
Abstract
Gene expression profiles contain the expression level of thousands of genes. Depending on the issue under investigation, this large amount of data makes analysis impractical. Thus, it is important to select subsets of relevant genes to work with. This paper investigates different metrics for gene selection. The metrics are evaluated based on their ability in selecting genes whose expression profile provides information to distinguish between tumor and normal tissues. This evaluation is made by constructing classifiers using the genes selected by each metric and then comparing the performance of these classifiers. The performance of the classifiers is evaluated using the error rate in the classification of new tissues. As the dataset has few tissue samples, the leave-one-out methodology was employed to guarantee more reliable results. The classifiers are generated using different machine learning algorithms. Support Vector Machines (SVMs) and the C4.5 algorithm are employed. The experiments are conduced employing SAGE data obtained from the NCBI web site. There are few analysis involving SAGE data in the literature. It was found that the best metric for the data and algorithms employed is the metric logistic.
gene selection; machine learning; gene expression; sage
BIOINFORMATICS
RESEARCH ARTICLE
Evaluation of gene selection metrics for tumor cell classification
Katti FaceliI; André C.P.L.F. de CarvalhoI; Wilson A. Silva JrII
IUniversidade de São Paulo, Instituto de Ciências Matemáticas e de Computação, São Carlos, SP, Brazil
IIFaculdade de Medicina de Ribeirão Preto, Departamento de Genética, Centro de Terapia Celular, Laboratório de Genética Molecular e Bioinformática, Ribeirão Preto, SP, Brazil
Correspondence Correspondence to André C.P.L.F. de Carvalho Universidade de São Paulo, Instituto de Ciências Matemáticas e de Computação, Departamento de Ciências de Computação e Estatística Avenida Trabalhador Sancarlense 400 Centro, Caixa Postal 668 13566-590 São Carlos, SP, Brazil Email: andre@icmc.usp.br
ABSTRACT
Gene expression profiles contain the expression level of thousands of genes. Depending on the issue under investigation, this large amount of data makes analysis impractical. Thus, it is important to select subsets of relevant genes to work with. This paper investigates different metrics for gene selection. The metrics are evaluated based on their ability in selecting genes whose expression profile provides information to distinguish between tumor and normal tissues. This evaluation is made by constructing classifiers using the genes selected by each metric and then comparing the performance of these classifiers. The performance of the classifiers is evaluated using the error rate in the classification of new tissues. As the dataset has few tissue samples, the leave-one-out methodology was employed to guarantee more reliable results. The classifiers are generated using different machine learning algorithms. Support Vector Machines (SVMs) and the C4.5 algorithm are employed. The experiments are conduced employing SAGE data obtained from the NCBI web site. There are few analysis involving SAGE data in the literature. It was found that the best metric for the data and algorithms employed is the metric logistic.
Key words: gene selection, machine learning, gene expression, sage
Introduction
This investigation was developed as part of the FAPESP/LIRC Clinical Genomics Project, that involves several Brazilian research groups. The main goal of this project is to study gene expression in neoplasias and develop approaches that may be relevant to clinical applications, either to identify disease markers or to define profiles related to clinical evolution or outcome. This paper investigates metrics to support the biologists in the selection of genes that could be related to the occurrence of some neoplasias, based on their expression profiles.
Today, there are several methods to monitor the expression level of a large amount of genes simultaneously. These large-scale gene expression analysis methods can be summarized in two main groups: the tag counting methods (SAGE, MPSS) and the hybridization-based methods (cDNA, oligonucleotide microarray). There are also several analyses that can be carried out with gene expression data, involving pairwise or multiple condition analysis (Claverie, 1999).
