Abstracts
Chromosomal 15q11-q13 region is of great interest in Human Genetics because many structural rearrangements have been described for it (deletions, duplications and translocations) leading to phenotypes resulting in conditions such as the Prader-Willi (PWS) and Angelman (AS) syndromes which were the first human diseases found to be related to the differential expression of parental alleles (genomic imprinting). Contrary to Mendelian laws where the parental inheritance of genetic information does not influence gene expression, genomic imprinting is characterized by DNA modifications that produce different phenotypes depending on the parental origin of the mutation. Clinical manifestation of PWS appears when the loss of paternally expressed genes occurs and AS results from the loss of a maternally expressed gene. Different genetic mechanisms can lead to PWS or AS, such as deletions, uniparental disomy or imprinting mutation. In AS patients an additional class occurs with mutations on the UBE3A gene. Studies of PWS and AS patients can help us to understand the imprinting process, so that other genomic regions with similar characteristics can be located, and different syndromes can have their genetic mechanisms elucidated.
O segmento cromossômico 15q11-q13 é de grande interesse em Genética Humana uma vez que diversos rearranjos estruturais têm sido descritos nessa região (deleções, duplicações e translocações) resultando em fenótipos diferentes como os das síndromes de Prader-Willi (PWS) e Angelman (AS), que foram as primeiras doenças humanas a serem relacionadas com a expressão diferencial de alelos parentais (imprinting genômico). Contrário às leis de Mendel onde a herança parental da informação genética não influencia a expressão gênica, o imprinting genômico é caracterizado por modificações no DNA que produzem diferentes fenótipos dependendo da origem parental da mutação. A manifestação clínica da PWS aparece quando ocorre a perda de genes com expressão exclusivamente paterna, e a AS resulta da perda do gene com expressão exclusivamente materna. Diferentes mecanismos genéticos podem resultar em PWS ou AS como deleções, dissomia uniparental ou mutações no processo de imprinting. Em pacientes com AS existe uma classe adicional de pacientes com mutação no gene UBE3A. Estudos de pacientes com PWS e AS podem ajudar no entendimento do processo de imprinting e, assim, outras regiões do genoma com características similares podem ser localizadas, e diferentes síndromes podem ter seus mecanismos genéticos elucidados.
REVIEW ARTICLE
Genomic imprinting: genetic mechanisms and phenotypic consequences in Prader-Willi and Angelman syndromes
Cintia Fridman and Célia P. Koiffmann
Departamento de Biologia, Instituto de Biociências, USP, Caixa Postal 11.461, 05422-970 São Paulo, SP, Brasil.
Send correspondence to C.F. Fax: +55-11-818-7419. E-mail: cfridman@ib.usp.br
ABSTRACT
Chromosomal 15q11-q13 region is of great interest in Human Genetics because many structural rearrangements have been described for it (deletions, duplications and translocations) leading to phenotypes resulting in conditions such as the Prader-Willi (PWS) and Angelman (AS) syndromes which were the first human diseases found to be related to the differential expression of parental alleles (genomic imprinting). Contrary to Mendelian laws where the parental inheritance of genetic information does not influence gene expression, genomic imprinting is characterized by DNA modifications that produce different phenotypes depending on the parental origin of the mutation. Clinical manifestation of PWS appears when the loss of paternally expressed genes occurs and AS results from the loss of a maternally expressed gene. Different genetic mechanisms can lead to PWS or AS, such as deletions, uniparental disomy or imprinting mutation. In AS patients an additional class occurs with mutations on the UBE3A gene. Studies of PWS and AS patients can help us to understand the imprinting process, so that other genomic regions with similar characteristics can be located, and different syndromes can have their genetic mechanisms elucidated.
INTRODUCTION
Genomic imprinting is defined as the differential marking of parental alleles in gametogenesis, resulting in the differential expression of maternal and paternal genes during development. Imprinting is an epigenetic process and requires three main steps, i.e., gamete DNA marking, maintenance of the marking in embryo and adult somatic tissues and resetting of marking at the beginning of gametogenesis (Hall, 1990).
Since imprinting seems to be a functional inactivation process and inactive chromatin regions have been associated with hypermethylation of CpG islands, DNA methylation has been suggested as candidate to the main mechanism responsible for the marking and differentiation of individual parental alleles (Peterson and Sapienza, 1993; Razin and Cedar, 1994; Glenn et al., 1996, 1997; Brannan and Bartolomei, 1999), although it is not known if DNA methylation is involved in the maintenance or in the establishment of imprinting.
The Prader-Willi (PWS) and Angelman (AS) syndromes are neurobehavioral disorders with completely different phenotypes and were the first human diseases found to be related to genomic imprinting. These syndromes are caused by the loss of function of imprinted genes in the 15q11-q13 region, and differential methylation patterns between normal, PWS and AS individuals can be detected in this region (Driscoll et al., 1992; Clayton-Smith et al., 1993; Dittrich et al., 1993; Reis et al., 1994). This is the first evidence of an epigenetic event and suggests that the DNA methylation may have an important function in the genomic imprinting mechanism (Nicholls, 1993a).
Besides the methylation difference found in the 15q11-q13 region, asynchronous DNA replication also occurs in imprinted gene clusters (Izumikawa et al., 1991; Knoll et al., 1994), with non-imprinted alleles replicating earlier than imprinted alleles. It is known that condensed inactive chromatin initiates a heterochromatic state and that, in general, heterochromatin replicates later, and genes that replicate later are not able to compete for transcription factors with earlier replicating euchromatin. However, it is not known whether this asynchronous replication phenomenon is related to regulation of imprinted gene expression or if it is just a consequence of imprinting (Glenn et al., 1997).
CLINICAL FEATURES
Prader-Willi syndrome
PWS was first described in 1956 (Prader et al., 1956) and today is one of the most common microdeletion syndromes and the most frequent form of genetically caused obesity in humans (Cassidy, 1997).
The syndrome is characterized by neonatal hypotonia, hypothermia, hypogenitalism, feeding problems during infancy and some facial anomalies (Cassidy, 1984; Butler et al., 1986; Butler, 1990). When hypotonia improves, appetite improves and weight gain increases. Obesity starts to occur between 1 and 6 years of age (Butler, 1990; Holm et al., 1993) and marks the second phase of the syndrome, characterized by neuropsycomotor delay, hyperphagia with obesity, short stature, short hands and feet and mental retardation with learning disabilities (Figure 1A) (q.v. Fridman, 1997). Some individuals present hypopigmentation, high pain threshold (Ledbetter et al., 1981; Butler, 1990), obsessive-compulsive disorder, depression, violent episodes and inappropriate social behavior (Holm et al., 1993; Cassidy, 1997). Einfeld et al. (1999) found that anti-social and psychopathological behavior was more frequent in PWS individuals than in a control group of mentally deficient patients. PWS patients show a higher degree of irritability, as well as stubbornness, a tendency to be argumentative, possessive, and persevering, and have a tendency to steal and to lie.
Although many clinical manifestations of PWS seem to be the result of hypothalamic deficit, no structural defect in the brain have been found, so any such deficit must be functional, but its exact nature remains unknown (Cassidy, 1997).
