nova TC de crânio foi realizada, evidenciando infarto no território da artéria cerebral média esquerda (Figura 1). Pela evidência de infarto em evolução, foi iniciada anticoagulação. Foi realizada também arteriografia cérvico-torácica, que mostrou, além dos achados do primeiro exame, estenose crítica no tronco inominado, artéria vertebral direita hipoplásica com estenose crítica na sua origem (vertebral esquerda dominante) e fluxo retrógrado na artéria vertebral esquerda. O exame de Doppler transcraniano evidenciou distúrbio hemodinâmico na circulação intracraniana, caracterizado por fluxo colateral em todos os vasos estudados, e provável exaustão distal em artéria cerebral média esquerda. Ecocardiograma mostrou hipertensão arterial pulmonar (HAP) grave (pressão sistólica na AP de 86 mmHg) e discreta hipertrofia do ventrículo direito. Realizada, então, arteriografia de circulação pulmonar (Figura 2), que mostrou múltiplas estenoses nas artérias do pulmão direito e esquerdo, com pressão sistólica no tronco da AP de 58 mmHg. Visando avaliar parede da artéria foi realizada uma angioressonância da aorta torácica com mensuração de sua espessura. O exame mostrou acentuada redução do calibre nos segmentos proximais do tronco-braquiocefálico, artérias carótidas comuns e subclávias (Figura 3).
A paciente recebeu alta com ajuste da dose de prednisona e metotrexato, além de programa e acompanhamento multidisciplinares. Após três meses, a paciente, ainda em acompanhamento multidisciplinar e ambulatorial, apresenta melhora parcial da afasia e da hemiparesia e melhora progressiva da dispnéia associada à melhora hemodinâmica cardiopulmonar e à redução da pressão sistólica da AP, avaliada pelo ecocardiograma.
DISCUSSÃO
Clinicamente, a apresentação da AT é heterogênea. Sintomas constitucionais (febre, perda de peso, astenia, anemia, mialgia e artralgia) geralmente precedem envolvimento clínico vascular. É uma vasculite granulomatosa de grandes artérias elásticas, especificamente a aorta e seus ramos principais, podendo afetar também as coronárias e as artérias pulmonares. O dano inflamatório na parede do vaso leva a áreas de destruição da camada muscular lisa e elástica da artéria, resultando em estenoses e aneurismas(2, 9). A oclusão completa dos vasos afetados resulta em perda do pulso, sendo também denominada "doença sem pulso". Diversos sistemas podem ser acometidos, e pode-se encontrar claudicação de mandíbula e de extremidades. Achados neurológicos (60%) devem-se à isquemia ou hemorragia cerebral, cegueira transitória, cefaléia, convulsões e síndrome do roubo da subclávia. Alterações cardiovasculares (50%) compreendem regurgitação aórtica, angina, hipertensão arterial sistêmica, insuficiência cardíaca congestiva e arritmias. Manifestações pulmonares (40%) incluem HAP (freqüentemente assintomática), hemoptise, pleurite e alteração das provas de função pulmonar. Ainda podem ser encontrados glomerulonefrite e depósitos amilóides nos rins, além de comprometimento cutâneo através de eritema multiforme, lesões papulares, ulceradas e necróticas(1).
O envolvimento das artérias pulmonares é bem descrito na AT, geralmente associado ao acometimento arterial sistêmico. Hayashi et al(3) relatam dois casos de AT nos quais a AP foi inicialmente acometida, com diagnóstico inicial de embolismo crônico pulmonar. Na mesma linha de pesquisa, Brugiere et al(4) descrevem um paciente com acometimento isolado de artérias pulmonares e AT. Os sinais e sintomas apresentados, na época, eram similares aos encontrados na doença tromboembólica. Arteriografia demonstrou estenoses e oclusão arterial pulmonar bilateral, enquanto não foram verificadas outras alterações arteriográficas em outros territórios. O paciente faleceu após hemoptise maciça e o exame anatomopatológico confirmou lesões características de AT pulmonares e ausência de lesão aórtica e ramos. Acredita-se que a freqüência desta apresentação seja subestimada, dadas as dificuldades diagnósticas e a similaridade com doença tromboembólica.
