Abstracts
Mental retardation (MR) is a definition which comprises a series of conditions whose common feature is an intellectual handicap that develops before the age of 18, afflicting 2-3% of the world's population. The classification of MR into different categories is determined by the extent of the handicap instead of its cause, which often remains unrecognized. Sometimes, MR runs in a family, characterizing familial MR, and those cases permit an in-depth look into the genetic causes and consequences of the problem. However, almost no work is available on the prevalence of familial MR among the registered MR cases, possibly because familial MR is a term with no clear definition. The scope of this work is to review the topic and discuss the implications of different genetic and environmental factors, which characterize particular categories of familial cases, suggesting a practical classification of familial MR, which is important for epidemiologic studies and also for counseling in the clinic. Some of the aspects are discussed under the perspective of a newly-developed country like Brazil.
Mental retardation; Public Health; Genetics; Epidemiology
Retardo mental (RM) é uma definição que compreende uma série de condições cuja característica em comum é um déficit intelectual que se desenvolve antes dos 18 anos, afetando 2-3% da população mundial. A classificação do RM em diferentes categorias é determinada pela gravidade do déficit ao invés de sua causa, que com frequência permanece obscura. O RM pode segregar na família, caracterizando RM familiar, e estes casos permitem um olhar mais aprofundado para as causas genéticas e as consequências do problema. Porém, praticamente não existem dados disponíveis sobre a prevalência do RM familiar dentre os casos registrados, possivelmente por ser um termo sem definição clara. O presente trabalho objetiva rever o tópico e discutir as implicações de diferentes fatores genéticos e ambientais que caracterizam categorias particulares de casos familiares, sugerindo uma classificação prática para o RM familiar, importante para estudos epidemiológicos e também na clínica, para aconselhamento. Alguns dos aspectos são discutidos na perspectiva de um país emergente, como o Brasil.
Retardo mental; Saúde Pública; Genética; Epidemiologia
REVIEW REVISÃO
Familial mental retardation: a review and practical classification
Retardo mental familiar: revisão e classificação prática
Angelica Francesca Maris 1; Ingrid Tremel BarbatoII; Alexis TrottIII; Marco Aurélio Echart MontanoIII
ICentro de Ciências Biológicas, Departamento de Biologia Celular, Embriologia e Genética, Universidade Federal de Santa Catarina (UFSC). 88040-900 Florianopolis SC. afmaris@gmail.com
IILaboratório de Análises Genéticas Neurogene
IIIÁrea de Ciências Biológicas e da saúde, Universidade do Oeste de Santa Catarina
ABSTRACT
Mental retardation (MR) is a definition which comprises a series of conditions whose common feature is an intellectual handicap that develops before the age of 18, afflicting 2-3% of the world's population. The classification of MR into different categories is determined by the extent of the handicap instead of its cause, which often remains unrecognized. Sometimes, MR runs in a family, characterizing familial MR, and those cases permit an in-depth look into the genetic causes and consequences of the problem. However, almost no work is available on the prevalence of familial MR among the registered MR cases, possibly because familial MR is a term with no clear definition. The scope of this work is to review the topic and discuss the implications of different genetic and environmental factors, which characterize particular categories of familial cases, suggesting a practical classification of familial MR, which is important for epidemiologic studies and also for counseling in the clinic. Some of the aspects are discussed under the perspective of a newly-developed country like Brazil.
Key words Mental retardation, Public Health, Genetics, Epidemiology
RESUMO
Retardo mental (RM) é uma definição que compreende uma série de condições cuja característica em comum é um déficit intelectual que se desenvolve antes dos 18 anos, afetando 2-3% da população mundial. A classificação do RM em diferentes categorias é determinada pela gravidade do déficit ao invés de sua causa, que com frequência permanece obscura. O RM pode segregar na família, caracterizando RM familiar, e estes casos permitem um olhar mais aprofundado para as causas genéticas e as consequências do problema. Porém, praticamente não existem dados disponíveis sobre a prevalência do RM familiar dentre os casos registrados, possivelmente por ser um termo sem definição clara. O presente trabalho objetiva rever o tópico e discutir as implicações de diferentes fatores genéticos e ambientais que caracterizam categorias particulares de casos familiares, sugerindo uma classificação prática para o RM familiar, importante para estudos epidemiológicos e também na clínica, para aconselhamento. Alguns dos aspectos são discutidos na perspectiva de um país emergente, como o Brasil.
Palavras-chave Retardo mental, Saúde Pública, Genética, Epidemiologia
Introduction
Mental retardation (MR) is one of the most challenging and possibly most frustrating subjects for medical researchers, since it cruelly exposes the limits of modern medicine. As stated by others: "mental retardation is an idea, a condition, a syndrome, a symptom, and a source of pain and bewilderment "1. It comprises a vast amount of heterogeneous conditions caused by so many different factors that the sole attempt of grouping all those together under one common definition is audacious2. However, they all have one thing in common: they cause a disturbance in a developing brain, before, during, or after birth, impairing intellectual function. This disturbance is caused by genetic or environmental factors (including toxic products, congenital infections, nutritional deficiencies, trauma) and may originate either an isolated disability in mental function (non-syndromic MR, or nonspecific MR), or a disorder where a broad range of developmental systems is affected (syndromic MR), resulting in associated physical and functional abnormalities. The intellectual disability itself may be the consequence of gross anatomic changes in brain structure3, or reflect less obvious disturbances in pathways of the brain metabolism which may lead to altered dendrite spine morphology4, inadequate neuronal interactions5, or to an accumulation of toxic metabolites6 influencing neuronal plasticity or survival. The identification of the altered structures and of the malfunctioning pathways allows insights into their importance for normal cognitive brain function, this being one of the main reasons still justifying the grouping together of these heterogeneous conditions. For some types of inborn errors of metabolism, the development of MR can be avoided with special diets or supplements and for others treatment can reduce the severity of the symptoms7.
After mental retardation is established, there is usually no way to revert the situation. Although there are reports suggesting that Fragile-X (FRAXA) deficits could (at least partially) be reverted in the future8-12, the present role of the clinician is to manage symptoms and search for causes.
Despite modern technologies, in about 30-60% of the cases the etiology of MR remains obscure (therefore called idiopathic MR)13,14. Array comparative genomic hybridization (aCGH), a technique enabling high-resolution and genome-wide screening of segmental genomic copy number variations (CNVs), is revealing the presence of rare "de novo" duplications or deletions in approximately 15% of the idiopathic MR cases and is being accepted as first choice for the investigation of MR, complementing or replacing more traditional cytogenetic methods even in pre-natal diagnostics15-17.