Genetically, PWS is caused by the absence of paternal alleles that are normally active in the 15q11-q13 region, with the maternal alleles normally being inactive due to the genomic imprinting. Paternal deletion of the 15q11-q13 region (70% of cases) or maternal uniparental disomy (UPD) (20-25% of cases) can lead to the absence of paternal alleles (Robinson et al., 1991; Mascari et al., 1992). Translocations or other structural anomalies of chromosome 15 are found in about 5% of cases, resulting in paternal deletion or UPD15 (Rivera et al., 1990; Smeets et al., 1992; Qumsiyeh et al., 1992; Donnai, 1993; Freeman et al., 1993; Reeve et al., 1993; Smith et al., 1993, 1994; Vickers et al., 1994). About 1-5% of cases, including families, have imprinting center (IC) mutations (Reis et al., 1994; Sutcliffe et al., 1994; Buiting et al., 1995; Dittrich et al., 1996; Saitoh et al., 1996, 1997). The absence of patients with point mutations in a single gene suggests that PWS is a contiguous gene syndrome, caused by the loss of two or more paternally expressed genes (Nicholls, 1993b, 1994; Ohta et al., 1996).
Clinical comparisons between deleted and UPD patients have not revealed significant differences, although hypopigmentation occurs in patients with deletion and increased maternal age is a factor in UPD cases (Butler, 1989; Robinson et al., 1991; Gillessen-Kaesbach et al., 1995) due to the known correlation between maternal age and nondisjunction events (Robinson et al., 1991, 1993). The non-imprinted P gene responsible for oculocutaneous albinism (OCA2) maps on the distal part of the 15q11-q13 and is deleted in the majority of PWS cases with deletion (Spritz et al., 1997).
Angelman syndrome
AS is characterized by developmental delay, severe mental retardation, speech impairment, seizures, ataxia, hyperactivity, macrostomia and an apparently happy demeanor with outburst of laughter (Figure 1B) (Angelman, 1965; Robb et al., 1989; Fryburg et al., 1991; Clayton-Smith and Pembrey, 1992; Williams et al., 1995; Fridman, 1997). The behavioral features are very important for diagnosis in young children because the facial anomalies are not so prominent and the phenotype shows wide variability.
The genetic mechanisms responsible for this syndrome are complex and about 2/3 of the patients show a 4-Mb maternal deletion in the 15q11-q13 region (Magenis et al., 1987; Knoll et al., 1989), which is the same segment deleted in PWS cases (Magenis et al., 1990). Paternal UPD is present in 2-3% of cases (Nicholls, 1993a), a small group of patients show IC mutations (Sutcliffe et al., 1994; Buiting et al., 1995; Saitoh et al., 1996), and about 8% of cases present mutations in the UBE3A gene (Kishino et al., 1997; Matsuura et al., 1997).
It has been pointed out that AS patients with UPD have a milder phenotype compared to patients with deletions (Bottani et al., 1994; Gillessen-Kaesbach et al., 1995; Fridman et al., 2000), possibly because in UPD cases only the AS gene is lost while the more severe phenotype seen in patients with deletion could be caused by the loss of both the AS gene and other contiguous genes. These differences in AS severity could also be explained by recessive mutations in hemizigosity (Bottani et al., 1994). UPD cases can also manifest characteristics caused by the homozygosity state of some loci.
Summary of genetic mechanisms
It therefore appears that there are 3 main classes of PWS patients, i.e., paternal deletion, maternal UPD and IC mutations and 4 classes of AS patients, i.e., maternal deletion, paternal UPD, IC mutations and UBE3A mutations. There are also about 5% of PWS and 15% of AS patients with no detectable cytogenetic or molecular abnormalities for whom the genetic mechanism of the condition is unknown, making genetic counseling very difficult.
THE 15q11-q13 REGION
The 15q11-q13 region is about 5 Mb and many genes and transcripts have been mapped to this region, which can be divided into 3 subregions comprising a distal portion containing non-imprinted genes, a central portion containing the only maternally expressed gene (UBE3A-E6), and a proximal portion containing paternally expressed genes (Figure 2).
Some paternally expressed genes and trancripts mapping at the PWS/AS region are SNRPN, ZNF127, NDN, IPW, PAR1 and PAR5 (Özcelik et al., 1992; Glenn et al., 1993; Reed and Leff, 1994; Sutcliffe et al., 1994; MacDonald and Wevrick, 1997), as well as the maternally expressed transcript UBE3A which is expressed exclusively in brain tissue (Sutcliffe et al., 1997; Kishino and Wagstaff, 1998). The imprinting of these genes seems to be under the control of the IC, since familial and sporadic cases of PWS and AS with microdeletions in this region are consistent with a mutation in the imprinting process (Reis et al., 1994; Sutcliffe et al., 1994; Buiting et al., 1995; Saitoh et al., 1996).
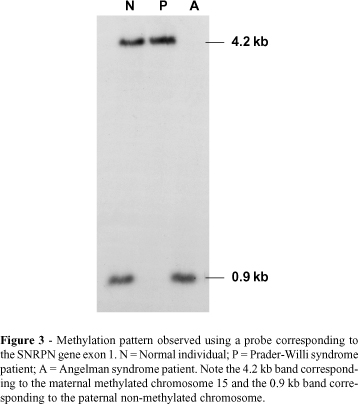
The small nuclear ribonucleoprotein polypeptide N (SNRPN) gene is imprinted both in mice (Cattanach et al., 1992) and humans (Glenn et al., 1993; Nakao et al., 1994; Reed and Leff, 1994), and is expressed only by the paternal chromosome. In humans this gene is mapped in the shortest region of overlap (SRO) of deletion in PWS (Figure 2) (Özçelik et al., 1992) and was the first candidate gene involved in the etiology of PWS (Glenn et al., 1996). This gene has 10 exons extending over 30 kb of genomic DNA, with exon 1 and the promoter being located in a CpG island (Sutcliffe et al., 1994; Glenn et al., 1996) and showing a differential methylation pattern between the paternal and maternal alleles in all somatic tissues (Glenn et al., 1996, 1997). SNRPN methylation analysis is the best test for the diagnosis of PWS and AS, identifying patients with deletion, UPD and imprinting mutation (Figure 3) (Glenn et al., 1993; Sutcliffe et al., 1994; ASHG/ACMG, 1996; Kubota et al., 1996; Saitoh et al., 1996). The gene also encodes a protein (SmN) involved in pre-mRNA splicing, but its exact role is still unknown (Özçelik et al., 1992; Glenn et al., 1996).
Sequence analysis and evolutionary studies of the upstream exons (exons 1, 2 and 3) of the SNRPN gene have indicated that there might also exist another open reading frame (ORF) in the SNRPN mRNA (Glenn et al., 1996, 1997) in addition to the ORF containing exons 4 to 10 previously described by Schmauss et al. (1992). This second ORF, confirmed by cDNA analysis, was named SNURF ("SNRPN upstream reading frame") and corresponds to the first 3 SNRPN exons (Gray et al., 1999). The finding that two independent proteins (SmN and SNURF) are encoded by a single mRNA renamed it as the bicistronic SNURF-SNRPN locus and suggested that the SNURF cistron may participate in the imprint regulation of the 15q11-q13 region, since it is mapped in the IC (Gray et al., 1999).