In spite of the method employed to acquire the gene expression data, their analysis involves several aspects that have been addressed in the literature (Brazma and Vilo, 2000; Dopazo et al., 2001). One of these aspects is gene selection. Gene expression profiles present the expression level of thousands of genes. Depending on the issue under investigation, this large amount of data makes the analysis impractical. Besides, and more importantly, large changes in a particular phenotype can be due to changes in the expression of a small subset of its genes. Thus, it is important to select subsets of relevant genes to work with. In summary, the interest in a small set of genes can be motivated by financial, personal workload or experimental reasons. Gene selection gives the biologists a small set of genes to make more specific, complex and usually expensive investigations.
There are several methods reported in the literature that have been applied to gene selection (Slonim et al., 2000; Golub et al., 1999; Zhang and Wong, 2001; Ben-Dor et al., 2000; Ben-Dor et al., 2002; Jaeger et al., 2003; Claverie, 1999). Most of them are feature-ranking techniques, frequently called scores. In this work, these techniques will be called metrics. Some of these metrics measure the similarity between the gene expression vector and the class vector (correlation metrics). Other metrics evaluate the difference between the same vectors (distance metrics). Additionally, there are methods described in the literature that are based on genetic algorithms (Liu et al., 2001), feature selection (Inza et al., 2002) and Support Vector Machines (Guyon et al., 2002; Zhang and Wong, 2001).
Many researchers use filtering rules based on fold difference criteria to select subsets of genes. For example, Schena et al. (1996) selected only those genes for which the ratios between the expression intensities of the two conditions being investigated were higher than twofold (Dopazo et al., 2001). However, the application of a simple rule, fold-based, can lead to a high number of false positives (Claverie, 1999). Slonim et al. (2000) employed a number of metrics to rank oligonucleotide microarray data, such as the Pearson correlation coefficient and Euclidean distance, and proposed a correlation metric that emphasizes the "signal-to noise" ratio in using the gene as a predictor, called here Golub's score. Ben-Dor et al. (2000) examined several scoring methods for mining relevant genes. They employed the TNoM and INFO scores, a score based on Logistic regression and the Golub's score. For their analysis, they employed two datasets obtained from oligonucleotide microarrays and one data set obtained from cDNA microarrays.
In the present paper, the authors investigate six metrics commonly used to select genes from microarray data, to select genes based on their expression level obtained with the SAGE technique. The metrics employed here represent a combination of those employed by Schena et al. (1996), Slonim et al. (2000) and Ben-Dor et al. (2000). The metrics employed in this paper are evaluated according to their ability in selecting predictive genes. This evaluation is made by constructing classifiers using as attributes the genes selected by each metric and then comparing the performance of these classifiers. The classifiers are generated using Support Vector Machines (SVMs) (Cristianini and Shawe-Taylor, 2000) and C4.5 (Quinlan, 1993).
In contrast to this paper, the papers found in the literature that describe metrics for gene selection usually do not compare a large number of metrics and apply the metrics to microarray data. There are few analyses involving SAGE data described in the literature and none with all the metrics evaluated here. The comparison of all these metrics and their application to SAGE data are the main contributions of this work. The comparison of two well-known learning algorithms for the analysis of the metrics using SAGE data can also be considered a contribution.
Material and Methods
Data set
The data set employed in the paper was obtained from the Cancer Genome Anatomy Project - CGAP1 1 http://cgap.nci.nih.gov/ . The authors selected 40 libraries containing expression profiles from normal and cancerous brain tissues. Each library represents a different tissue or condition and contains the tag, the frequency of the tag, the associated unigene number and an annotation about the gene. In order to create the data set used in the classification experiments, the libraries were combined in a unique file, with the rows representing a gene (each row corresponds to a tag-unigene combination) and the columns representing the tissues or conditions. This file contains seven columns of normal tissues and 33 columns of cancerous tissues. When a tag is not found in a library, its frequency is set to 0.