The evaluation of intellectual functioning has been a controversial issue ever since Alfred Binet developed the tool which has come to be known as IQ tests. These tests typically include a range of items that assess the individual's general knowledge, vocabulary, problem-solving skills and reasoning ability. The normal IQ should score above 85. An IQ of 70 is usually considered the breaking point between borderline mental skills and mild mental retardation (MMR), considered as IQ 70-49. IQ 50-35 defines moderate MR and IQ 34-20 severe MR. Intelligence below regular testing conditions is considered IQ under 20 and defines profound MR. Despite being contentious, IQ tests still are the most frequent tool used internationally to assess cognitive ability. Allied to the evaluation of adaptive functioning, to the extent of needed support and considering cultural differences, they usually allow a relatively reliable picture of the magnitude of the mental handicap in an affected individual1,2.
For practical purposes, throughout this paper we are going to use the still most widely employed term "mental retardation" and, like others2,18-21, the following classification of MR: mild mental retardation (MMR), IQ 70-50, and serious mental retardation (SMR), at an IQ below 50 (which then encompasses moderate, severe, and profound MR).
Considered a frequent developmental disability, the overall prevalence of MR in the world population is estimated at between 1 and 3%, being one of the major causes for referral to pediatric neurologists and clinical geneticists2,22,23.
Considering only the population under 18, the World Health Organization (WHO) estimates that 0.5% to 2.5% has some degree of MR (from mild to serious) in developed countries and around 4,8% in developing countries24. However, reported prevalence rates diverge widely among different studies2,18,19,21,25-29. Most of the usually higher rates of MR in developing countries are attributed to the multifactorial (mild) familial MR the type of MR whose occurrence is most impacted by the socio-economic status of the population30. Nonetheless, there is an obvious contribution of serious MR to these higher rates.
Reviewing 33 studies conducted between 1963 and 1977, mainly in Australia and other western industrialized countries, Roeleveld and coworkers found prevalence rates from 0.28% to 0.73% for serious MR and 0.32% to 7.9% for mild MR20. Stein and coworkers, in a pilot study which assessed around 1000 children between the ages of 3 and 9 in communities of eight developing countries, including Brazil, found prevalence rates of 0.5% to 1.6% for serious MR (0.67% for Brazil)21. King and coworkers (2009) found that among those with ID, mild, moderate, severe, and profound mental retardation affects about 85%, 10%, 4%, and 2% of the population, respectively31. It is estimated that over 60% of the MR cases are of genetic etiology32 and close to 300 MR genes had been identified up to 200433. The actual number of genes implicated in MR is largely unknown. To date a little over ninety genes for X-linked MR (which are responsible for 10-12% of male MR) are identified; autosomal recessive MR genes are estimated to run into the thousands34.
Methods
The publications used for this article are from several sources. Most of them were scientific articles selected from the SciELO, MEDLINE, LILACS, PubMed and Scopus databases. The search was conducted in English, Spanish and Portuguese utilizing the keywords "mental retardation" or "intellectual disability" in combination with "familial", "hereditary", "recurrent", "causes", "X-linked", "prevalence", "epidemiology", "dominant", "recessive", among others. Review articles and original research conducted on mental retardation, prioritarily those published in the last 12 years, were selected based on relevance by the authors. Some book references and documents from other reliable sources were also included.
Familial Mental Retardation
Familial mental retardation (familial MR) is a term which, in scientific articles, has been applied to families with two or more members affected by MR presumably caused by the same etiological factor(s), irrespective of being mild or serious, having narrow or wider familial implications. Analyzing the prevalence of familial MR cases among the cohort of a day-care institution from Santa Catarina, Brazil, we encountered extreme difficulties in obtaining epidemiological data on familial MR cases to relate our findings, except for the so-called "familial-cultural MR"30 and for X-linked MR35,36. Expanding the survey to three other institutions for daily care of seriously mentally handicapped individuals in the Middle West of Santa Catarina, Brazil, revealed that 15-20% of the individuals are cases which may be of familial recurrence (preliminary results).
Familial MR as a broad category is not considered in any more recent epidemiological study, probably because of the absence of practical definition of the term. According to Zigler and Hodapp30 people with MR could be divided into meaningful groups for research and intervention purposes. One is considering the level of functioning, based on the IQ scale, as described before; the other one divides people with MR into two sub-groups, based on etiology. The latter, so-called "two-group approach", was defined by Edward Ziegler in the late sixties and comprised familial cultural MR (with no obvious "organic causes"), and organic MR, whose prevalence was estimated around 25% to 50% of all MR cases30. The familial cultural MR still remains the only academic definition of familial MR that could be found searching in articles, books and public databases. Meanwhile, Ziegler's familial definition as a sole category is obsolete. The group he referred to represents the multifactorial ("cultural familial" or "psychosocial") familial cases, mainly in the upper half of mild MR (IQ range 60-70) and we actually have no idea of the overall prevalence of the "non-cultural" familial MR. Precisely the MR which is caused by one defined factor and follows a predictable inheritance pattern, a knowledge which is a key aspect to plan preventive health measures and to direct public funding, particularly in developing countries, where public resources for more expensive diagnostic procedures are scarce.
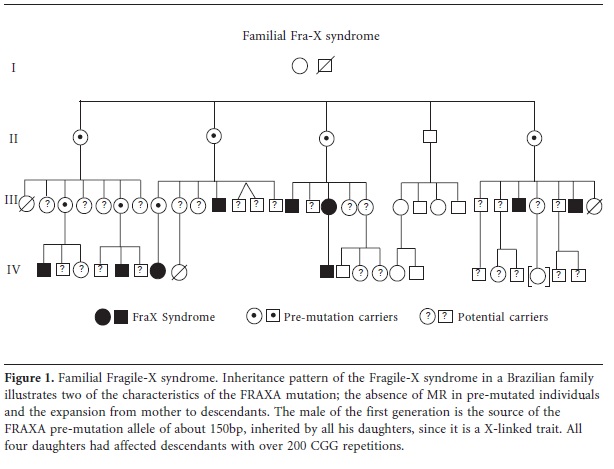
Familial is not equal to hereditary, as it comprises only the cases were familial recurrence is found. This may eventually lead to the identification of rare familial cases where the main etiology is environmental. Then again, not all hereditary cases will result in familial cases. Despite the regular use of the term "familial MR" in articles and publications, there is more to familial MR than usually considered. Irrespective of being mild or severe, familial cases are the ones where more suffering under the burden of MR occurs. Families where the fate of MR strikes again and again do not have an equal-opportunity chance in emotional, social, cultural, and financial aspects. They live with members that will need lifelong care, sometimes an extremely intense one, and often without any outside help. These problems are worse in the developing world when compared to industrialized countries were families have more support of the public system. On the other hand, it is precisely where familial MR hits, that the etiological factor has a better chance of being uncovered. Inheritance patterns allow a better insight in the etiology without the need of a wide array of sophisticated techniques and, when available, the necessary genetic test can be required very specifically, often also with the possibility of genetic diagnostics for other family members. Those are the cases in which public health services with limited resources should at least invest in diagnostic and genetic counseling. These families are aware there is something running in the family, although they do not know exactly what it is and whom it may hit next. In our experience we found that in families like that most pregnancies (often unplanned ones) are stressful events, a stress that may vanish only months after the birth because many non-syndromic MR and some syndromic conditions, like Fragile X Syndrome or Angelman Syndrome, illustrated by pedigrees of Brazilian families on Figures 1 and 2, only show identifiable signs several months after birth and the affected family already is aware of this. It is not uncommon for members of these families to stay childless or to adopt children instead of having their own, as can be seen in this family with Fragile-X Syndrome, which had no previous diagnostic for the condition and therefore no way to foresee who was at risk (Figure 1).