Another gene within the PWS/AS region, denominated NECDIN (NDN) was identified (Jay et al., 1997; MacDonald and Wevrick, 1997) and is orthologous to mouse Necdin (Ndn). This gene is transcribed only by the paternal allele in fibroblast, amniocytes, and brain and placenta tissues, indicating that NDN is imprinted in these tissues (Jay et al., 1997; MacDonald and Wevrick, 1997). The NDN gene encodes a nuclear protein (NECDIN) expressed in all central nervous system post-mitotic neurons (Watrin et al., 1997). In agreement with the observation that imprinted genes have few and small introns (Hurst et al., 1996), the human NDN is formed by only one exon (MacDonald and Wevrick, 1997) and its cDNA has 1,592 kb. Watrin et al. (1997) have also reported that the NDN gene is imprinted, paternally expressed with the maternal allele methylated, and replicates asynchronously. The loss of expression of this gene could lead to anomalies, especially in brain development (Jay et al., 1997; MacDonald and Wevrick, 1997), and contributes to the PWS phenotype.
It has been suggested that the UBE3A mutation is the main cause of AS (Kishino et al., 1997; Matsuura et al., 1997; Malzac et al., 1998; Fang et al., 1999). This gene maps within the critical region for AS and has 16 exons extending over 120 kb of genomic DNA, with transcription being oriented from telomere to centromere (Burke et al., 1996; Kishino and Wagstaff, 1998). How mutations in the maternally inherited UBE3A gene cause AS and whether mutations in this gene can account for all AS patients in the biparental class remain to be determined (Glenn et al., 1997).
Many types of mRNA (isoforms) produced by the UBE3A gene through alternative splicing of pre-mRNA have been detected (Rougeulle et al., 1997; Yamamoto et al., 1997; Kishino and Wagstaff, 1998). Recent studies in mice and humans (Albrecht et al., 1997; Rougeulle et al., 1997) have shown imprinting and maternal-only expression of this gene in specific brain regions, especially the hippocampus and cerebellum, and it is the first gene with maternal-only expression mapped within the PWS/AS region, supporting the idea that it is a candidate gene for AS (Rougeulle et al., 1997).
The UBE3A gene encodes an E3 ubiquitin-protein ligase (E6AP) that acts in the ubiquitination process (Huibregtse et al., 1995; Yamamoto et al., 1997), the two main substrates of UBE3A being p53 and HHRAD23A, but it is not known if they are the target substrates in AS (Malzac et al., 1998).
Rearrangements within the 15q11-q13 segment such as deletions, marker chromosomes, duplications, triplications and translocations have been detected. Knoll et al. (1990) classified the AS patients with deletion into two groups according to the breakpoints located in the proximal region of 15q11-q13 (the terminal breakpoint is common to the two groups). Later it was also observed in PWS patients with deletion (Christian et al., 1995). These breakpoints (BP1, BP2 and BP3) (Figure 2) were characterized by Christian et al. (1999) as DNA regions containing pseudogenes, called duplicons. Studies using YACs have revealed the presence of 2 in tandem oriented duplicon copies at BP3 and 1 copy at BP2.
Buiting et al. (1992, 1998a) isolated a gene family (MN7) presenting many copies in the 15q11-q13 and 16p11.2 regions. The presence of many of these copies in the proximal 15q region may be related to the instability of this region. Amos-Landgraf et al. (1999) have identified new duplicons mapped at BP1, BP2 and BP3 that include MN7 sequences and represent large duplications of a new gene mapped within the 15q11-q13 region, the HERC2 (Ji et al., 1999).
All these data suggest that duplication of large DNA segments within the pericentromeric region may create the instability that is responsible for the high level of chromosomal rearrangements due to the unequal crossing over event.
THE IMPRINTING CENTER
Biparental inheritance disclosed by microsatellite analysis and uniparental methylation pattern can identify PWS and AS patients with imprinting mutations (Reis et al., 1994; Buiting et al., 1995; Dittrich et al., 1996; Glenn et al., 1993, 1997; Saitoh et al., 1996, 1997). Half of the known cases are familial and, like the sporadic cases, show the classical PWS or AS phenotype (Reis et al., 1994; Buiting et al., 1995; Dittrich et al., 1996; Saitoh et al., 1996, 1997). Most of these patients inherited microdeletions in the 5' region of SNRPN gene, indicating that this region is responsible for the imprinting process. This region, the IC (Buiting et al., 1995; Dittrich et al., 1996; Saitoh et al., 1996, 1997), maps between the loci D15S63 (Dittrich et al., 1992) and SNRPN loci (Figure 2) (Glenn et al., 1996), and is responsible for the differential allele marking, spanning its signal bidirectionally over a segment of about 2 Mb where the group of imprinted genes is located.
This IC region has been intensively studied and some transcripts are now known, including alternative SNRPN exons, mapped on the 5' end of this gene (Dittrich et al., 1996; Färber et al., 1999). Mutations in these alternative exons occur in PWS and AS patients with imprinting mutation (Dittrich et al., 1996; Färber et al., 1999). RT-PCR analysis has shown that these transcripts can use two alternative start sites, are subject to alternative splicing and use SNRPN exons at their 3' end (Dittrich et al., 1996). Färber et al. (1999) have found that the presence of more than one start site can assure IC transcript expression even if deletions occur at one of these sites.
The analysis of informative families has allowed the delineation of the IC microdeletion map, and has shown that deletions in AS patients are centromeric while deletions in PWS are more telomeric. In this way, the SRO of deletions in AS has been established and corresponds to IC exon 3 (IC3), spanning 2 kb (Saitoh et al., 1996; Ohta et al., 1999a). This seems to be the element responsible for the paternal to maternal imprinting switch (pat®mat) in the female gametogenesis. In PWS, the SRO is defined as the SNRPN exon 1 and the promoter region, corresponding to 4 kb of genomic DNA. This region is responsible for the maternal to paternal (mat®pat) imprinting switch during spermatogenesis (Figure 2) (Buiting et al., 1995; Ohta et al., 1999b). The IC seems to be a bipartite structure and the gene transcription in patients with IC mutation is altered due to a failure in the imprinting erasure and reestablishment (Reis et al., 1994; Buiting et al., 1995; Dittrich et al., 1996; Saitoh et al., 1996).
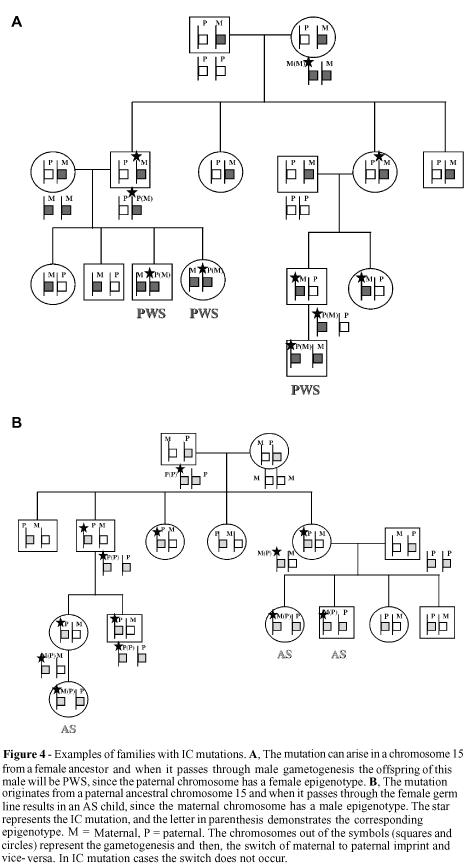
Figure 4 shows how the transmission of IC mutations occurs in males and females, and can be summarized as follow. When a male transmits a maternal imprinting mutation, the maternal epigenotype is not erased and a paternal imprinting during male gametogenesis is not established. This mutation blocks the mat®pat switch, resulting in the transmission of maternal imprinting to half of the gametes. The individual who inherits this abnormal epigenotype will be homozygous for the maternal epigenotype and, therefore, present PWS. On the other hand, the transmission of a paternal mutation by a woman results in failure of the pat®mat switch, leading to the inheritance of an abnormal paternal epigenotype. The woman's offspring will be homozygous for the paternal imprint and, therefore, present AS (Nicholls et al., 1998).
