Resumos
A displasia diafisária progressiva ou doença de Camurati-Engelmann é uma doença rara, caracterizada por dor e fraqueza muscular dos membros afetados e espessamento progressivo da cortical diafisária dos ossos longos. Os autores descrevem o caso clínico de um doente com manifestações iniciais da doença na infância, mas cujo diagnóstico só foi estabelecido durante a idade adulta, depois de evoluída a doença e após surgirem as mesmas manifestações em um dos filhos. Salienta-se a dificuldade no diagnóstico e a relevância do diagnóstico diferencial com outras doenças que cursam com osteoesclerose e/ou hiperostose. Na literatura é rara a descrição da sua evolução ao longo dos anos.
doença de Camurati-Engelmann; displasia diafisária progressiva
Camurati-Engelmann Disease or progressive diaphyseal dysplasia is a rare disease, characterized by limb pain and muscular weakness, and cortical thickening of the diaphyses of long bones. The authors report a case of a male patient with manifestations since his childhood, whose diagnosis was established later on, when he was an adult, with the disease already progressed, and when the same manifestations began in one of his sons. The importance of the differential diagnosis regarding other diseases concurrent with osteosclerotic and/or hyperostotic changes is emphasized here. Description of its evolution along the years is rarely found in the literature.
Camurati-Engelmann disease; progressive diaphyseal dysplasia
RELATO DE CASO
Doença de Camurati-Engelmann: manifestações típicas de uma doença rara
Mónica BogasI; Vanessa BogasII; Frederico PintoIII
IMédica-Residente do Internato Complementar de Reumatologia do Serviço de Reumatologia do Centro Hospitalar do Alto Minho, Ponte de Lima, Portugal
IIDoutoranda em Ciências Forenses, Departamento de Biologia e Genética Forense, Delegação do Centro do Instituto Nacional de Medicina Legal, Universidade de Coimbra, Portugal
IIIMédico Fisiatra do Serviço de Medicina Física e Reabilitação do Hospital de Matosinhos, Portugal
Endereço para correspondência Endereço para correspondência: Centro Hospitalar do Alto Minho 04990-041 - Ponte de Lima. Telefone: 25-8909-500. Fax: 25-9909-501. E-mail: monica.bogas@sapo.pt
RESUMO
A displasia diafisária progressiva ou doença de Camurati-Engelmann é uma doença rara, caracterizada por dor e fraqueza muscular dos membros afetados e espessamento progressivo da cortical diafisária dos ossos longos. Os autores descrevem o caso clínico de um doente com manifestações iniciais da doença na infância, mas cujo diagnóstico só foi estabelecido durante a idade adulta, depois de evoluída a doença e após surgirem as mesmas manifestações em um dos filhos. Salienta-se a dificuldade no diagnóstico e a relevância do diagnóstico diferencial com outras doenças que cursam com osteoesclerose e/ou hiperostose. Na literatura é rara a descrição da sua evolução ao longo dos anos.
Palavras-chave: doença de Camurati-Engelmann; displasia diafisária progressiva.
INTRODUÇÃO
A displasia diafisária progressiva ou doença de Camurati-Engelmann é uma doença hereditária do metabolismo ósseo, autossômica dominante, rara, caracterizada pelo espessamento progressivo da cortical diafisária dos ossos longos. A marcha "bamboleante" de características miopáticas e a dor nos membros atingidos são as manifestações clínicas mais frequentes. Dada a sua evolução e raridade, o seu diagnóstico é frequentemente estabelecido com atraso e dificuldade. Neste artigo são descritas a evolução de um doente, a dificuldade no diagnóstico da doença de Camurati-Engelmann e a relevância de se considerar o diagnóstico de displasia óssea na dor inespecífica dos membros e de outras doenças que cursam com osteoesclerose e/ou hiperostose no seu diagnóstico diferencial.