The file generated contains 285,723 tags representing the genes. In this file, a normalization operation was applied to the frequencies, to adjust for libraries to have the same total number of tags. All libraries were adjusted to have a total of 200,000 tags (new frequency = original frequency * 200,000 / total number of tags). Next, a filter was applied to the data to remove tags that contain errors and imprecisions due to SAGE. The genes with expression level (frequency) smaller than 24 in all libraries were removed from the file. This filtering kept only 7,888 genes. Table 1 shows a portion of the file generated.
Gene selection
The choice of the approach for gene selection depends very much on the properties the researcher wants to measure (Dopazo et al., 2001). Most of these approaches described in the literature are feature ranking techniques, also called scores or metrics. In this work, these techniques will be called metrics. Some of these metrics measure the similarity (correlation metrics) and others measure the difference (distance metrics) between the gene expression vector and the class vector.
The metrics or scores can be divided into parametric and nonparametric scores. The parametric scores make assumptions about the form of the statistical distribution of the scores within each group, while the nonparametric scores do not make such assumptions, and are more robust (Ben-Dor et al., 2002).
The nonparametric metrics generally specify a hypothesis in terms of population distributions, rather than parameters like means and standard deviations. These metrics are almost as capable of detecting differences among populations as the parametric scores when normality and other assumptions need to be satisfied. Nonparametric scores may be, and often are, more powerful in detecting population differences when these assumptions are not satisfied.
In the present work, the authors compare the fold change criteria (FC), the difference (Diff), the Golub's, TNoM and INFO scores, a score based in Logistic regression (Logistic), the Euclidean distance (Euclidean) and the Pearson correlation coefficient (Pearson). All these metrics are detailed in the next subsection.
For the analysis performed, the authors decided to evaluate subsets of 100, 10 and 4 genes. The gene selection was carried out in the following way. First, all metrics were calculated for each gene of the SAGE data set previously described. Next, the data were sorted according to the ranking provided by each metric. Subsets with the 100, 10 and 4 genes with the best values for each metric were then selected (for each metric, three data sets were produced - with 100, 10 and 4 genes). The best value for the Euclidean distance, TNoM, INFO and Logistic metrics means the lowest values. For the FC, Diff, Golub and Pearson metrics, the highest positive values were chosen as representing the most hyper-expressed genes and the smallest negative values were chosen as representing the most hypo-expressed genes in tumor tissues. Half of the genes selected were hyper-expressed and the other half were hypo-expressed genes. The genes selected generated the data sets employed later in the training of the machine learning algorithms. The three data sets generated for each metric are composed of 40 tissues (conditions) as samples and 100, 10 and 4 genes as attributes or features. Table 2 contains a summary description of these datasets.
The selection process resulted in 24 data sets (8 metrics x 3 number of genes selected). With these data sets, the authors generated SVM and C4.5 classifiers. The metrics were evaluated according to the performance of the classifiers. The performance for each dataset in the classification of new tissues was obtained by performing leave-one-out crossvalidation (Mitchell, 1997). A good metric is one that selects the best set of genes to distinguish the classes (normal and tumor tissues), and a good class distinction is detected by low error rates in the classification of new tissues.
The SVMs were trained with linear kernels. Only this kernel was employed because results of previous similar works show the worst results were attained with the Gaussian and polinomial kernels.
Metrics
The metrics FC and Diff refer to the comparison of two conditions. The dataset analyzed has multiple conditions for each type of tissue (several normal and several tumor tissue libraries). For these metrics, the authors considered the libraries of each type of tissue as a pool. The expression level of each gene in a pool is the mean of the expression levels of the gene in each library.
Let m be the number of genes and n be the number of samples or tissues. Each gene in the dataset, or gene expression matrix, can be represented by a gene expression vector g Î Rn. This work centers the discussion in the case where there are two groups of conditions to be compared. In this case, there are two classes in which the data samples can be separated. These classes will be represented here as -1, or neg (for example, a control condition, such as normal tissues) and +1, or pos (for example, the experimental condition being investigated, such as cancerous tissues). A class vector c Î {-1, 1}n represents the two-class distinction presented in the data. The within-class mean, µx, is the mean of the expression levels of the samples in class x for a particular gene. The within-class standard deviation, sx, is the standard deviation of the expression levels of the samples in the class x for a particular gene.