In these PWS and AS cases, the IC mutations have no direct effect on the carrier, are silenced and transmitted through the same sex of the origin of the mutation, with manifestation only occurring when the mutation passes through the gametogenesis process of the opposite sex.
A model of the functioning of IC (Dittrich et al., 1996) and how mutations can result in PWS or AS individuals has been proposed by several authors (Buiting et al., 1998b; Nicholls et al., 1998; Ohta et al., 1999a,b). The bipartite IC structure consists of two elements, the imprintor (IC exon 3) and the switch initiation site (SIS) (SNRPN exon 1). Only in the paternal chromosome does the imprintor encode a transcript that acts in cis form on the SIS, possibly by the introduction of modifications in chromatin structure. This change allows a trans factor, present only in the female germ line, to act and complete the pat®mat switch, spreading the imprint bidirectionally over the 2-Mb region of imprinted genes. This signal inactivates the imprintor activity since it now has the maternal imprint. In the male germ line, with the absence of the trans factor, the SIS erases the maternal imprint and the paternal imprint is established, reactivating the imprintor activity (Dittrich et al., 1996).
In this way, mutations in imprintor (or IC3) lead to a failure in the erasure of the paternal imprint and to the establishment of the maternal imprint, producing an AS child, while mutations in the SIS lead to a failure in the erasure of the maternal imprint and to the establishment of the paternal imprint, resulting in a PWS child.
Both imprinting mutation cases with deletion in the IC (familial and sporadic) and sporadic cases with imprinting mutation and no structural alteration in the IC have been detected (Bürger et al., 1997; Buiting et al., 1998b).
Patients with IC deletion present the typical AS phenotype (Williams et al., 1995), however, some characteristics distinguish these patients from those with 15q11-q13 deletion, including absent crisis or milder seizures, absence of microcephaly, larger neonatal length, better motor control and normal pigmentation (Ohta et al., 1999a). These differences can be explained because the non-imprinted genes, when in a haploinsufficiency state (deletion), can contribute to the complete AS phenotype (Nicholls et al., 1998), while IC mutation or UPD15 patients may present a milder phenotype due to incomplete imprinting of the UBE3A gene in the brain (Albrecht et al., 1997; Rougeulle et al., 1997; Vu and Hoffman, 1997), and then a double dose of paternal expression of this gene occurs.
GENETIC COUNSELING
Genetic counseling for PWS and AS is complex since the parents of deleted or UPD patients have a low recurrence risk (<1%); but when an IC mutation is detected it is necessary to verify whether or not there is a deletion and if it is inherited or sporadic. If the deletion is de novo or there are IC mutations without deletion the risk is low (<1%). The IC deletion group includes familial (up to 50% risk) and sporadic cases, and all non-IC deletion cases are sporadic (Bürger et al., 1997; Saitoh et al., 1997; Buiting et al., 1998b). AS patients with the UBE3A mutation can be sporadic, with a low recurrence risk, or familial with a 50% risk of inheritance from a carrier mother (Stalker and Williams, 1998).
MOUSE MODEL
The central part of mouse chromosome 7 (chr 7C) is homologous with the 15q11-q13 human region, with the genes arranged in the same order but inverted in relation to the centromere (Albrecht et al., 1997; MacDonald and Wevrick, 1997; Wevrick and Francke, 1997).
Some genes with only paternal expression mapped at the PWS/AS region show homologous genes that are also imprinted in the mouse, such as Snrpn (Leff et al., 1992), Ipw (Wevrick and Francke, 1997), Ndn (MacDonald and Wevrick, 1997), and Zfp127 (review in Gabriel et al., 1998). The Ube3a gene, also mapped in the 7C, shows only maternal expression in some brain tissues, as in humans (Albrecht et al., 1997; Rougeulle et al., 1997; Vu and Hoffmann, 1997). So, not only the physical characteristics of 15q11-q13 human segment are conserved in the mouse but also the imprinting features (Gabriel et al., 1998).
Working with balanced chromosome translocations, Cattanach et al. (1992, 1997) obtained paternal and maternal Chr 7C UPD mice. Studying the phenotype of the animals they concluded that the presence of two maternal copies of this region results in the complete absence of the paternally expressed gene and that the mice died between the 2nd and 8th post-natal days because of developmental delay. These mice were considered as a model for PWS. The paternal Chr 7C UPD resulted in mice with developmental retardation, ataxia and hyperactivity - all characteristics present in AS patients, although the mouse phenotypes were milder.
Jiang et al. (1998) showed that the maternal deletion of Ube3a is sufficient to produce animals with motor delay and abnormal electroencephalogram (EEG), a phenotype similar to that of AS children (seizures, hyperactivity and learning disabilities) suggesting that it could be used as a mouse model of AS (Nicholls, 1998).
Some studies have been conducted to better define the role of Snrpn in imprinting regulation. This gene was characterized in mice and is very similar to the SNRPN human gene, containing 10 exons spanning over 22 kb of genomic DNA with the promoter region showing only paternal expression (Cattanach et al., 1992; Leff et al., 1992).
To evaluate the role of SNRPN and IC in PWS, Yang et al. (1998) described mice with either intragenic deletion in the Snrpn region or a large deletion which included a small part of the Snrpn and the distal part of the putative mouse IC. The animals with the intragenic deletion were phenotypically normal, suggesting that SNRPN mutations are not sufficient to cause PWS. The mice with deletion in the putative IC died a few days after birth, were small and showed developmental delay with weight loss due to feeding difficulties. These animals also presented loss of expression of other paternally expressed genes such as Zfp127 (homologous to ZNF127), Ndn and Ipw, showing that the IC position and its role in imprinting regulation are conserved between humans and mice (Yang et al., 1998).
Gabriel et al. (1999) described a mouse lineage with deletion of the homologous PWS/AS region and demonstrated that the paternal deletion transmission resulted in loss of paternally imprinted gene expression while the maternal deletion transmission resulted in loss of the Ube3a expression, a situation similar to that of patients who have a 4-Mb deletion in the 15q11-q13 region. A similar phenotype including developmental delay and neonatal death was obtained in the three PWS mouse models (Cattanach et al., 1992; Yang et al., 1998; Gabriel et al., 1999).
The development of mouse models for Prader-Willi and Angelman syndromes using knockout techniques is very important, allowing investigations of gene function and the future development of therapeutic strategies for individuals with these syndromes.
ACKNOWLEDGMENTS
Publication supported by FAPESP.
RESUMO
O segmento cromossômico 15q11-q13 é de grande interesse em Genética Humana uma vez que diversos rearranjos estruturais têm sido descritos nessa região (deleções, duplicações e translocações) resultando em fenótipos diferentes como os das síndromes de Prader-Willi (PWS) e Angelman (AS), que foram as primeiras doenças humanas a serem relacionadas com a expressão diferencial de alelos parentais (imprinting genômico). Contrário às leis de Mendel onde a herança parental da informação genética não influencia a expressão gênica, o imprinting genômico é caracterizado por modificações no DNA que produzem diferentes fenótipos dependendo da origem parental da mutação. A manifestação clínica da PWS aparece quando ocorre a perda de genes com expressão exclusivamente paterna, e a AS resulta da perda do gene com expressão exclusivamente materna. Diferentes mecanismos genéticos podem resultar em PWS ou AS como deleções, dissomia uniparental ou mutações no processo de imprinting. Em pacientes com AS existe uma classe adicional de pacientes com mutação no gene UBE3A. Estudos de pacientes com PWS e AS podem ajudar no entendimento do processo de imprinting e, assim, outras regiões do genoma com características similares podem ser localizadas, e diferentes síndromes podem ter seus mecanismos genéticos elucidados.