RELATO DE CASO
O paciente tem 46 anos, é trabalhador aeroportuário, filho de pais não consanguíneos e natural de Moçambique. Desde a infância, sentia dores nos membros inferiores, de predomínio proximal, descrevendo como de origem óssea e muscular, e progressiva diminuição da força muscular adquirindo marcha bamboleante de características miopáticas. As dores tinham um ritmo misto e um componente noturno importante. Ao longo do tempo, as queixas álgicas passaram a ser menos intensas e a adaptação física e psicológica ao quadro foi permitindo ter uma vida dentro dos parâmetros considerados normais na sociedade, tendo uma profissão normal, casando-se e tendo filhos. Foi aprendendo técnicas para o levante e para o disfarce da marcha, negando limitações na realização das tarefas diárias e profissionais. No início do quadro clínico e durante alguns anos, procurou orientação médica, mas toda a investigação foi inconclusiva, não existindo, no estudo efetuado, dados que apoiassem o diagnóstico de doença neuromuscular ou indícios de malignidade. Quatro décadas depois do início dos sintomas, a evidência de alterações radiográficas nas diáfises femurais, nunca antes detectadas, fez despertar de novo o interesse no esclarecimento do quadro clínico. Ao exame objetivo, eram descritas alterações relevantes, como diminuição da força proximal dos membros inferiores com manobra de Gowers positiva e atrofia muscular das pernas. Os reflexos osteo-tendinosos eram normais e não havia evidência de alterações cognitivas, do tônus ou de comprometimento articular. No estudo analítico, não existiam alterações, nomeadamente do hemograma, velocidade de sedimentação, função renal, metabolismo fosfocálcico e fosfatase alcalina. Nessa altura, o seu filho mais velho, com 5 anos de idade, começou a apresentar um quadro clínico semelhante, tendo a etiologia hereditária passado a fazer parte do raciocínio médico. O diagnóstico de doença de Camurati-Engelmann foi então considerado e confirmado em ambos por detecção da mutação C.652C > T (p.Arg218Cys) em heterozigotia no éxon 4 do gene TGF²1. Quando interrogado, o doente negava antecedentes familiares semelhantes. Na sua outra filha, assintomática, não foi detectada alteração genética compatível com a doença.
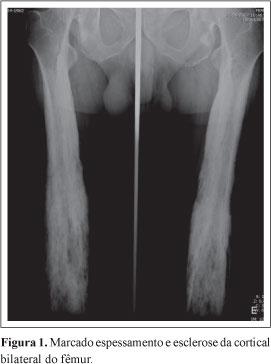
Mais recentemente, o doente sofreu fratura do colo femural e trocanter direitos após acidente de viação, sendo submetido a uma cirurgia para encavilhamento endomedular proximal do fêmur e orientado para tratamentos de reabilitação funcional. O espessamento e esclerose da cortical, alterações radiológicas típicas da doença, foram encontrados nos ossos longos dos membros inferiores e superiores de forma bilateral e simétrica, sem alterações das mãos ou pés. (Figuras 1,2,3,4 e 5) O estudo por tomografia axial computorizada mostrou estreitamento do canal medular dos ossos envolvidos. (Figura 6) A radiografia do crânio era normal.
O doente não apresentou complicações no pós-operatório e recuperou a marcha autônoma com os tratamentos de fisioterapia. Manteve-se a vigilância na consulta externa de fisiatria durante cinco meses.