Focused diagnostics will help with management aspects and/or relieve parental and extended familial anguish, especially when clarifying inheritance risks/safety. For sporadic cases, prevention is more difficult and, except for more common conditions, the identification of a causative factor is rare and found more often by research than by regular diagnostics. For the benefit of the most easily preventable cases, i.e. the familial ones, a focus on the family must always exist when assessing MR cases, be it for diagnostic purposes or for epidemiological surveys. For this, it is essential to gather as much information as possible on prevalence (of the overall familial cases in a survey study and of the specific conditions within each family), gender and when possible, detailed familial relationship of the affected persons and the often pleiotropic (varying) presentation of these cases within the family. In the clinic, a no less than three generation pedigree is essential; for epidemiologic studies the degree to which familial matters can be asserted depends on the size of the population, but should at least include the information if someone else in the family has a similar condition, the number of affected, if among siblings, if parent-child, or others. Publications from the late nineteen fifties to seventies concentrated very much on registering familial associations; however, in the era of molecular genetics those more "traditional" aspects seem not considered worth publishing, outshined by the molecular aspects. Revising literature, when cohort studies indeed list cases as familial, information regarding the familial relationship of the affected individuals is rarely investigated (or reported) in detail. This makes comparisons and information gathering for familial MR epidemiology extremely difficult.
Underscoring the importance of familial cases, J-P Fryns, who conducted and participated in extensive surveys within institutionalized populations, most of them with serious MR, reveals that, in the idiopathic MR group (exclusion of known genetic and acquired etiology) of those surveys, approximately 50% represented a familial form of MR; about 80% of them having at least one affected sibling and nearly 20% of them having one or both parents with MR (J-P Fryns, personal communication). Large epidemiological studies on the prevalence of mental retardation2,19,21,25-28,37 hardly mention familial cases at all and when, then never depict familial relationship at any degree.
Considering the visible mode of transmission of MR in families, it seems worth to classify familial mental retardation into at least three broad categories for practical use: multifactorial familial mental retardation (multifactorial intellectual disability), sibling mental retardation (sibling intellectual disability), and hereditary familial mental retardation (hereditary familial intellectual disability). They differ considerably not only in their social and familial significance, but also in the approach to etiological conclusions, for genetic counseling and possible prevention. Designed as simple categories, they are very straightforward, require no previous genetic or causal knowledge and allow easy application to population-wide prevalence studies being of help in the general clinic, especially for non-geneticists. If cohort and larger epidemiological studies apply the suggested sub-classification when collecting and reporting information about the prevalence of MR, they will produce substantial genetically relevant information on the subject, expanding the possibilities for the development of adequate public health measures and for estimates on the epidemiology of MR within families of defined populations.
A description of the suggested (broad) categorization of familial cases of MR for practical purposes, mentioning some of the genetic aspects of each class and the pattern of inheritance which can be observed, is given bellow.
Multifactorial familiar mental retardation
In earlier work also denominated cultural-familial MR, socio-cultural MR, psychosocial MR, or just familial MR, it presents no clear-cut inheritance pattern, is found with increased frequency in families with low or borderline IQ and low socio-economic background30 and is believed to present the major cause of MR in developing countries accounting for 75-95% of all MR cases. Individuals within this IQ range (borderline to upper part of the IQ range of mild MR) are rarely identified before school age, and about 50% of them may attend regular schools without ever being identified as MR cases2,38. Although experiencing much more difficulty in reaching the required intellectual standards and obtaining relatively low grades at school, they are not the only ones with low achievement because several individuals with normal-range IQ get similar results38.
The etiology of this type of familial MR is polygenic and multifactorial, caused by the combined inheritance of several gene-allele variants with sub-optimal roles in the development of intelligence (probably of gene loci whose products are involved in structural brain development and maintenance or with importance for neuronal plasticity), added to unfavorable economic conditions and intellectually non-stimulating social and cultural environment. Possibly the contribution of genetic factors could be circumvented by early stimulation, good health care and a positive educational background to enhance adaptive skills and eventually increase the IQ score in several cases. However, post-birth intervention therapies have little effect on pre-birth events, in particular the lack of adequate food supply during pregnancy or maternal alcohol consumption. Overall, this is the type of MR which is most strongly impacted by amelioration of the socio-economic conditions of a population and most developed countries have eliminated the key factors on the road toward that form of mild MR (poor nutrition, lack of formal education, low incomes), and now are more concerned about the prevention of "organic" (mild and serious) MR cases38. In the late 90s, the World Health Organization estimated that that the contribution of genetic and environmental factors to MR is 15 and 36%, respectively, in developing countries, and 47 and 17% in developed countries39, highlighting well the effect of environmental improvements.
Familial mental retardation in siblings or just sibling mental retardation
It may seem an odd category (why not simply autosomal recessive MR?), but was felt to be needed for practical purposes, to be applied to familial cases where the recurrence of MR is only found among the offspring of one particular couple of an extended family, with no other registered cases. Previous work suggests that up to 25% of the cases referred to developmental pediatricians or child neurologists are such cases40. Although most of them will be caused by autosomal recessive gene defects41, the term "sibling MR" instead of "autosomal recessive MR", was found more adequate because there are other situations which cause recurrent cases in only one couple of a family, like germline mosaicism or environmental factors, like systematic alcohol or other teratogenic/genotoxic exposure by the parents. As the etiological factor often cannot be determined, "sibling MR" is an appropriate category to be used in epidemiological surveys, yielding genetically useful information.
Appearing as recurrent cases usually affecting only children of one couple in a family, sibling MR hits limited but hard. The risk of recurrence for the couple concerned is 25% when the cause is recessive inheritance and may be even higher for germline mutations/germline mosaicism or fetal alcohol exposure, as described below. The consequences often are moderate to profound MR of unknown etiology.