REFERENCES
(Received June 15, 2000)
- Albrecht, U., Sutcliffe, J.S., Cattanach, B.M., Beechey, C.V., Armstrong, D., Eichele, G. and Beaudet,A.L. (1997). Imprinted expression of the murine Angelman syndrome gene, Ube3a, in hippocampal and Purkinje neurons. Nat. Genet. 17: 75-78.
- Amos-Landgraf, J.M., Ji, Y., Gottlieb, W., Depinet, T., Wandstrat, A.E., Cassidy, S.B., Driscoll, D.J., Rogan, P.K., Schwartz, S. and Nicholls, R.D. (1999). Chromosome breakage in the Prader-Willi and Angelman syndromes involves recombination between large, transcribed repeats at proximal and distal breakpoints. Am. J. Hum. Genet. 65: 370-386.
- Angelman, H. (1965). "Puppet" children: a report on three cases. Develop. Med. Child. Neurol. 7: 681-688.
- ASHG/ACMG (1996). Report diagnostic testing for Prader-Willi and Angelman syndromes: report of the ASHG/ACMG test and technology transfer committee. Am. J. Hum. Genet. 58: 1085-1088.
- Bottani, A., Robinson, W.P., DeLozier-Blanchet, C.D., Engel, E., Morris, M.A., Schmitt, B., Thun-Hohenstein, L. and Schinzel, A. (1994). Angelman syndrome due to paternal uniparental disomy of chromosome 15: A milder phenotype? Am. J. Med. Genet. 51: 35-40.
- Brannan, C.I. and Bartolomei, M.S. (1999). Mechanisms of genomic imprinting. Curr. Opin. Genet. Dev. 9: 164-170.
- Buiting, K., Greger, V., Brownstein, B.H., Mohr, R.M., Voiculescu, I., Winterpacht, A., Zabel, B. and Horsthemke, B. (1992). A putative gene family in 15q11-13 and 16p11.2: possible implications for Prader-Willi and Angelman syndromes. Proc. Natl. Acad. Sci. USA 89: 5457-5461.
- Buiting, K., Saitoh, S., Gross, S., Dittrich, B., Schwartz, S., Nicholls, R.D. and Horsthemke, B. (1995). Inherited microdeletions in the Angelman and Prader-Willi syndromes define an imprinting center on human chromosome 15. Nat. Genet. 9: 395-400.
- Buiting, K., Grob, S., Ji, Y., Senger, G., Nicholls, R.D. and Horsthemke, B. (1998a). Expressed copies of the MN7 (D15F37) gene family map close to the common deletion breakpoints in the Prader-Willi/Angelman syndromes. Cytogenetic. Cell Genet. 81: 247-253.
- Buiting, K., Dittrich, B., Grob, S., Lich, C., Färber, C., Buchholz, T., Smith, E., Reis, A., Bürger, J., Nöthen, M.M., Barth-Witte, U., Janssen, B., Abeliovich, D., Lerer, I., van den Ouweland, A.M.W., Halley, D.J.J., Schrander-Stumpel, C., Smeets, H., Meinecke, P., Malcolm, S., Gardner, A., Lalande, M., Nicholls, R.D., Friend, K., Schulze, A., Matthijs, G., Kokkonem, H., Hilbert, P., Maldergem, L.V., Glover, G., Carbonell, P., Willems, P., Gillessen-Kaesbach, G. and Horsthemke, B. (1998b). Sporadic imprinting defects in Prader-Willi syndrome and Angelman syndrome: implications for imprint-switch models, genetic counselling, and prenatal diagnosis. Am. J. Hum. Genet. 63: 170-180.
- Bürger, J., Buiting, K., Dittrich, B., Grob, S., Lich, C., Sperling, K., Horsthemke, B. and Reis, A. (1997). Different mechanisms and recurrence risks of imprinting defects in Angelman syndrome. Am. J. Hum. Genet. 61: 88-93.
- Burke, L.W., Wiley, J.E., Glenn, C.C., Driscoll, D.J., Loud, K.M., Smith, A.J.W. and Kushnik, T. (1996). Familial cryptic translocation resulting in Angelman syndrome: implications for imprinting or location of the Angelman gene? Am. J. Hum. Genet. 58: 777-784.
- Butler, M.G. (1989). Hypopigmentation: a common feature of Prader-Labhart-Willi syndrome. Am. J. Hum. Genet. 45: 140-146.
- Butler, M.G. (1990). Prader-Willi syndrome: current understanding of cause and diagnosis. Am. J. Hum. Genet. 35: 319-332.
- Butler, M.G., Meaney, F.J. and Palmer, C.G. (1986). Clinical and cytogenetic survey of 39 individuals with Prader-Labhart-Willi syndrome. Am. J. Med. Genet. 23: 793-809.
- Cassidy, S.B. (1984). Prader-Willi syndrome. Curr. Probl. Pediatr. 14: 1-55.
- Cassidy, S.B. (1997). Prader-Willi syndrome. J. Med. Genet. 34: 917-923.
- Cattanach, B.M., Barr, J.A., Evans, E.P., Burtenshaw, M., Beechey, C.V., Leff, S.E., Brannan, C.I., Copeland, N.G., Jenkins, N.A. and Jones, J. (1992). A candidate mouse model for Prader-Willi syndrome which shows an absence of Snrpn expression. Nat. Genet. 2: 270-274.
- Cattanach, B.M., Barr, J.A., Beechey, C.V., Martin, J., Noebels, J. and Jones, J. (1997). A candidate model for Angelman syndrome in the mouse. Mamm. Genome 8: 472-478.
- Christian, S.L., Robinson, W.P., Huang, B., Mutirangura, A., Line, M.R., Nakao, M., Surti, U., Chakravarti, A. and Ledbetter, D.H. (1995). Molecular characterization of two proximal deletion breakpoint regions in both Prader-Willi and Angelman syndrome patients. Am. J. Hum. Genet. 57: 40-48.
- Christian, S.L., Fantes, J.A., Mewborn, S.K., Huang, B. and Ledbetter, D.H. (1999). Large genomic duplicons map to sites of instability in the Prader-Willi/Angelman syndrome chromosome region (15q11-q13). Hum. Mol. Genet. 8: 1025-1037.
- Clayton-Smith, J. and Pembrey, M.E. (1992). Angelman syndrome. J. Med. Genet. 29: 412-415.
- Clayton-Smith, J., Driscoll, D.J., Waters, M.F., Webb, T., Andrews, T., Malcolm, S., Pembrey, M.E. and Nicholls, R.D. (1993). Difference in methylation patterns within the D15S9 region of chromosome 15q11-13 in first cousins with Angelman syndrome and Prader-Willi syndrome. Am. J. Med. Genet. 47: 683-686.