To calculate the TNoM, INFO and Logistic metrics, the software scoreGenes2 2 http://www.cs.huji.ac.il/labs/compbio/scoregenes/ was employed. Next, each metric used is briefly described.
Fold change
This metric involves the calculation of a ratio relating the expression level of a gene under two experimental conditions. These conditions are, usually, a control and an under investigation condition, such as normal and tumor tissue samples. An arbitrary ratio (usually 2-fold) is then selected as being significant. The cDNA microarray data are already represented as ratio, because most of cDNA microarrays involves two-color fluorescence competitive assays. But when analyzing SAGE or oligonucleotide microarrays the ratio (or FC) has to be calculated.
When only two experiments are compared, the ratio can be calculated directly. However, in some cases, there are several experiments for each of the two classes of interest. In these cases, the mean of the expression values can be calculated for each class and the ratio can be taken from these values. The authors are employing the last case, so the formulas using the mean as the expression values for each class will be shown.
The metric involving the ratio can be expressed in three different ways: the ratio itself, the fold change way and the log of the ratio. Their formulas can be observed in the Equations 1, 2 and 3, respectively.

The shortcoming of R is that the hyper-expression, or induction, and hypo-expression, or repression, are represented by values of different magnitude. For example, a two-fold induction will have more weight than a one-half repression in any comparison. (Dopazo et al., 2001). The FC and LR metrics overcome this problem. In these metrics, the induction and repression have the same magnitude. The graphics of the three kinds of ratio can be observed in Figure 1. Equation 3 represents a log2 transformation, but other logarithms can be applied. In this case, a twofold induction is indicated by the value 1 and a one-half repression by the value -1.
Difference
This metric is the difference between the mean of the expression values of the tumor samples and the mean of the expression values of the normal samples, as shown by Equation 4.

Euclidean distance
This metric measures the absolute distance between two points in space. These points can be two profiles, or, as in this case, one profile and the class vector. Usually, this metric does not require the data to be normalized, and considers profiles of genes with the same magnitude to be similar. However, sometimes, one is looking for genes expressed at different levels, but with the same overall expression. For this purpose, the data should be re-scaled and normalized. In the experiments described in this work, when a gene expression profile is being compared to the class vector, the expression values need to be re-scaled to be comparable to the class vector. The formula of the Euclidean distance is shown in Equation 5. In this work, g_norm is the gene expression vector re-scaled to the interval [-1, 1] and ci is the class vector where -1 represents tumor tissue and 1 represents normal tissue.

Pearson correlation coefficient
The formula of Pearson correlation coefficient can be seen in Equation 6, where g_norm is the gene expression vector normalized to have zero mean and variance 1 and ci is the class vector, where -1 represents tumor tissue and 1 represents normal tissue.

This metric usually does not need any transformation to be applied to the data. But, in this case, the gene expression vector is normalized because it is being compared to the class vector, employing a simplified equation. The values resulting from this metric lie between -1, meaning a negative correlation, and 1, meaning a positive correlation. Thus, a value of 0 indicates no correlation between the gene and the class vector.
Golub
This is a correlation metric proposed by Golub et al. (Golub et al., 1999; Slonim et al., 2000). Its formula can be seen in Equation 7. It measures relative class separation. This metric reflects the difference between the classes relative to the standard deviation within the classes. Large values of |Golub| indicate a strong correlation between gene expression and class distinction. The sign of Golub corresponds to g being more highly expressed in the class pos or neg. The values of this metric are not confined to the range [-1,1].