Autosomal recessive MR: In most of the American and West European world, autosomal recessive MR, despite being hereditary, is not classically familial they do not "run in the family". When no consanguinity among the parents is present, other members of the family normally have nothing to worry about, unless for more common known conditions. Exemplified by the inborn errors of the metabolism, several of them causing serious MR, recessive disorders may be the underlying cause of up to 30% of sporadic undiagnosed cases of mental retardation, congenital anomalies, and dysmorphism41. The incidence of autosomal recessive MR may vary widely among populations. There are highly consanguineous populations in the world, mainly in North and sub-Saharan Africa as well as West, Central and South Asia, where 20% to over 50% of marriages are consanguineous42. The countries with the highest consanguinity rates include Saudi Arabia, Iraq, Pakistan and Nigeria, with 50% up to 88% (rural population of Pakistan) of consanguineous marriages25,43,44. In those regions, recurrent autosomal recessive MR occurs within large consanguineous kindreds, actually "running in the family", in contrast to the usual concept where recessive conditions only affect the offspring of one union, offering the possibility to trace the gene trough inheritance. Up to 2006 only three genes for autosomal recessive MR had been identified. In the last years, thanks to coordinated efforts and collaboration of large consanguineous families from Iran and elsewhere, the number of identified autosomal recessive genes implicated in MR raised to more than 7045.
Germline mosaicism: A rare but not neglectable cause of Sibling MR may be due to the presence of dominant mutations (monogenic or a chromosome abnormality) in the germline of one parent46. Germline mutations or germline mosaicism is often the only explanation for the recurrence of a highly detrimental penetrant dominant gene mutation or chromosomal abnormality (which usually is found only as de novo mutation) among siblings of unaffected parents with apparently normal karyotype. Sometimes, the examination of a wider sample of lymphocytes or buccal mucosa cells can reveal that one parent presents mosaicism not only in the germline. The risk of recurrence in cases with mosaicism is difficult to establish, as it depends on the type of mutation, the degree of the germline mosaicism (which can be very different of the degree of mosaicism found in blood or other tissues47, as well as on the fecundation competence of the gametes carrying the mutation and the developmental capacity of the mutated zygote (a history of difficulty in getting pregnant and miscarriages may be common)46.
Fetal alcohol syndrome (FAS): Albeit missing statistics, it probably is a non-neglectable cause of sibling MR. FAS is considered the leading cause of non-genetic (environmental) MR in the Western world, affecting at least 1 per 1000 newborns, probably more48. In Brazil, there is no statistic in this regard, but it is one of the leading alcohol consuming populations, and therefore children with FAS are quite common in the clinical practice. A preventable cause of MR, FAS represents the upper extreme of a large spectrum of effects caused by fetal alcohol exposure. The hallmarks of FAS are primarily central nervous system damage, with cognitive impairment and a medium IQ of 60 to 65; characteristic facial dysmorphologies and growth deficiency49. The risk of conceiving a child with FAS depends on the amount, frequency and timing of prenatal alcohol exposure, however there are no safe consumption limits and the risk varies widely among women. Other factors influencing alcohol metabolism, including the genetic background of the mother and of the embryo/fetus do influence the outcome of fetal alcohol exposure50, illustrated by the fact that monozygotic twins are more likely to suffer the same effects than fraternal twins51, and by the fact that heavy drinkers often have children without FAS. Because of this, the risk of having another child with FAS is considered very high (much higher than for women with no previous FAS history in the family) for those mothers who continue alcohol consumption during subsequent pregnancies. Recurrence risk for FAS is zero if the mother does not drink alcohol during her pregnancies.
Hereditary familial mental retardation
In contrast to autosomal recessive MR, which outside consanguineous unions only affects siblings of one couple of a kindred, the term "hereditary familial MR" should be applied when the problem affects several members of different generations of a non consanguineous family when it is noticed that MR "runs in the family" and is serious. It differs from the multifactorial (cultural-) familial type, because it is mainly caused by one single definable genetic defect (a monogenic or a contiguous gene disorder) that follows a specific, though sometimes intricate, hereditary pattern. This type of familiar MR is especially apparent when MR is serious, but can also be found in mild MR cases22,52. Undoubtedly, the most heterogeneous class of familial MR, the type of gene defects that give rise to hereditary familial MR is diverse and appears to be caused predominantly by recessive X-linked disorders, some chromosomal abnormalities - mainly rearrangements (obvious or micro-rearrangements, often deletions, duplications or subtelomeric translocations), disorders of epigenetic nature (like imprinting defects or defects in imprinted genes), unstable mutations (like trinucleotide repeat disorders), and autosomal dominant gene defects with reduced penetrance or considerable phenotypic expression variability. While "running in the family" and afflicting several generations, this MR phenotype generally does not segregate as a classic Mendelian trait, and the inheritance pattern may be somewhat unusual for clinicians not familiar with genetics. However, when 3 - 4 generations of an extended family are analyzed, a hereditary pattern usually can be identified. Because of their peculiar segregation, some of the main causes for hereditary familial MR are represented below and may be of help for non-geneticist clinicians.
Recessive X-linked MR (XLMR): Representing at least 10% of all MR cases, most of the hereditary familial cases are attributed to recessive X-linked disorders36, which affect more males than females and follow an easily recognizable hereditary pattern. The high frequency of recessive XLMR is explained by the tendency of such mutations passing unnoticed through females to manifest themselves in hemizygosis in males, in addition to the exceptionally high density of brain-expressed genes on the X chromosome36,52. Transmitted through unaffected females, they cause MR in nearly 50% of their sons. About 50% of the daughters of one carrier mother are carriers themselves, a third of them symptomatic (more often with milder symptoms) because of varying X-inactivation patterns, which often are skewed when MR mutations are present53. Therefore, monozygotic female twin sisters carrying the same mutation on the X-chromosome may present discordant phenotypes, a striking feature of this inheritance. The trait is never transmitted by a male to his sons, but an affected or permutated carrier male will transmit the X-linked trait to all of his daughters, most of them being asymptomatic carriers, as well illustrated in the pedigree of our Fragile-X family (Figure 1). The typical pedigree shows affected maternal uncles (brothers of the mother) of an affected and usually male - individual. The main gene affected in X-linked MR (15-20% of all cases) is the FMR1 gene54, resulting in a fragile site for the X-chromosome when the cells are grown in folate poor media (FRAXA)55. The remaining carry mutations in one of more than 91 clinically relevant XLMR genes34,36,56. When suspecting X-linked MR, karyotype analysis at a higher than 550 band resolution with fragile site investigation and mutation analysis for the typical trinucleotide (CGG) expansion (FRAXA) is essential52. For more detailed information, excellent recent reviews are available on X-linked MR22,52,54,56. Copy number variants on the X chromosome, as accessed by array-CGH, present up to 10% of X-linked MR34.