- Dittrich, B., Robinson, W.P., Knoblauch, H., Buiting, K., Schmidt, K., Gillessen-Kaesbach, G. and Horsthemke, B. (1992). Molecular diagnosis of Prader-Willi and Angelman syndromes by detection of parent-of-origin specific DNA methylation in 15q11-13. Hum. Genet. 90: 313-315.
- Dittrich, B., Buiting, K., Grob, S. and Horsthemke, B. (1993). Characterization of a methylation imprint in the Prader-Willi syndrome region. Hum. Mol. Genet. 2: 1995-1999.
- Dittrich, B., Buiting, K., Korn, B., Richard, S., Buxton, J., Saitoh, S., Nicholls, R.D., Poustra, A., Winterpacht, A., Zabel, B. and Horsthemke, B. (1996). Imprinting switching on human chromosome 15 may involve alternative transcripts of the SNRPN gene. Nat. Genet. 14: 163-170.
- Donnai, D. (1993). Robertsonian translocations: clues to imprinting. Am. J. Med. Genet. 46: 681-682.
- Driscoll, D.J., Waters, M.F., Williams, C.A., Zori, R.T., Glenn, C.C., Avidano, K.M. and Nicholls, R.D. (1992). A DNA methylation imprinting, determined by the sex of the parent, distinguishes the Angelman and Prader-Willi syndromes. Genomics 13: 917-924.
- Einfeld, S.L., Smith, A., Durvasula, S., Florio, T. and Tonge, B.J. (1999). Behavior and emotional disturbance in Prader-Willi syndrome. Am. J. Med. Genet. 82: 123-127.
- Fang, P., Lev-Lehman, E., Tsai, T.-F., Matsuura, T., Benton, C.S., Sutcliffe, J.S., Christian, S.L., Kubota, T., Halley, D.J., Meijers-Heijboer, H., Langlois, S., Graham Jr., J.M., Beuten, J., Willems, P.J., Ledbetter, D.H. and Beaudet, A.L. (1999). The spectrum of mutations in UBE3A causing Angelman syndrome. Hum. Mol. Genet. 8: 129-135.
- Färber, C., Dittrich, B., Buiting, K. and Horsthemke, B. (1999). The chromosome 15 imprinting centre (IC) region has undergone multiple duplication events and contains an upstream exon of SNRPN that is deleted in all Angelman syndrome patients with an IC microdeletion. Hum. Mol. Genet. 8: 337-343.
- Freeman, S.B., May, K.M., Pettay, D., Fernhoff, P.M. and Hassold, T.J. (1993). Paternal uniparental disomy in a child with a balanced 15,15 translocation and Angelman syndrome. Am. J. Med. Genet. 45: 625-630.
- Fridman, C. (1997). Síndrome de Prader-Willi e Síndrome de Angelman: Imprinting Genômico na Espécie Humana. Sociedade Brasileira de Genética, Série Monografias No.5, 1-55.
- Fridman, C., Varela, M.C., Kok, F., Diament, A. and Koiffmann, C.P. (2000). Paternal UPD15: further genetic and clinical studies in four Angelman syndrome patients. Am. J. Med. Genet. 92: 322-327.
- Fryburg, J.S., Breg, W.R. and Lindgren, V. (1991). Diagnosis of Angelman syndrome in infants. Am. J. Med. Genet. 38: 58-64.
- Gabriel, J.M., Gray, T.A., Stubbs, L., Saitoh, S., Ohta, T. and Nicholls, R.D. (1998). Structure and function correlations at the imprinted mouse Snrpn locus. Mamm. Genome. 9: 788-793.
- Gabriel, J.M., Merchant, M., Ohta, T., Ji, Y., Caldwell, R.G., Ramsey, M.J., Tucker, J.D., Longnecker, R. and Nicholls, R.D. (1999). A transgene insertion creating a heritable chromosome deletion mouse model of Prader-Willi and Angelman syndrome. Proc. Natl. Acad. Sci. USA 96: 9258-9263.
- Gillessen-Kaesbach, G., Albrecht, B., Passarge, E. and Horsthemke, B. (1995). Further patient with Angelman syndrome due to paternal disomy of chromosome 15 and a milder phenotype. Am. J. Med. Genet. 56: 328-329.
- Glenn, C.C., Porter, K.A., Jong, M.T.C., Nicholls, R.D. and Driscoll, D.J. (1993). Functional imprinting and epigenetic modification of the human SNRPN gene. Hum. Mol. Genet. 2: 2001-2005.
- Glenn, C.C., Saitoh, S., Jong, M.T.C., Filbrandt, M.M., Surti, U., Driscoll, D.J. and Nicholls, R.D. (1996). Gene structure, DNA methylation, and imprinted expression of the human SNRPN gene. Am. J. Hum. Genet. 58: 335-346.
- Glenn, C.C., Driscoll, D.J., Yang, T.P. and Nicholls, R.D. (1997). Genomic imprinting: potencial function and mechanisms revealed by the Prader-Willi and Angelman syndromes. Mol. Hum. Reprod. 3: 321-332.
- Gray, T.A., Saitoh, S. and Nicholls, R.D. (1999). An imprinted, mammalian bicistronic transcript encodes two independent proteins. Proc. Natl. Acad. Sci. USA 96: 5616-5621.
- Hall, J.G. (1990). Genomic imprinting: review and relevance to human diseases. Am. J. Hum. Genet. 46: 857-873.
- Holm, V.A., Cassidy, S.B., Butler, M.G., Hanchett, J.M., Greenswag, L.R., Whitman, B.Y. and Greenberg, F. (1993). Prader-Willi syndrome: consensus diagnostic criteria. Pediatrics 91: 398-402.
- Huibregtse, J.M., Scheffner, M., Beaudenon, S. and Howley, P.M. (1995). A family of proteins structurally and functionally related to the E6-AP ubiquitin-protein ligase. Proc. Natl. Acad. Sci. USA 92: 2563-2567.
- Hurst, L.D., McVean, G. and Moore, T. (1996). Imprinted genes have few and small introns. Nat. Genet. 12: 234-237.
- Izumikawa, Y., Natitomi, K. and Hirayama, K. (1991). Replication asynchrony between homologs 15q11.2: cytogenetic evidence for genomic imprinting. Hum. Genet. 87: 1-5.
- Jay, P., Rougeulle, C., Massacrier, A., Moncla, A., Mattei, M.-G., Malzac, P., Roëckel, N., Taviaux, S., Lefranc, J.-L.B., Cau, P., Berta, P., Lalande, M. and Muscatelli, F. (1997). The human necdin gene, NDN, is maternally imprinted and located in the Prader-Willi syndrome chromosomal region. Nat. Genet. 17: 357-361.
- Ji, Y., Walkowicz, M.J., Buiting, K., Johnson, D.K., Tarvin, R.E., Rinchik, E.M., Horsthemke, B., Stubbs, L. and Nicholls, R.D. (1999). The ancestral gene for transcribed, low-copy repeats in the Prader-Willi/Angelman region encodes a large protein implicated in protein trafficking, which is deficient in mice with neuromuscular and spermiogenic abnormalities. Hum. Mol. Genet. 8: 533-542.
- Jiang, Y.-h., Armstrong, D., Albrecht, U., Atkins, C.M., Noebels, J.L., Eichele, G., Sweatt, J.D. and Beaudet, A.L. (1998). Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron 21: 799-811.
- Kishino, T. and Wagstaff, J. (1998). Genomic organization of the UBE3A/E6-AP gene and related pseudogenes. Genomics 47: 101-107.