Referred to by Ben-Dor et al. (2000) as a Gaussian separation score, the Golub metric attempts to, using a Gaussian approximation, measure to what extent the pos and neg classes are separated. Intuitively, the separation between two groups of expression values is proportional to the distance between their mean. This distance has to be normalized by the standard deviation of the groups. A large standard deviation indicates points in the group far away from the mean value and thus the separation would not be strong.
TNoM score
The TNoM (Threshold Number of Misclassification) score (Ben-Dor et al., 2000; Ben-Dor et al., 2002) calculates a minimal error decision boundary and counts the number of misclassifications carried out with this boundary.
This score is based on the idea that a gene g is relevant to the tissue partition if it is over-expressed in one of the classes. This can be formalized by considering how g's expression levels in the class pos relates to its expression levels in the class neg. Let t be a vector of the ordered expression levels of g (t1 is the minimum and tn is the maximum expression level of g). A rank vector, v, of g is defined as a vector of length n where vi is the label associated with ti. If g is under-expressed in the class pos, then the pos entries of v are concentrated in the left hand side of the vector and the neg entries are concentrated at the right hand side. Similarly for the opposite situation. Thus, the relevance of g increases as the homogeneity within the left hand side and within the right hand side of v increases. On the other hand, if g is not informative with respect to the given labeling, the pos and neg in v are interleaved.
The TNoM score comes from a natural way of defining the homogeneity on the two sides and then combining them into one score. The score of v corresponds to the maximal combined homogeneity over all possible ways to break v in two parts, a prefix x, consisting of mostly pos and a suffix y, consisting of mostly neg, or vice versa. The TNoM score of v corresponds to the partition that best divides v into a homogeneous prefix x and a homogeneous suffix y.
The MinCardinality (MC) of a pos-neg vector x is the cardinality of the minority symbol in x, as can be seen in Equation 8, where #s(x) is the number of times a symbol s appears in the vector x. The TNoM score can be seen in the Equation 9.

INFO score
This score, similarly to TNoM, measures the level of homogeneity of the partitions of the rank vector of g. However, it does not count the number of misclassified samples. Instead, it uses the notion of conditional entropy. Let w be a vector composed of pos and neg samples, and let p denote the fraction of the pos entries in w. The entropy of w is defined according to Equation 10.
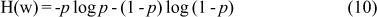
The entropy measures the information in the vector w. This quantity is non-negative, and equal to 0 if and only if p = 0 or p = 1, that is, if w is homogeneous. The maximal value of H(w) is 1 when w is composed of an equal number of pos and neg labels.
The INFO score of v is defined to be the minimal weighted sum of the entropies of a prefix-suffix division, as can be seen in Equation 11, where |.| is the length of the vector. This is the conditional entropy of the rank vector given the partition of the samples in two groups (x and y).
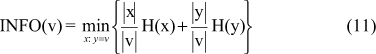
Logistic
This metric is based on Logistic regression, as described by Ben-Dor et al. (2000). The main idea of this metric is to have the probability of both labels close to 0.5, when the expression values are close to the decision threshold and confident for extreme expression values. The conditional probability is either 0 or 1. Such conditional probabilities can be represented by the logistic family:
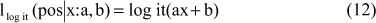
where logit(z) is the logistic function in Equation 13.

To score a gene using this metric, it is necessary to find the parameters a and b that minimize the logloss function. This can be carried out by gradient based non-linear optimization.
Learning algorithms employed
The algorithms employed in this work to classify the tissue samples produced by SAGE technique are Support Vector Machines (SVMs) (Cristianini and Shawe-Taylor, 2000) and the C4.5 algorithm (Quinlan, 1993).
SVMs represent a class of learning algorithms based on the Statistical Learning Theory. Their formulation results in a quadratic optimization problem and involves the principle of Structural Risk Minimization (SRM), which minimizes the "generalization" error. These characteristics give the SVMs algorithms an usually high generalization capacity. SVMs construct a separating hyperplane as the decision surface by maximizing the margin of separation between the positive and negative examples. In order to deal with nonseparable data, a kernel function is employed to map the original input space into a higher dimensional feature space. The separating hyperplane is constructed in this feature space. The classification of a new sample is obtained by projecting it from the input space to the feature space and classifying it based on its position relative to the separating hyperplane.
