Abstract
Butyrylcholinesterase (BChE) is a plasma enzyme that catalyzes the hydrolysis of choline esters, including the muscle-relaxant succinylcholine and mivacurium. Patients who present sustained neuromuscular blockade after using succinylcholine usually carry BChE variants with reduced enzyme activity or an acquired BChE deficiency. We report here the molecular basis of the BCHE gene underlying the slow catabolism of succinylcholine in a patient who underwent endoscopic nasal surgery. We measured the enzyme activity of BChE and extracted genomic DNA in order to study the promoter region and all exons of the BCHE gene of the patient, her parents and siblings. PCR products were sequenced and compared with reference sequences from GenBank. We detected that the patient and one of her brothers have two homozygous mutations: nt1615 GCA > ACA (Ala539Thr), responsible for the K variant, and nt209 GAT > GGT (Asp70Gly), which produces the atypical variant A. Her parents and two of her brothers were found to be heterozygous for the AK allele, and another brother is homozygous for the normal allele. Sequence analysis of exon 1 including 5'UTR showed that the proband and her brother are homozygous for -116GG. The AK/AK genotype is considered the most frequent in hereditary hypocholinesterasemia (44%). This work demonstrates the importance of defining the phenotype and genotype of the BCHE gene in patients who are subjected to neuromuscular block by succinylcholine, because of the risk of prolonged neuromuscular paralysis.
hereditary hypocholinesterasemia; butyrylcholinesterase; succinylcholine BCHE gene; DNA polymorphism
SHORT COMMUNICATION
Biochemical and genetic analysis of butyrylcholinesterase (BChE) in a family, due to prolonged neuromuscular blockade after the use of succinylcholine
Daniel Fantozzi GarciaI, II, III; Ticiano G. OliveiraI, IV; Greice A. MolfettaII; III; Luiz V. GarciaI, IV; Cristiane A. FerreiraII, III; Adriana A. MarquesII, III; Wilson Araujo Silva Jr.I, II, III
IHospital das Clínicas, Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP, Brazil
IIDepartamento de Genética, Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP, Brazil
IIICentro Regional de Hemoterapia de Ribeirão Preto, Instituto Nacional de Ciência e Tecnologia em Terapia Celular e Centro de Terapia Celular, Ribeirão Preto, SP, Brazil
IVDepartamento de Biomecânica, Serviço de Anestesiologia, Medicina e Reabilitação do Aparelho Locomotor, Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP, Brazil
Send correspondence to Send correspondence to: Wilson Araújo da Silva Junior Centro Regional de Hemoterapia de Ribeirão Preto Rua Tenente Catão Roxo 2501, Monte Alegre 14051-140 Ribeirão Preto, SP, Brazil. E-mail: wilsonjr@usp.br.
ABSTRACT
Butyrylcholinesterase (BChE) is a plasma enzyme that catalyzes the hydrolysis of choline esters, including the muscle-relaxant succinylcholine and mivacurium. Patients who present sustained neuromuscular blockade after using succinylcholine usually carry BChE variants with reduced enzyme activity or an acquired BChE deficiency. We report here the molecular basis of the BCHE gene underlying the slow catabolism of succinylcholine in a patient who underwent endoscopic nasal surgery. We measured the enzyme activity of BChE and extracted genomic DNA in order to study the promoter region and all exons of the BCHE gene of the patient, her parents and siblings. PCR products were sequenced and compared with reference sequences from GenBank. We detected that the patient and one of her brothers have two homozygous mutations: nt1615 GCA > ACA (Ala539Thr), responsible for the K variant, and nt209 GAT > GGT (Asp70Gly), which produces the atypical variant A. Her parents and two of her brothers were found to be heterozygous for the AK allele, and another brother is homozygous for the normal allele. Sequence analysis of exon 1 including 5'UTR showed that the proband and her brother are homozygous for -116GG. The AK/AK genotype is considered the most frequent in hereditary hypocholinesterasemia (44%). This work demonstrates the importance of defining the phenotype and genotype of the BCHE gene in patients who are subjected to neuromuscular block by succinylcholine, because of the risk of prolonged neuromuscular paralysis.
Key words: hereditary hypocholinesterasemia, butyrylcholinesterase, succinylcholine BCHE gene, DNA polymorphism.
Butyrylcholinesterase or pseudocholinesterase is a serine hydrolase that catalyses the hydrolysis of choline esters, including the muscle-relaxant succinylcholine and mivacurium. Currently, succinylcholine is the drug of choice for tracheal intubation in rapid-sequence inductions, as in emergency situations, and for patients at high risk of gastroesophageal reflux (Caldwell, 2004; Miller, 2004).
After the introduction, in 1951, of suxamethonium chloride as a muscle relaxant drug, it was noticed that subjects with butyrylcholinesterase deficiency to which this drug was given suffered from prolonged neuromuscular blockade, requiring continuous artificial respiratory support (Evans et al., 1952). Later on, BChE deficiency was identified as an inherited condition in which the plasma enzyme was unable to hydrolyze the drug efficiently that could be identified and classified by biochemical analysis (Kalow and Genest, 1957).
The phenotypic classification of BChE is made by measuring the enzyme activity and by using specific enzyme inhibitors that produce phenotype-specific patterns of 'inhibitor numbers'. The silent phenotype is characterized by the complete absence of BChE activity or by an enzyme activity < 10% of the average levels of the usual phenotype. The atypical (A) variant resists in vitro to the inhibition by dibucaine. The K variant (named in honor of Dr. Werner Kalow) is associated with a reduction in BChE activity (Rubinstein et al., 1978), and is a variant that can only be identified by inhibition methods when it occurs in heterozygosis with the atypical variant allele (Goodall, 2004), although sometimes the phenotype classification may not correspond to the DNA analysis (Bartels et al., 1992).