- Kishino, T., Lalande, M. and Wagstaff, J. (1997). UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet. 15: 70-73.
- Knoll, J.H.M., Nicholls, R.D., Magenis, R.E., Graham Jr., J.M., Lalande, M. and Latt, S.A. (1989). Angelman and Prader-Willi syndromes shares a common chromosome 15 deletion but differ in parental origin of the deletion. Am. J. Med. Genet. 32: 285-290.
- Knoll, J.H.M., Nicholls, R.D., Magenis, R.E., Glatt, K., Graham Jr., J.M., Kaplan, L. and Lalande, M. (1990). Angelman syndrome: Three molecular classes identified with chromosome 15q11-q13 specific DNA markers. Hum. Genet. 47: 149-155.
- Knoll, J.H.M., Cheng, S.D. and Lalande, M. (1994). Allele specificity of DNA replication timing in the Angelman/Prader-Willi syndrome imprinted chromosomal region. Nat. Genet. 6: 41-46.
- Kubota, T., Sutcliffe, J.S., Aradhya, S., Gillessen-Kaesbach, G., Christian, S.L., Horsthemke, B., Beaudet, a.l. and Ledbetter, D.H. (1996). Validation studies of SNRPN methylation as a diagnostic test for Prader-Willi syndrome. Am. J. Med. Genet. 66: 77-80.
- Ledbetter, D.H., Riccardi, V.M., Airhart, S.D., Strobel, R.J., Keenan, B.S. and Crawford, J.D. (1981). Deletions of chromosome 15 as a cause of the Prader-Willi syndrome. N. Engl. J. Med. 304: 325-329.
- Leff, S.E., Brannan, C.I., Reed, M.L., Özçelik, T., Francke, U., Copeland, N.G. and Jenkins, N.A. (1992). Maternal imprinting of the mouse Snrpn gene and conserved linkage homology with the human Prader-Willi syndrome region. Nat. Genet. 2: 259-264.
- MacDonald, H.R. and Wevrick, R. (1997). The necdin gene is deleted in Prader-Willi syndrome and is imprinted in human and mouse. Hum. Mol. Genet. 6: 1873-1878.
- Magenis, R.E., Brown, M.G., Lacy, D.A., Budden, S. and LaFranchi, S. (1987). Is Angelman syndrome an alternative result of del (15)(q11q13)? Am. J. Med. Genet. 28: 829-838.
- Magenis, R.E., Toth-Fejel, S., Allen, L.J., Black, M., Brown, M.G., Budden, S., Cohen, R., Friedman, J.M., Kalousek, D., Zonana, J., Lacy, D., LaFranchi, S., Lahr, M., Macfarlane, J. and Williams, C.P.S. (1990). Comparison of the 15q deletions in Prader-Willi and Angelman syndromes: specific regions, extent of deletions, parental origin, and clinical consequences. Am. J. Med. Genet. 35: 333-349.
- Malzac, P., Webber, H., Moncla, A., Graham Jr., J.M., Kukolich, M., Williams, C., Pagon, R.A., Ramsdell, L.A., Kishino, T. and Wagstaff, J. (1998). Mutation analysis of UBE3A in Angelman syndrome patients. Am. J. Hum. Genet. 62: 1353-1360.
- Mascari, M.J., Gottlieb, W., Rogan, P.K., Butler, M.G., Waller, D.A., Armour, J.A.L., Jeffreys, A.J., Ladda, R.L. and Nicholls, R.D. (1992). The frequency of uniparental disomy in Prader-Willi syndrome. N. Engl. J. Med. 326: 1599-1607.
- Matsuura, T., Sutcliffe, J.S., Fang, P., Galjaard, R.-J., Jiang, Y.-h., Benton, C.S., Rommens, J.M. and Beaudet, A.L. (1997). De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat. Genet. 15: 74-77.
- Nakao, M., Sutcliffe, J.S., Durtschi, B., Mutirangura, A., Ledbetter, D.H. and Beaudet, A.L. (1994). Imprinting analysis of three genes in the Prader-Willi/Angelman region: SNRPN, E6-associated protein, and PAR-2 (D15S225E). Hum. Mol. Genet. 3: 309-315.
- Nicholls, R.D. (1993a). Genomic imprinting and uniparental disomy in Angelman and Prader-Willi syndromes: a review. Am. J. Med. Genet. 46: 16-25.
- Nicholls, R.D. (1993b). Genomic imprinting and candidate genes in the Prader-Willi and Angelman syndromes. Curr. Opin. Genet. Dev. 3: 445-456.
- Nicholls, R.D. (1994). New insights reveal complex mechanisms involved in genomic imprinting. Am. J. Hum. Genet. 54: 733-740.
- Nicholls, R.D. (1998). Strange bedfellows? Protein degradation and neurological dysfunction. Neuron 21: 647-649.
- Nicholls, R.D., Saitoh, S. and Horsthemke, B. (1998). Imprinting in Prader-Willi and Angelman syndromes. TIG 14: 194-200.
- Ohta, T., Rogan, P.K., Saitoh, S., Deng, G., Buiting, K., Horsthemke, B., Driscoll, D.J., Weksberg, R., Ishikawa, T., Butler, M.G., Gabriel, J.M. and Nicholls, R.D. (1996). Minimal definition and function of the imprinting center for Prader-Willi syndrome. Am. J. Hum. Genet. 59 (Suppl.): A283 (Abstract).
- Ohta, T., Buiting, K., Kokkonen, H., McCandless, S., Heeger, S., Leisti, H., Driscoll, D.J., Cassidy, S.B., Horsthemke, B. and Nicholls, R.D. (1999a). Molecular mechanism of Angelman syndrome in two large families involves an imprinting mutation. Am. J. Hum. Genet. 64: 385-396.
- Ohta, T., Gray, T.A., Rogan, P.K., Buiting, K., Gabriel, J.M., Saitoh, S., Muralighar, B., Bilenska, B., Krajewska-Walasek, M., Driscoll, D.J., Horsthemke, B., Butler, M.G. and Nicholls, R.D. (1999b). Imprinting-mutation mechanisms in Prader-Willi syndrome. Am. J. Hum. Genet. 64: 397-413.
- Özçelik, T., Leff, S., Robinson, W., Donlon, T., Lalande, M., Sanjines, E., Schinzel, A. and Francke, U. (1992). Small nuclear ribonucleoprotein polypeptide N (SNRPN), an expressed gene in Prader-Willi syndrome critical region. Nat. Genet. 2: 265-269.
- Peterson, K. and Sapienza, C. (1993). Imprinting the genome: imprinted genes, imprinting genes, and a hypothesis for their interaction. Annu. Rev. Genet. 27: 7-31.
- Prader, A., Labhart, A. and Willi, H. (1956). Ein syndrom von Adipositas, kleinwuchs, kryptochismus und ologophrenie nach myotonieartigem zustand in neugeborenalter. Schweiz. Med. Wochenschr. 86: 1260-1261.
- Qumsiyeh, M.B., Dalton, J.D., Gordon, P.L., Wilroy, R.S. and Tharapel, A.T. (1992). Deletion of chromosome 15pter-q11.2 due to t(Y,15) in a boy with Prader-Willi syndrome. Am. J. Med. Genet. 42: 109-111.
- Razin, A. and Cedar, H. (1994). DNA methylation and genomic imprinting. Cell 77: 473-476.