C4.5 is a learning algorithm that generates models in the form of decision trees. It builds a decision tree from training data by applying a divide-and-conquer strategy and employing a greedy approach that uses a gain ratio as its guide. It chooses an attribute for the root of the tree, divides the training instances into subsets corresponding to the values of the attribute and test the gain ratio on this attribute. This process is repeated for all input attributes of the training patterns. C4.5 chooses the attribute that gains the most information to be at the root of the tree. The algorithm is applied recursively to form sub-trees, terminating when a given subset contains instances of only one class (Quinlan, 1993).
Results
Table 3 presents the error rates obtained in the evaluation of the classifiers generated with each metric and number of genes selected. From this table, it can be observed that most of the lowest errors were achieved with the Logistic metric (with 100 and 4 genes). In these cases, the lowest errors were 2.5% for SVM and 5% for C4.5. For 10 genes, the metric Golub presented the lowest error (2.5%), for both SVM and C4.5. An error of 2.5% means that only one tissue was wrongly classified. Although Golub has presented the best results with 10 genes, Logistic also presented low error rate (5%). In most of the cases, the results obtained with C4.5 were worse than those obtained with SVM.
For the cases of 100 and 4 genes, other metrics presented the same level of error of Logistic as the algorithm C4.5 (100 genes: Golub and TNoM, 4 genes: TNoM). In most of the cases, the TNoM metric showed an error of 5% (2 samples wrongly classified), and in the other cases an error of 10%.
Discussion
The analysis performed in this work encompasses the metrics employed in the works of Schena et al. (1996), Slonim et al. (2000) and Ben-Dor et al. (2000). This work compares the metrics in the context of SAGE data, while these authors employed them for microarray data.
Each of these metrics has its shortcomings. The choice of the best metric for an analysis should take into account the data to be analyzed.
The selection of an arbitrary threshold for the fold change metric results in low specificity and low sensitivity. The low specificity is related to the false positives, particularly with low-abundance transcripts or when a data set is derived from a divergent comparison. The low sensitivity is related to the false negatives, particularly with high-abundance transcripts or when a data set is derived from a closely linked comparison. This metric has several shortcomings and it is now accepted that its use should be discontinued (Murphy, 2002). It also should be noted that this kind of metric removes all information about the absolute gene expression levels.
The Golub metric works well for data normally distributed in each class.
The TNoM score provides partial information about the quality of the predictions made by the best rules. For example, there is no distinction between a rule that makes k one-sided errors (for example, all the errors are samples of class pos predicted as neg) and a rule that makes k/2 errors of the second kind. This distinction is important, since the performance of a rule, such as the one initially described, is very poor for one of the classes. The INFO score makes such distinctions finer.
In the present work, the metrics are compared according to the error rate of the classifiers generated with the genes selected by the metrics.
As expected, the simplest metrics, FC and Diff, presented high error rates. Although these high errors did not occur in all cases, these results suggest that relying only on these metrics to select genes is not appropriate. Such a conclusion just confirms the characteristics of the metrics presented.
In a few cases, the classifiers generated presented an error superior to the majority error. These classifiers did not learn the class separation, and were equivalent to a classifier that just assigns all samples to the tumor class. Metrics that produced such classifiers in at least one case were the FC, Diff, Euclidean, Golub and INFO metrics.
The metric Golub showed unstable behavior. It presented the best result in some cases, the worst result in another and an intermediate result in other cases.
The most stable metrics were the Logistic and TNoM metrics. These metrics presented the same error for the C4.5, but the Logistic metric was more accurate when SVM was employed.
Looking at the results obtained, it is not possible to establish the influence of the number of genes selected in the classification.