DISCUSSÃO
A displasia diafisária progressiva ou doença de Camurati-Engelmann é uma doença hereditária rara, autossômica dominante, caracterizada pelo espessamento progressivo da cortical diafisária dos ossos longos.1
Uma mutação no gene TGF²1 no cromossoma 19q13, que se traduz no aumento da atividade dessa proteína, parece estar na origem dessa entidade clínica.2 O quadro clínico compreende dor nos membros atingidos, fraqueza muscular, marcha anormal semelhante à miopática e fadiga fácil.3 A doença tem início na infância, manifestando-se geralmente antes dos 30 anos.1,3 A idade de início não é previsível, mas parece haver tendência para ser mais precoce e/ou com um fenótipo mais grave nas gerações sucessivas.4 No entanto, a evolução da doença parece poder ser variável, podendo o quadro clínico apresentar um percurso indolente ou mesmo parecer tornar-se quiescente, dificultando o diagnóstico mesmo na presença de alterações radiográficas evoluídas.5 Geralmente, o comprometimento ósseo é bilateral e tem início nas diáfises femorais e tibiais, progredindo lentamente para os perôneos, úmeros, rádios e cúbitos, com deformidade óssea progressiva.1,6-8 Embora menos frequentemente, o crânio e a bacia também podem ser envolvidos.1,6,9 O aumento da atividade osteoblástica na região afetada pode ser detectado precocemente por cintigrafia do esqueleto.1,6 As epífises e os espaços articulares são geralmente poupados, embora na evolução possam estar envolvidos secundariamente.7 O quadro clínico pode, ocasionalmente, acompanhar-se de manifestações sistêmicas, como anemia, leucopenia e hepatoesplenomegalia, por estenose e comprometimento medular.1,10,11 A elevação da velocidade de sedimentação globular ou da fosfatase alcalina podem ocorrer.6,10 Embora possa ser sugerido pela observação, o comprometimento neuromuscular não faz parte do quadro clínico.3,12
Pensa-se que as alterações na remodelação óssea induzidas afetem tanto o processo de reabsorção osteoclástica quanto o de formação osteoblástica. Isso estaria de acordo com o papel da proteína TGFb1 na estimulação e supressão da reabsorção óssea. Apesar disso, as alterações dos marcadores clássicos de formação ou reabsorção óssea não são consistentes.13
Na doença de Camurati-Engelmann, ao contrário de outras doenças do metabolismo ósseo, as fraturas de baixo impacto são raras.7 As alterações microscópicas do osso são inespecíficas, sendo a biópsia útil apenas para excluir outras causas.7 A cirurgia ortopédica, associada a complicações peri-operatórias em outras doenças do metabolismo ósseo (por exemplo, doença de Paget), tem sido raramente descrita nos doentes com Camurati-Engelmann, conhecendo-se pouco a sua evolução e prognóstico. No caso descrito, não houve qualquer intercorrência durante e no pós-operatório num período de quatro meses de vigilância na consulta externa de fisiatria e ortopedia.
No diagnóstico diferencial dessa doença, as displasias pertencentes ao grupo das hiperostoses craniotubulares, como a síndrome de Van Buchem, a osteosclerose e a esclerostose, além de entidades como a displasia craniodiafisária e a hiperfosfatasemia familiar, deverão ser consideradas.7,14 Em virtude de poder ocorrer alguma latência temporal entre o aparecimento dos sintomas num membro e no outro contralateral, deverão ser consideradas outras situações que envolvem dor, tumefação de ossos longos e alterações radiográficas semelhantes, como o osteossarcoma, a osteomielite e a doença de Paget.7,14 Dada a gravidade de algumas dessas doenças e a necessidade de tratamento específico, o estudo diagnóstico deverá iniciar-se rapidamente.
Os efeitos secundários dos corticosteroides, considerados adversos na maioria das situações, podem ser aproveitados para o tratamento da doença de Camurati-Engelmann.1,15 A sua capacidade de indução da apoptose e interferência na proliferação e diferenciação dos osteoblastos e dos osteócitos são responsáveis pela diminuição da densidade mineral óssea, útil nessa situação. A dexametasona, a prednisolona e o deflazacort, em doses correspondentes a 0,5-1 mg/kg/dia de prednisolona, foram usados com sucesso no tratamento da dor e sintomas constitucionais.1,15 Esses fármacos não alteram, no entanto, o curso da doença.
A terapêutica com anti-inflamatórios não esteroides tem apenas como objetivo a analgesia, não tendo qualquer efeito no metabolismo ósseo.1 O uso de bisfosfonatos é controverso: há descrições distintas de melhoria e de agravamento sintomático com a sua administração.1,16 O tratamento fisioterápico pode ser instituído com o principal objetivo de aumento da força muscular e de evitar retrações tendinosas.1
A cirurgia poderá estar indicada no alargamento do canal medular e na descompressão neurológica.17-19
Recebido em 22/07/2008.
Aprovado, após revisão, em 18/01/2009.
Declaramos a inexistência de conflitos de interesse.
Trabalho realizado no Serviço de Medicina Física e Reabilitação do Hospital de Matosinhos, Portugal.
-
1Janssens K, Vanhoenacker F, Bonduelle M, Verbruggen L, Van Maldergem L, Ralston S et al Camurati-Engelmann disease: review of the clinical, radiological, and molecular data of 24 families and implications for diagnosis and treatment. J Med Genet 2006;43(1):1-11.