- Reed, M.L. and Leff, S.E. (1994). Maternal imprinting of human SNRPN, a gene deleted in Prader-Willi. Nat. Genet. 6: 163-167.
- Reeve, A., Norman, A., Sinclair, P., Whittington-Smith, R., Hamey, Y., Donnai, D. and Read, A. (1993). True telomeric translocation in a baby with the Prader-Willi phenotype. Am. J. Med. Genet. 47: 1-6.
- Reis, A., Dittrich, B., Greger, V., Buiting, K., Lalande, M., Gillessen-Kaesbach, G., Anvret, M. and Horsthemke, B. (1994). Imprinting mutations suggested by abnormal DNA methylation patterns in familial Angelman and Prader-Willi syndromes. Am. J. Hum. Genet. 54: 741-747.
- Rivera, H., Zuffardi, O. and Gargantini, L. (1990). Nonreciprocal and jumping translocations of 15q1-qter in Prader-Willi syndrome. Am. J. Med. Genet. 37: 311-317.
- Robb, S.A., Pohl, K.R.E., Baraitser, M., Wilson, J. and Brett, E.M. (1989). The 'happy puppet' syndrome of Angelman: review of the clinical features. Arch. Dis. Child. 64: 83-86.
- Robinson, W.P., Bottani, A., Yagang, X., Balakrishman, J., Binkert, F., Machler, M., Prader, A. and Schinzel, A. (1991). Molecular cytogenetic, and clinical investigations of Prader-Willi syndrome patients. Am. J. Hum. Genet. 49: 1219-1234.
- Robinson, W.P., Bernasconi, F., Mutirangura, A., Ledbetter, D.H., Langlois, S., Malcolm, S., Morris, M.A. and Schinzel, A.A. (1993). Nondisjunction of chromosome 15: origin and recombination. Am. J. Hum. Genet. 53: 740-751.
- Rougeulle, C., Glatt, H. and Lalande, M. (1997). The Angelman syndrome candidate gene, UBE3A/E6-AP, is imprinted in brain. Nat. Genet. 17: 14-15.
- Saitoh, S., Buiting, K., Rogan, P.K., Buxton, J.L., Driscoll, D.J., Arnemann, J., Konig, R., Malcolm, S., Horsthemke, B. and Nicholls, R.D. (1996). Minimal definition of the imprinting center and fixation of a chromosome 15q11-q13 epigenotype by imprinting mutations. Proc. Natl. Acad. Sci. USA 93: 7811-7815.
- Saitoh, S., Buiting, K., Cassidy, S.B., Conroy, J.M., Driscoll, D.J., Gabriel, J.M., Gillessen-Kaesbach, G., Glenn, C.C., Greenswag, L.R., Horsthemke, B., Kondo, I., Kuwajima, K., Niikawa, N., Rogan, P.K., Schwartz, S., Seip, J., Williams, C.A. and Nicholls, R.D. (1997). Clinical spectrum and molecular diagnosis of Angelman and Prader-Willi syndrome imprinting mutation patients. Am. J. Med. Genet. 68:195-206.
- Schmauss, C., Brines, M.L. and Lerner, M.R. (1992). The gene encoding the small nuclear ribonucleoprotein-associated protein N is expressed at high levels in neurons. J. Biol. Chem. 267: 8521-8529.
- Smeets, D.F.C.M., Hamel, B.C.J., Nelen, M.R., Smeets, H.J.M., Bollen, J.H.M., Smits, A.P.T., Ropers, H.-H. and van Oost, B.A. (1992). Prader-Willi syndrome and Angelman syndrome in cousins from a family with translocation between chromosomes 6 and 15. N. Engl. J. Med. 326: 807-811.
- Smith, A., Robson, L., Neumann, A., Mulcahy, M., Chabros, V., Deng, Z.-M., Woodage, T. and Trent, R.J. (1993). Fluorescence in situ hybridisation and molecular studies used in the characterisation of a Robertsonian translocation (13q15q) in Prader-Willi syndrome. Clin. Genet. 43: 5-8.
- Smith, A., Deng, Z.M., Beran, R., Woodage, T. and Trent, R.J. (1994). Familial unbalanced translocation t(8,15)(p23.3,q11) with uniparental disomy in Angelman syndrome. Hum. Genet. 93: 471-473.
- Spritz, R.A., Bailin, T., Nicholls, R.D., Lee, S.-T., Park, S.-K., Mascari, M.J. and Butler, M.G. (1997). Hypopigmentation in the Prader-Willi syndrome correlates with P gene deletion but not with haplotype of the hemizygous P allele. Am. J. Med. Genet. 71: 57-62.
- Stalker, H.J. and Williams, C.A. (1998). Genetic counseling in Angelman syndrome: the challenges of multiple causes. Am. J. Med. Genet. 77: 54-59.
- Sutcliffe, J.S., Nakao, M., Christian, S., Örstavik, K.H., Tommerup, N., Ledbetter, D.H. and Beaudet, a.l. (1994). Deletions of a differentially methylated CpG island at the SNRPN gene define a putative imprinting control region. Nat. Genet. 8: 52-58.
- Sutcliffe, J.S., Jiang, Y.-H., Galjaard, R.J., Matsuura, T., Fang, P., Kubota, T., Christian, S.L., Bressler, J., Cattanach, B., Ledbetter, D.H. and Beaudet, A.L. (1997). The E6-AP ubiquitin-protein ligase (UBE3A) gene is localized within a narrowed Angelman syndrome critical region. Genome Res. 7: 368-377.
- Vickers, S., Dahlitz, M., Hardy, C., Kilpatrick, M. and Webb, T. (1994). A male with a de novo translocation involving loss of 15q11q13 material and Prader-Willi syndrome. J. Med. Genet. 31: 478-481.
- Vu, T.H. and Hoffman, A.R. (1997). Imprinting of the Angelman syndrome gene, UBE3A, is restricted to brain. Nat. Genet. 17: 12-13.
- Watrin, F., Roëckel, N., Lacroix, L., Mignon, C., Mattei, M.-G., Disteche, C. and Muscatelli, F. (1997). The mouse Necdin gene is expressed from the paternal allele only and lies in the 7C region of the mouse chromosome 7, a region conserved synteny to the human Prader-Willi syndrome region. Eur. J. Hum. Genet. 5: 324-332.
- Wevrick, R. and Francke, U. (1997). An imprinted mouse transcript homologous to the human imprinted in Prader-Willi syndrome (IPW) gene. Hum. Mol. Genet. 6: 325-332.
- Williams, C.A., Angelman, H., Clayton-Smith, J., Driscoll, D.J., Hendrickson, J.E., Knoll, J.H.M., Magenis, R.E., Schinzel, A., Wagstaff, J., Whidden, E.M. and Zori, R.T. (1995). Angelman syndrome: consensus for diagnostic criteria. Am. J. Med. Genet. 56: 237-238.
- Yamamoto, Y., Huibregtse, J.M. and Howley, P.M. (1997). The human E6-AP gene (UBE3A) encodes three potential protein isoforms generated by differential splicing. Genomics 41: 263-266.
- Yang, T., Adamson, T.E., Resnick, J.L., Leff, S., Wevrick, R., Francke, U., Jenkins, N.A., Copeland, N.G. and Brannan, C.I. (1998). A mouse model for Prader-Willi syndrome imprinting-centre mutations. Nat. Genet. 19: 25-31.
Publication Dates
-
Publication in this collection
13 Nov 2001 -
Date of issue
Dec 2000
History
-
Received
15 June 2000