Autosomal chromosomal abnormalities: When one of the parents is the carrier of a balanced chromosomal translocation or inversion (no part of the genome missing, only misplaced), the carrier usually shows no sign of a phenotypic abnormality, but may have a higher probability of having affected children inheriting unbalanced combinations (missing chromosomes or chromosome parts). They usually are serious syndromic MR cases. For instance, about 5% of Down syndrome cases are caused by a non-balanced translocation of chromosome 21 to another acrosomic chromosome, like chromosomes 14 (most cases), 13, 15, 22, or even another chromosome 2157. The gametes produced by carriers of a balanced translocation are diverse and so is their probability to originate a viable gestation, which depends not only of the chromosomal defect, but also of the gender of the individual which originates the gamete. For instance, the female carrier of a translocation of chromosomes 14 and 21 has the empirical probability of 10% to generate a live-born child with Down syndrome, while for the male carrier this chance is below 1%58. Translocation carriers often have more problems getting pregnant and experience a higher rate of miscarriages57. Although most of the children born to balanced translocation carriers are normal, a number of them will themselves be carriers of the balanced translocation.
The pedigree of a translocation or other abnormal chromosome inheritance is unlikely to show a clear pattern. It may be suspected when families show an uncommon clustering of serious or syndromic MR in comparison with the general population, a higher than normal rate of miscarriages, no comprehensible inheritance pattern and no history of consanguineous unions. Regular karyotyping will reveal the chromosomal abnormality unless it is on a micro scale, where only more refined techniques, like fluorescence in situ hybridization, multiplex ligation dependent probe amplification or molecular array karyotyping can reveal the abnormality. Subtelomeric rearrangements were found in approximately 3 4% of unexplained, not X-linked familial mental retardation and present important familial cases59,60.
Dominant autosomal MR: Potentially hereditary genetic mutations may be caused by monogenic or contiguous gene disorders (CGD). CGD are micro chromosomal abnormalities, like micro duplications or deletions of less than 5-10 Mb in size, involving a few closely linked genes whose inheritance pattern may mimic a monogenic disorder.
Serious MR dominant mutations (monogenic or contiguous gene disorders) as a rule occur as isolated cases caused by new mutations and not as familial cases, since the affected person is unlikely to have descendants. Still, serious dominant mutations may be transmitted generating hereditary familial diseases when they show reduced penetrance (not all carriers having symptoms) and/or when they exhibit variable expression (from mild to severe), depending on interacting effects with environmental or other genetic factors. Well-known examples of familial dominant MR occur with neurofibromatosis type 1 (incidence of about 1/4000) or the tuberous sclerosis complex (incidence of 1/6000), which in some cases have a very mild phenotypic expression and in others cause MR61-63.
The pedigree of a dominant MR-causing mutation shows a dominant inheritance pattern of MR cases of different severity, often skipping a generation. This will have to be considered when MR cases of varying expression cluster in a family with no consanguineous unions and no X-linked segregation pattern.
Defects on imprinted genes or imprinting defects: Another important and possibly still underestimated cause for familial MR may be mutations in imprinted genes. An imprinted gene implies that, for certain autosomal genes, only one of the two alleles which each person inherits from its parents is active, depending on its parental origin. The other allele is silenced by mechanisms not yet entirely clarified in a pattern that is gender-specific and involves differential histone and DNA methylation, as well as histone desacetylation64. Depending on the gene, it may be imprinted on all cells of the body or only on specific cell lines, but possibly developmental stage-specific imprinting may exist as well. The expression of an imprintable gene will be hemizygous, relying on only one allele and the MR is usually transmitted by an unaffected parent. The inheritance pattern of imprinted genes is peculiar and non-geneticist clinicians are rarely familiar with it.
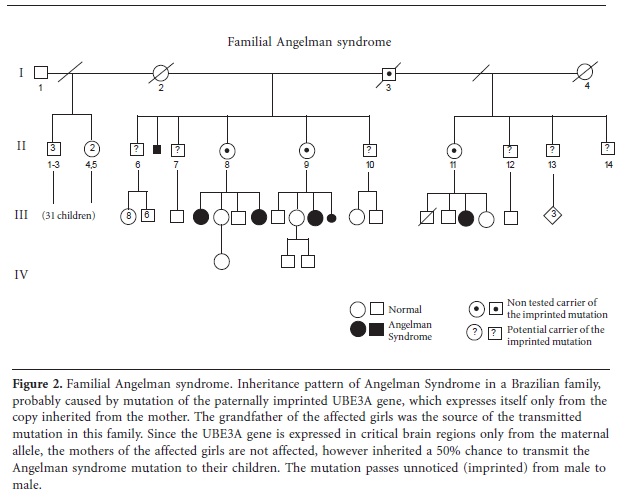
The only well-known cases of MR caused by imprinted genes are the Prader-Willy and Angelman syndromes. Usually, those syndromes are caused by deletions of a specific region of chromosome 15, which are sporadic events, but familial cases can occur when a gene-defect occurs in the genetic portion responsible for establishing the imprinting pattern. Familial Angelman syndrome also may occur due to a maternally inherited mutation within the coding region of the critical imprinted gene, the UBE3A gene65. Thus, mutations which impair maternal UBE3A expression cause MR, while the same mutation - when inherited from the father - does not cause symptoms. As a result, the mutation for Angelman syndrome may be transmitted through several generations from male to male without ever becoming apparent. However, daughters of these males have a 50% chance of being unaffected carriers and if they are they have a 50% risk of having children with Angelman syndrome. One example of the familial transmission of a defect in an imprinted gene (UBE3A mutation) can be seen in the pedigree of familial Angelman syndrome from Brazil (Figure 2).
Trinucleotide repeat expansion disorders: They correspond to a group of unstable mutations, which often cause difficulties in interpreting pedigrees, affecting recessive X-linked genes, as is the case with the fragile X-syndrome, or autosomal dominant genes like the Steinert myotonic dystrophy gene.
Trinucleotide repeats exist within translated or non-translated regions of certain genes and in some of them have the ability to amplify66. They were found to be responsible for a considerable amount of human genetic diseases most of them affecting neurological systems. Depending on the gene, a certain amount of amplified repetitions is well tolerated, but after a specific threshold they impair or alter the expression or the function of the encoded gene-product. The effect may be a threshold effect alone (function or non-function after a defined number of repetitions) or may also show a proportional effect in which the symptoms increase in severity as the number of trinucleotide repeats increases.