-
2Janssens K, ten Dijke P, Ralston SH, Bergmann C, Van Hul W. Transforming growth factor-beta 1 mutations in Camurati-Engelmann disease lead to increased signaling by altering either activation or secretion of the mutant protein. J Biol Chem 2003;278(9):7718-24.
-
3Bondestam J, Pihko H, Vanhanen SL, Brander A, Toiviainen-Salo S, Marttinen E et al Skeletal dysplasia presenting as a neuromuscular disorder - report of three children. Neuromuscul Disord 2007;17(3):231-4.
-
4Saraiva JM. Anticipation in progressive diaphyseal dysplasia. J Med Genet 2000;37(5):394-5.
-
5Naveh Y, Kaftori JK, Alon U, Ben-David J, Berant M. Progressive diaphyseal dysplasia: genetics and clinical and radiologic manifestations. Pediatrics 1984;74(3):399-405.
-
6Kumar B, Murphy WA, Whyte MP. Progressive diaphyseal dysplasia (Engelmann disease): scintigraphic-radiographic-clinical correlations. Radiology 1981;140(1):87-92.
-
7Kaftori JK, Kleinhaus U, Naveh Y. Progressive diaphyseal dysplasia (Camurati-Engelmann): radiographic follow-up and CT findings. Radiology 1987;164(3):777-82.
-
8Vanhoenacker FM, Janssens K, Van Hul W, Gershoni-Baruch R, Brik R, De Schepper AM. Camurati-Engelmann disease. Review of radioclinical features. Acta Radiol 2003;44(4):430-4.
-
9Moumoulidis I, De R, Ramsden R, Moffat D. Unusual otological manifestations in Camurati-Engelmann's disease. J Laryngol Otol 2006;120(10):892-5.
-
10Crisp AJ, Brenton DP. Engelmann's disease of bone—a systemic disorder? Ann Rheum Dis 1982;41(2):183-8.
-
11Mondal RK, Karmakar B, Chandra PK, Mukherjee K. Ghosal type hemato-diaphyseal dysplasia: a rare variety of Engelmann's disease. Indian J Pediatr 2007;74(3):291-3.
-
12Yoshioka H, Mino M, Kiyosawa N, Hirasawa Y, Morikawa Y, Kasubuchi Y et al Muscular changes in Engelmann's disease. Arch Dis Child 1980;55(9):716-9.
-
13Hernández MV, Peris P, Guañabens N, Alvarez L, Monegal A, Pons F et al Biochemical markers of bone turnover in Camurati-Engelmann disease: a report on four cases in one family. Calcif Tissue Int 1997;61(1):48-51.
-
14Seeger LL, Hewel KC, Yao L, Gold RH, Mirra JM, Chandnani VP et al Ribbing disease (multiple diaphyseal sclerosis): imaging and differential diagnosis. Am J Roentgenol 1996;167(3):689-94.
-
15Minford AMB, Hardy GJ, Forsythe WI, Fitton JM, Rowe VL. Engelmann's disease and the effect of corticosteroids. J Bone Joint Surg Br 1981;63(4):597-600.
-
16Inaoka T, Shuke N, Sato J, Ishikawa Y, Takahashi K, Aburano T et al Scintigraphic evaluation of pamidronate and corticosteroid therapy in a patient with progressive diaphyseal dysplasia (Camurati-Engelmann disease). Clin Nucl Med 2001;26(8):680-2.
-
17Lazzarone C, Cartesegna M, Crova M, Calorio D. Progressive diaphyseal dysplasia: Camurati-Engelmann's disease. Ital J Orthop Traumatol 1983;9(1):109-14.
-
18Simpson RK Jr, Fisher DK, Gall GK, Rose JE. Fatal cerebellar herniation secondary to Camurati-Englemann's disease. J Neurol Neurosurg Psychiatry 1988;51(10):1349-52.
-
19Raffaelli P, Ronzini MF. Camurati-Engelmann's disease. A case report. Ital J Orthop Traumatol 1988;14(2):267-71.
Datas de Publicação
-
Publicação nesta coleção
07 Jul 2009 -
Data do Fascículo
Jun 2009
Histórico
-
Recebido
22 Jul 2008 -
Aceito
18 Jan 2009