Vanoli et al(5) estudando 104 pacientes com AT na Itália relataram que, do total avaliado, nove pacientes foram submetidos à arteriografia pulmonar demonstrando estenoses em dois deles e oclusão completa em dos ramos da AP em um paciente. Os autores acreditam que a taxa de 30% de envolvimento pulmonar subestima a taxa real.
Nunes et al(6) descrevem o acometimento pulmonar na AT entre 14 e 56%. Entretanto, o desenvolvimento de HAP grave é um evento extremamente raro no curso natural da doença. Os autores relatam um caso no qual o paciente apresentava insuficiência cardíaca congestiva grave de início recente e que a avaliação seqüencial sugeriu a presença de HAP de etiologia desconhecida. A ausência de pulso radial esquerdo, ao exame físico, levou-os a realizar arteriografia no paciente que confirmou o diagnóstico de AT com envolvimento pulmonar e HAP grave.
Bletry et al(7) publicaram três casos de envolvimento arterial pulmonar grave em pacientes com AT e concluem que o acometimento pulmonar nesta doença ocorre, preferencialmente, no ramo principal direito da AP, podendo, também, ser mais distal e danificar arteríolas pulmonares. Além disso, afirmam que as principais complicações desse padrão de apresentação são a HAP e a possibilidade de hemoptise maciça.
O envolvimento das artérias carótidas e das vertebrais causam diminuição do fluxo sanguíneo cerebral, desenvolvendo vertigem, síncope, cefaléia, convulsão e demência. Diminuição da acuidade visual é manifestação tardia e é resultado da isquemia cerebral(12). A AT é causa bem conhecida de doença cerebrovascular nos países orientais, sendo pouco descrita nos países ocidentais. Ringleb et al(8) avaliaram um grupo de 17 pacientes europeus com AT, através de exame clínico neurológico e imagens de ressonância magnética. O autor encontrou evidência de doença intracraniana em sete pacientes, incluindo infarto, isquemias transitórias recorrentes e tontura. Além disso, afirma que o curso das manifestações neurológicas é benigno quando o paciente portador da vasculite é tratado e bem assistido.
Historicamente, AT era uma doença devastadora, mas o diagnóstico precoce, tratamento agressivo e o acompanhamento multidisciplinar têm levado a um melhor prognóstico. Complicações cardiovasculares, incluindo aceleração da aterosclerose e doenças isquêmicas, são as principais causas de óbito. Relatos recentes mostram que o desenvolvimento de HAP grave é evento de apresentação variável na evolução da doença(6). A recanalização das estenoses das artérias elásticas pulmonares (também chamada "vasos sanguíneos no vaso sanguíneo") parece estar envolvida com a hipertensão arterial pulmonar(2), através do remodelamento das camadas da parede da artéria, bem como pelo aumento da resistência hemodinâmica vascular, associados ou não a eventos tromboembólicos.
Lie(10) relata cinco pacientes, dois homens e três mulheres, com AT que foram submetidos à cirurgia para ressecção pulmonar ou para reconstrução/bypass das artérias pulmonares obstruídas. Histologicamente, foram observadas lesões vasculares incluindo a clássica arterite granulomatosa de células gigantes de grandes vasos e um tipo peculiar de trombos organizados com marcante recanalização e neoangiogênese, sugerindo achados histopatológicos próprios do acometimento das artérias pulmonares em pacientes com AT. A associação dos eventos acima descritos influencia a morbi-mortalidade, a longo prazo, desses pacientes.