The most common form of hereditary intellectual disability is the Fragile-X syndrome67, whose molecular cause was found to be the expansion of an unstable, non-coding CGG repeat sequence in the 5' untranslated region (UTR) of the FMR1gene which is hypermethylated in affected individuals68, resulting in the total absence of expression of the FMR1 gene. The FMR1 alleles can be divided into four categories according to the number of CGG repeats: normal (6-24 repeats), intermediate or gray zone (25-54), premutation (55-199) and fully mutated (200 to over 1000 repeats) which causes MR69. The full mutation shows a typical threshold effect, since more than 200 repeats of the CGG triplet result in the complete absence of FMR1 expression. The inheritance risk depends on the alleles which a given person carries68. Alleles are stable in the normal zone (not amplifying when transmitted), may be unstable in the gray zone and are extremely unstable in the premutation range where, in one single generation, they can expand to the full mutation through a still not fully understood mechanism. This amplification occurs almost exclusively when transmitted from the mother, causing MR in her children, in particular the males (since the trait is X-linked)69. The premutated father, as a rule, transmits the premutation only to his daughters, with no further expansion, but turning all of them into carriers of the permutation with a high chance of having fully mutated sons11, which is well illustrated in the Fragile X syndrome pedigree of the Brazilian family, figure 1. Since from the grey zone on, the trinucleotide repeats are unstable they also may diminish.
Steinert myotonic dystrophy - type 1 (DM1), is another example of a trinucleotide repeat expansion disorder often accompanied by MR in the congenital form (over 1000 repeats)70,71. The disease is caused by the expansion of an unstable CTG repeat located in the 3' untranslated region of the dystrophia myotonica protein kinase (DMPK) gene on the long arm of chromosome 1972. It is transmitted as a dominant autosomal trait whose expression is highly variable, depending on the number of CTG repeats and on whether it was inherited from the mother or father73.
The CTG repeats in normal people are between 5 and 34. Analogous to the fragile X syndrome, there is also an unstable zone (premutation) of between 35 and 49 repeats within which the trinucleotide repeat can expand to full mutation in consecutive generations73. Affected people carry 50 to over 2000 CTG repeats. For Steinert myotonic dystrophy, the age of onset of the symptoms as well as the severity of the disease is affected by the size of the mutated trinucleotide expansion.
Conclusion
When intellectual disability is familial, the impact is quite different from isolated cases. Although isolated cases do not exclude a recessive mode of inheritance (in fact they often reflect unrecognized recessive gene defects) the occurrence of more than one case in a family makes it almost certain that something is wrong in the genes. In most countries throughout the world, gene array analysis or other sophisticated and highly technologic approach to every idiopathic MR case is not a viable option. However, when cases cluster in one family and the phenotypic spectrum can be assessed in more than one individual along with the observation of the mode of transmission, etiological conclusions may be drawn without the need of extensive often unaffordable molecular surveys. As mentioned before, the benefit of diagnosing familial cases is immense, as these families doubtless are in deep need of genetic counseling. However little data exists about the epidemiological relevance of familial MR. The above suggested categorization in at least three broad classes is not meant to yield error-free results, but when applied will help the gathering of data which can subside future studies in the topic.
The present paper is meant to draw attention to familial transmission which can be found in more than 15% of the moderately to severely intellectually disabled population in Brazilian institutions (personal data, from our surveys unpublished) and up to 50% of idiopathic MR (J. P. Fryns, personal communication). In that sense, epidemiologists and researchers are urged to explore and report the familial cases, classifying their study subjects into the suggested categories (multifactorial, sibling or hereditary familial MR) when illustrated family transmission (pedigrees) are not possible or adequate.
Contributors
AF Maris conceived the article, conducted the data analysis, and wrote the article. A Trott contributed to the research, data analysis, and elaboration of the article. IT Barbato and MAE Montano collaborated in the article's revision and critical analysis.
Acknowledgements
This research was supported by a research project grants from Fundação de Apoio a Pesquisa Científica e Tecnológica do Estado de Santa Catarina (FAPESC)/Ministério da Saúde/SES-SC/CNPq. The authors also thank Barbara Uecker who kindly revised the English manuscript.
Artigo apresentado em 25/10/2011
Aprovado em 20/01/2012
Versão final apresentada em 04/02/2012
- 1. Biasini FJ, Grupe L, Huffman L, Bray NW. Mental retardation: a symptom and a syndrome, in Comprehensive Textbook of Child and Adolescent Disorders. New York: Oxford University Press; 1999. p. 6-23.
- 2. Leonard H, Wen X. The epidemiology of mental retardation: challenges and opportunities in the new millennium. Ment Retard Dev Disabil Res Rev 2002; 8(3):117-134.
- 3. Gungor S, Yalnizoglu D, Turanli G, Saatci I, Erdogan-Bakar E, Topcu M. Malformations of cortical development: clinical spectrum in a series of 101 patients and review of the literature (Part I). Turk J Pediatr 2007; 49(2):120-130.
- 4. Kaufmann WE, Moser HW. Dendritic anomalies in disorders associated with mental retardation. Cereb Cortex 2000; 10(10):981-991.
- 5. Boda B, Alberi S, Nikonenko I, Node-Langlois R, Jourdain P, Moosmayer M, Parisi-Jourdain L, Muller D. The mental retardation protein PAK3 contributes to synapse formation and plasticity in hippocampus. J Neurosci 2004; 24(48):10816-10825.
- 6. Gray RG, Preece MA, Green SH, Whitehouse W, Winer J, Green A. Inborn errors of metabolism as a cause of neurological disease in adults: an approach to investigation. J Neurol Neurosurg Psychiatry 2000; 69(1):5-12.
- 7. van Karnebeek CD, Stockler S. Treatable inborn errors of metabolism causing intellectual disability: A systematic literature review. Mol Genet Metab 2011; 105(3):368-381.
- 8. Hayashi ML, Rao BS, Seo JS, Choi HS, Dolan BM, Choi SY, Chattarji S, Tonegawa S. Inhibition of p21-activated kinase rescues symptoms of fragile X syndrome in mice. Proc Natl Acad Sci USA 2007; 104(27):11489-11494.
- 9. Dolen G, Bear MF. Courting a cure for fragile X. Neuron 2005; 45(5):642-644.
- 10. Lauterborn JC, Rex CS, Kramar E, Chen LY, Pandyarajan V, Lynch G, Gall CM. Brain-derived neurotrophic factor rescues synaptic plasticity in a mouse model of fragile X syndrome. J Neurosci 2007; 27(40):10685-10694.
- 11. Penagarikano O, Mulle JG, Warren ST. The pathophysiology of fragile x syndrome. Annu Rev Genomics Hum Genet 2007; 8:109-129.
- 12. Yan QJ, Rammal M, Tranfaglia M, Bauchwitz RP. Suppression of two major Fragile X Syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology 2005; 49(7):1053-1066.