Conclusion
This paper investigated six metrics commonly used to select genes from microarray data, to select genes based on their expression level obtained with the SAGE technique. The metrics were evaluated based on their ability in selecting predictive genes. This evaluation was made by constructing classifiers using the genes selected and comparing their performance. The classifiers were generated using the SVM and C4.5 techniques.
The best classifiers were generated with the metrics Logistic in most of the cases. The lowest error rates, 2.5%, were achieved with at least one classifier generated with SVM for each number of genes and with one classifier generated with C4.5 for the case of 10 genes.
The comparison of all these metrics and their application to SAGE data are the main contributions of this work.
There are several other metrics for gene selection described in the literature. It would be interesting to integrate a few more common metrics in the present analysis as a future work. Another future step is the application of the same evaluation described in this paper to other data sets. It is important to evaluate the behavior of the metrics for microarray data too.
Acknowledgments
This research was supported in part by the Brazilian Research Councils FAPESP and CNPq.
Received: August 15, 2003; Accepted: August 20, 2004
- Ben-Dor A, Friedman N and Yakhini Z (2000) Scoring genes for relevance. Technical Report 2000-38, School of Computer Science and Engineering, Hebrew University.
- Ben-Dor A, Friedman N and Yakhini Z (2002) Overabundance analysis and class discovery in gene expression data. Technical Report AGL-2002-4, Agilent Laboratories.
- Brazma A and Vilo J (2000) Gene expression data analysis. FEBS Letters 480:1724.
- Claverie JM (1999) Computational methods for the identification of differential and coordinated gene expression. Hum Mol Genet 8:18211832.
- Cristianini N and Shawe-Taylor J (2000) An Introduction to Support Vector Machines and Other Kernel-Based Learning Methods. Cambridge University Press.
- Dopazo J, Zanders E, Dragoni I, Amphlett G and Falciani F (2001) Methods and approaches in the analysis of gene expression data. J Immunol Methods 250:93112.
- Golub T, Slonim D, Tamayo P, Huard C, Gaasenbeek M, Mesirov J, Coller H, Loh M, Downing J, Caligiuri M, Bloomfield C, and Lander E (1999) Molecular classification of cancer: Class discovery and class prediction by gene expression. Science 286:531537.
- Guyon I, Weston J, Barnhill S and Vapnik V (2002) Gene selection for cancer classification using support vector machines. Machine Learning 46:389422.
- Inza I, Sierra B, Blanco R and Larrañaga P (2002) Gene selection by sequential search wrapper approaches in microarray cancer class prediction. Journal of Intelligent and Fuzzy Systems 12:25-33.
- Liu J, Iba H and Ishizuka M (2001) Selecting informative genes with parallel genetic algorithms in tissue classification. Proceedings of the Genome Informatics Workshop, pp 1423.
- Mitchell T (1997) Machine Learning. McGraw Hill.
- Murphy D (2002) Gene expression studies using microarrays: Principles, problems, and prospects. Advan Physiol Educ 26:256270.
- Quinlan JR (1993) C4.5: Programs for Machine Learning. Morgan Kaufmann.
- Schena MD, Shalon R, Heller A, Chai PO, Brown and Davis RW (1996) Parallel human genome analysis: microarray-based expression monitoring of 1000 genes. Proc Natl Acad Sci USA 93:1061410619.
- Slonim D, Tamayo P, Mesirov J, Golub T and Lander E (2000) Class prediction and discovery using gene expression data. Proceedings of the 4th Annual International Conference on Computational Molecular Biology, Tokyo, Japan, pp 263272.
- Zhang X and Wong WH (2001) Recursive sample classification and gene selection based on svm: Method and software description. Technical report, Department of Biostatistics, Harvard School of Public Health.
Publication Dates
-
Publication in this collection
14 Jan 2005 -
Date of issue
2004
History
-
Received
15 Aug 2003 -
Accepted
20 Aug 2004