Observamos após a discussão acima, que o caso descrito encontra semelhanças com os da literatura revisada. Acreditamos ser importante informar que AT ainda permanece um desafio diagnóstico e terapêutico. Apesar dos tratamentos disponíveis atualmente, a progressão da lesão vascular é freqüentemente contínua. Agentes imunossupressores como metotrexato, azatioprina e micofenolato de mofetil associados aos corticóides podem beneficiar muitos pacientes. Infelizmente, reativações da doença são comuns quando se tenta reduzir as doses das medicações prescritas. Conhecendo melhor a patogenia da AT, pesquisas têm sido desenvolvidas com agentes antagonistas do fator de necrose tumoral alfa (anti-TNF alfa) em pacientes com doença refratária, sendo os resultados iniciais animadores. Procedimentos de revascularização têm bons resultados com o uso de bypass convencionais. Lesões vasculares curtas podem ser tratadas através de angioplastia transluminal percutânea, apesar de o uso dos stents comuns não manter a patência do vaso a longo prazo, ocorrendo reestenose em até 40% dos casos.
A inflamação crônica e a disfunção endotelial colocam os portadores de AT em risco para desenvolvimento de aterosclerose precoce. Por esses motivos, sugerimos o controle dos fatores de risco modificáveis para aterosclerose para todos os pacientes. Futuramente, esperamos que novas tendências terapêuticas estejam disponíveis visando não só a inflamação, mas também a proliferação miointimal vascular observada na AT(13).
Declaramos a inexistência de conflitos de interesse.
Recebido em 08/04/06.
Aprovado, após revisão, em 28/09/06.
Departamento de Reumatologia do Hospital de Clínicas da Universidade Federal do Paraná (HC-UFPR).
-
1eMedicine - Takayasu Arteritis: Article by Julie Beales, 2005. Acesso em 15/08/2006. <http://www.emedicine.com/med/topic2232.htm>.
-
2Matsubara O, Yoshimura N, Tamura A, et al: Pathological features of the pulmonary artery in Takayasu arteritis. Heart vessels Suppl 7: 18-25, 1992.
-
3Hayashi K, Nagasaki M, Matsunaga N: Initial Pulmonary Artery Involvement in Takayasu Arteritis'. Radiology 159: 401-3, 1986.
-
4Brugiere O, Mal H, Sleiman, et al: Isolated pulmonary arteries involvement in a patient with Takayasu's arteritis. Eur Respir J 11: 767–70, 1998.
-
5Vanoli M, Daina E, Salvarani C, et al: Takayasu's Arteritis: A Study of 104 Italian Patients. Arthritis Rheum 53: 100-7, 2005.
-
6Nunes H, Ramos JM, Valerio L, et al: Pulmonary artery involvement in Takayasu Arteritis. A case of right ventricular failure as presentation form. Rev Port Cardiol 11: 775-80, 1992.
-
7Bletry O, Kieffer E, Herson S, et al: Severe pulmonary involvement of Takayasu Arteritis. 3 cases and review of the literature. Arch Mal Coeur Valss 84: 817-22, 1991.
-
8Ringleb PA, Strittmatter EI, Loewer M, et al: Cerebrovascular manifestations of Takayasu arteritis in Europe. Rheumatology (Oxford) 44: 1012-5, 2005.
-
9Klippel J: Primer on the Rheumatic Diseases, 12.a ed, Atlanta, Editora Arthritis Foundation, 2001.
-
10Lie JT: Isolated pulmonary Takaysu Arteritis: clinicopathologic characteristics. Mod Pathol 9: 469-74, 1996.
-
11Numano F: The story of Takayasu arteritis. Rheumatology 41: 103–6, 2002.
-
12Up To Date – Takayasu Arteritis: Article by Gene G Hunder, 2006. Acesso em 15/08/2006.<http://www.utdol.com/utd/content/topic.do?topicKey=vasculit/ 7733&type=A&selectedTitle=1~25>.
-
13Liang P, Hoffman GS: Advances in the medical and surgical treatment of Takayasu arteritis. Curr Opin Rheumatol 17: 16-24, 2005.
Datas de Publicação
-
Publicação nesta coleção
09 Fev 2007 -
Data do Fascículo
Dez 2006
Histórico
-
Recebido
08 Abr 2006 -
Aceito
28 Set 2006