- 13. Battaglia A, Bianchini E, Carey JC. Diagnostic yield of the comprehensive assessment of developmental delay/mental retardation in an institute of child neuropsychiatry. Am J Med Genet 1999; 82(1):60-66.
- 14. Rauch A, Hoyer J, Guth S, Zweier C, Kraus C, Becker C, Zenker M, Hüffmeier U, Thiel C, Rüschendorf F, Nürnberg P, Reis A, Trautmann U. Diagnostic yield of various genetic approaches in patients with unexplained developmental delay or mental retardation. Am J Med Genet A 2006; 140(19):2063-2074.
- 15. Pfundt R, Veltman JA. Structural genomic variation in intellectual disability. Methods Mol Biol 2012; 838:77-95.
- 16. Girirajan S, Brkanac Z, Coe BP, Baker C, Vives L, Vu TH, Shafer N, Bernier R, Ferrero GB, Silengo M, Warren ST, Moreno CS, Fichera M, Romano C, Raskind WH, Eichler EE. Relative burden of large CNVs on a range of neurodevelopmental phenotypes. PLoS Genet 2011; 7(11):e1002334.
- 17. Brady PD, Devriendt K, Deprest J, Vermeesch JR. Array-based approaches in prenatal diagnosis. Methods Mol Biol 2012; 838:151-171.
- 18. Boyle CA, Yeargin-Allsopp M, Doernberg NS, Holmgreen P, Murphy CC, Schendel DE. Prevalence of selected developmental disabilities in children 3-10 years of age: the Metropolitan Atlanta Developmental Disabilities Surveillance Program, 1991. MMWR CDC Surveill Summ 1996; 45(2):1-14.
- 19. Durkin MS, Khan NZ, Davidson LL, Huq S, Munir S, Rasul E, Zaman SS. Prenatal and postnatal risk factors for mental retardation among children in Bangladesh. Am J Epidemiol 2000; 152(11):1024-1033.
- 20. Roeleveld N, Zielhuis GA, Gabreels F. The prevalence of mental retardation: a critical review of recent literature. Dev Med Child Neurol 1997; 39(2): 125-132.
- 21. Stein Z, Durkin M, Belmont L. "Serious" mental retardation in developing countries: an epidemiologic approach. Ann N Y Acad Sci 1986; 477:8-21.
- 22. Ropers HH, Hamel BC. X-linked mental retardation. Nat Rev Genet 2005; 6(1):46-57.
- 23. Maulik PK, Mascarenhas MN, Mathers CD, Dua T, Saxena S. Prevalence of intellectual disability: a meta-analysis of population-based studies. Res Dev Disabil 2011; 32(2):419-436.
-
24Pan American Health Organization (PAHO). Mental Health. Main Disorders, in World Health Day. Washington: WHO; 2001.
- 25. Durkin MS, Hasan ZM, Hasan KZ. Prevalence and correlates of mental retardation among children in Karachi, Pakistan. Am J Epidemiol 1998; 147(3):281-288.
- 26. Croen LA, Grether JK, Selvin S. The epidemiology of mental retardation of unknown cause. Pediatrics 2001; 107(6):E86.
- 27. Heikura U, Linna SL, Olsen P, Hartikainen AL, Taanila A, Jarvelin MR. Etiological survey on intellectual disability in the northern Finland birth cohort 1986. Am J Ment Retard 2005; 110(3):171-180.
- 28. Stromme P. Aetiology in severe and mild mental retardation: a population-based study of Norwegian children. Dev Med Child Neurol 2000; 42(2):76-86.
- 29. Zlotogora J. Genetic disorders among Palestinian Arabs: 1. Effects of consanguinity. Am J Med Genet 1997; 68(4):472-475.
- 30. Zigler E, Hodapp RM. Behavioral functioning in individuals with mental retardation. Annu Rev Psychol 1991; 42:29-50.
- 31. King BH, Toth KE, Hodapp RM, Dykens EM. Intellectual disability. In: Sadock BJ, Sadock VA, Ruiz P. Comprehensive textbook of psychiatry. Philadelphia: Lippincott Williams & Wilkins; 2009. p. 3444-3474.
- 32. Moser HW. Genetic causes of mental retardation. Ann N Y Acad Sci 2004; 1038:44-48.
- 33. Inlow JK, Restifo LL. Molecular and comparative genetics of mental retardation. Genetics 2004; 166: 835-881.
- 34. Ropers HH. Genetics of early onset cognitive impairment. Annu Rev Genomics Hum Genet 2010; 11: 161-187.
- 35. Mandel JL, Chelly J. Monogenic X-linked mental retardation: is it as frequent as currently estimated? The paradox of the ARX (Aristaless X) mutations. Eur J Hum Genet 2004; 12(9):689-693.
- 36. Ropers HH. X-linked mental retardation: many genes for a complex disorder. Curr Opin Genet Dev 2006; 16(3):260-269.
-
37Instituto Brasileiro de Geografia e Estatística (IBGE). Censo 2000 Características Gerais da População. Rio de Janeiro: IBGE; 2003.
- 38. Stein Z, Susser M. Mutability of Intelligence and Epidemiology of Mild Mental Retardation. Rev Educ Res 1970; 40(1):29-67.
- 39. World Health Organization (WHO). Primary prevention of mental, neurological and psychosocial disorder. Geneva: WHO; 1998.
- 40. Higgins JJ, Pucilowska J, Lombardi RQ, Rooney JP. Candidate genes for recessive non-syndromic mental retardation on chromosome 3p (MRT2A). Clin Genet 2004; 65(6):496-500.
- 41. Hamamy HA, Masri AT, Al-Hadidy AM, Ajlouni KM. Consanguinity and genetic disorders. Profile from Jordan. Saudi Med J 2007; 28(7):1015-1017.
- 42. Yunis K, Rafei RE, Mumtaz G. International Perspectives: Consanguinity: Perinatal Outcomes and Prevention - A View from the Middle East. Neoreviews 2008; 9(2):59-65.
- 43. Ropers H-H. Molekulare Ursachen genetish bedingter kognitiver Störungen, in Tätigkeitsbericht. Berlin: Max Plank Gesellschaft; 2004.
- 44. Mokhtari R, Bagga A. Consanguinity, genetic disorders and malformations in the Iranian population. Acta Biol Szeged 2003; 47(1-4):47-50.
- 45. Najmabadi H, Hu H, Garshasbi M, Zemojtel T, Abedini SS, Chen W, Hosseini M, Behjati F, Haas S, Jamali P, Zecha A, Mohseni M, Püttmann L, Vahid LN, Jensen C, Moheb LA, Bienek M, Larti F, Mueller I, Weissmann R, Darvish H, Wrogemann K, Hadavi V, Lipkowitz B, Esmaeeli-Nieh S, Wieczorek D, Kariminejad R, Firouzabadi SG, Cohen M, Fattahi Z, Rost I, Mojahedi F, Hertzberg C, Dehghan A, Rajab A, Banavandi MJ, Hoffer J, Falah M, Musante L, Kalscheuer V, Ullmann R, Kuss AW, Tzschach A, Kahrizi K, Ropers HH. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature 2011; 478(7367):57-63.
- 46. Zlotogora J. Germ line mosaicism. Hum Genet 1998; 102(4):381-386.
- 47. Koolen DA, Dupont J, de Leeuw N, Vissers LE, van den Heuvel SP, Bradbury A, Steer J, de Brouwer AP, Ten Kate LP, Nillesen WM, de Vries BB, Parker MJ. Two families with sibling recurrence of the 17q21.31 microdeletion syndrome due to low-grade mosaicism. Eur J Hum Genet 2012; 20(7):729-733.
- 48. May PA, Fiorentino D, Coriale G, Kalberg WO, Hoyme HE, Aragón AS, Buckley D, Stellavato C, Gossage JP, Robinson LK, Jones KL, Manning M, Ceccanti M. Prevalence of children with severe fetal alcohol spectrum disorders in communities near Rome, Italy: new estimated rates are higher than previous estimates. Int J Environ Res Public Health 2011; 8(6):2331-2351.
- 49. Spohr HL, Willms J, Steinhausen HC. Fetal alcohol spectrum disorders in young adulthood. J. Pediatr 2007; 150(2):175179.
- 50. Warren KR, Li T-K. Genetic polymorphisms: Impact on the risk of fetal alcohol spectrum disorders. Birth Defects Res A Clin Mol Teratol 2005; 73(4):195-203.
- 51. Streisguth AP, Dehaene P. Fetal alcohol syndrome in twins of alcoholic mothers: Concordance of diagnosis and IQ. Am J Med Genet 1993; 47(6):857-861.
- 52. Raymond FL. X linked mental retardation: a clinical guide. J Med Genet 2006; 43(3):193-200.
- 53. Plenge RM, Stevenson RA, Lubs HA, Schwartz CE, Willard HF. Skewed X-chromosome inactivation is a common feature of X-linked mental retardation disorders. Am J Hum Genet 2002; 71(1):168-173.
- 54. Kleefstra T, Hamel BC. X-linked mental retardation: further lumping, splitting and emerging phenotypes. Clin Genet 2005; 67(6):451-467.
- 55. Sutherland GR. Heritable fragile sites on human chromosomes I. Factors affecting expression in lymphocyte culture. Am J Hum Genet 1979; 31(2):125-135.
- 56. Chiurazzi P, Schwartz CE, Gecz J, Neri G. XLMR genes: update 2007. Eur J Hum Genet 2008; 16(4): 422-434.
- 57. Sergovich FR, Soltan HC, Carr DH. A 13-15/21 translocation chromosome in carrier father and mongol son. Can Med Assoc J 1962; 87:852-858.
- 58. Gardner RJM, Sutherland GR. Chromosome Abnormalities and Genetic Counseling New York: Oxford University Press; 2004.
- 59. Bocian E, Helias-Rodzewicz Z, Suchenek K, Obersztyn E, Kutkowska-Kazmierczak A, Stankiewicz P, Kostyk E, Mazurczak T. Subtelomeric rearrangements: results from FISH studies in 84 families with idiopathic mental retardation. Med Sci Monit 2004; 10(4):CR143-151.
- 60. 60. Sogaard M, Tumer Z, Hjalgrim H, Hahnemann J, Friis B, Ledaal P, Pedersen VF, Baekgaard P, Tommerup N, Cingöz S, Duno M, Brondum-Nielsen K. Subtelomeric study of 132 patients with mental retardation reveals 9 chromosomal anomalies and contributes to the delineation of submicroscopic deletions of 1pter, 2qter, 4pter, 5qter and 9qter. BMC Med Genet 2005; 6:21.
- 61. Sanchez-Diaz A, Morales-Peydro C, Madrigal-Bajo I. Autosomal dominant mental retardation. Rev Neurol 2006; 42(Supl.):S33-37.
- 62. Schwartz RA, Fernandez G, Kotulska K, Jozwiak S. Tuberous sclerosis complex: advances in diagnosis, genetics, and management. J Am Acad Dermatol 2007; 57(2):189-202.
- 63. Acosta MT, Gioia GA, Silva AJ. Neurofibromatosis type 1: new insights into neurocognitive issues. Curr Neurol Neurosci Rep 2006; 6(2):136-143.
- 64. Biliya S, Bulla LA. Genomic imprinting: the influence of differential methylation in the two sexes. Exp Biol Med 2010; 235(2):139-147.
- 65. Chamberlain SJ, Lalande M. Neurodevelopmental disorders involving genomic imprinting at human chromosome 15q11-q13. Neurobiol Dis 2010; 39(1): 13-20.
- 66. Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci 2007; 30:575-621.
- 67. Turner G, Webb T, Wake S, Robinson H. Prevalence of fragile X syndrome. Am J Med Genet 1996; 64(1):196-197.
- 68. Sutcliffe JS, Nelson DL, Zhang F, Pieretti M, Caskey CT, Saxe D, Warren ST. DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum Mol Genet 1992; 1(6):397-400.
- 69. Zhong N, Ju W, Pietrofesa J, Wang D, Dobkin C, Brown WT. Fragile X "gray zone" alleles: AGG patterns, expansion risks, and associated haplotypes. Am J Med Genet 1996; 64(2):261-265.
- 70. Ekstrom AB, Hakenas-Plate L, Samuelsson L, Tulinius M, Wentz E. Autism spectrum conditions in myotonic dystrophy type 1: a study on 57 individuals with congenital and childhood forms. Am J Med Genet B Neuropsychiatr Genet 2008; 147B(6):918-926.
- 71. Modoni A, Silvestri G, Vita MG, Quaranta D, Tonali PA, Marra C. Cognitive impairment in myotonic dystrophy type 1 (DM1): a longitudinal follow-up study. J Neurol 2008; 255(11):1737-1742.
- 72. Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Jansen G, Neville C, Narang M, Barceló J, O'Hoy K. Myotonic dystrophy mutation: an unstable CTG repeat in the 3' untranslated region of the gene. Science 1992; 255:1253-1255.
- 73. Martorell L, Monckton DG, Sanchez A, Lopez De Munain A, Baiget M. Frequency and stability of the myotonic dystrophy type 1 premutation. Neurology 2001; 56:328-335.
Publication Dates
-
Publication in this collection
04 June 2013 -
Date of issue
June 2013
History
-
Received
25 Oct 2011 -
Accepted
04 Feb 2012 -
Reviewed
20 Jan 2012