Recently, DNA sequencing has made it possible to identify the BChE variants at the DNA level. It was demonstrated that the alleles BCHEIA and BCHEIK correspond, respectively, to the point mutations nt209 GAT > GGT (Asp70Gly) and nt1615 GCA > ACA (Ala539Thr), whereas different mutations have been described as being responsible for the silent phenotype (La Du et al., 1990; Bartels et al., 1992; Lockridge and Masson, 2000; Parmo-Folloni et al., 2008).
A mutation detection approach has already been proposed for the investigation and characterization of the BChE variants in individuals who present reduced plasmatic enzyme activity and need to undergo anesthetic procedures. What makes this approach advantageous is the fact that the phenotype is well defined, the gene is relatively small (73 kb; four exons interrupted by 3 introns with exon 1 not translated; coding region 1722 nucleotides), and the majority of abnormalities described are point mutations (Yen et al., 2003; Goodall, 2004).
In this study, we report the mutations in the BCHE gene responsible for the prolonged neuromuscular blockade presented by a patient subjected to an endoscopic nasal surgery at the Hospital das Clínicas, Faculdade de Medicina de Ribeirão Preto, USP, Ribeirão Preto, SP, Brazil. Afterwards, other members of the family were screened for the same mutation.
The patient, a woman, was 34 years old at the time of the study, weighed 90 kg, had untreated chronic arterial hypertension, presented recurring epistaxis for 24 h and was hospitalized in the Otolaryngology Department for etiological investigation. Nasal fibroscopy was performed, which showed active bleeding in the right sphenopalatine artery area. Homeostasis was tried with a nasal tampon. After two days, as the patient's bleeding continued, an endoscopic ligation of the sphenopalatine artery was performed, under general anesthesia, with an estimated duration of 30 to 60 min.
As preanesthetic medication 1.5 mg midazolam were used, and the patient was monitored with a pulse oximeter, electrocardiogram and an automatic noninvasive blood pressure monitor. Anesthesia was induced with remifentanil in continuous infusion, etomidate (0.2 mg/kg), suxamethonium (1.1 mg/kg) and, after the orotracheal intubation, atracurium (0.4 mg/kg). The anesthesia was maintained with continuous administration of propofol and remifentanil.
Although the patient had been fasting for over eight hours, rapid-sequence orotracheal intubation was chosen due to the presence of blood in the airways. The surgical procedure lasted 55 min and was accomplished without complications. Immediate drug suspension at the infusion pump and neuromuscular block reversion with atropine (1 mg) and neostigmine (2 mg, plus 1 mg) were done.
Fifteen minutes after the end of the procedure, the patient presented tachycardia, lacrimation and lack of motor muscular response. Due to the prolonged neuromuscular block, the patient was sedated and monitored with TOF (train of four, TOF-Guard INMT®). After succinylcholine administration, the first response in TOF appeared in 155 min, and spontaneous ventilation in 250 min. At this time, TOF was 40%. After 355 min, the patient had three consecutive TOF measurements over 90%. Sedation was discontinued, the airways were aspirated, and the patient extubated.
Due to the symptoms presented, the possibility of the patient having butyrylcholinesterase deficiency was raised. Plasma cholinesterase measured by the photocolorimeter method was 1,310 U/L (normal reference rate: 3,200 to 9,000 U/L) (Table 1). After the initial evaluation, it was suggested to continue the investigation through enzyme activity measurements and molecular biology studies of the patient and her family. They all signed informed consent forms for genetic studies.
Before the molecular analysis, butyrylcholinesterase enzyme activity was measured in the samples obtained, using the method described by Garry and Routh (1965). This is a colorimetric method for plasma or serum, using acetylthiocholine as substrate and 2 nitrobenzoic acid (5:5-dithiobis) (DTNB) as chromogenic agent. Plasma cholinesterase acts on acetylthiocholine releasing thiocholine which reacts with DTNB, producing a yellow color. The enzyme activity is measured after incubation for 3 min at 37 °C, and then the reaction is stopped by adding quinidine sulfate. The difference in absorbance between the test and the blank is measured at 412 nm in a spectrophotometer.
Genomic DNA was extracted from peripheral blood leukocytes, using a Super Quick-gene-rapid DNA isolation kit (Promega), following the manufacturer's instructions.
The BCHE promoter region and its four exons were amplified by PCR using specific primers as described by Levano et al. (2005) (Table 2).
All PCR reactions were standardized with the following conditions: 2.5 mM of each primer, 2.5 mM of dNTPs, 2.5 μL of 1x buffer (Biotools), 1U of Taq polymerase (Biotools); 200 ng of genomic DNA, and 14 μL of distilled water, in a final reaction volume of 25 μL. PCR was performed as follows: an initial denaturation step of 4 min, followed by 35 cycles consisting of 40 s at 94 °C (denaturation), 50 s at annealing temperatures (specific for each reaction) (Table 2), an extension of 50 s at 72 °C, and a final extension of 10 min at 72 °C.
The PCR-amplified DNA fragments were subjected to direct sequencing in an automatic capillary sequencing system (MegaBACE 1000 DNA Sequencing System®, Molecular Dynamics & Amersham Life Science), using a DYEnamic ET terminator Sequencing System® kit and the same PCR primers, following the manufacturer's instructions.
The sequencing results were analyzed using the FinchTV version 1.4.0 (Geospiza Inc. 2004-2006) and Polyphred (Geospiza Inc.) softwares; the sequences obtained were compared with reference sequences from GenBank (NM_000055). The nomenclature used to classify the genotypes was as proposed by La Du et al (1991).
The BChE enzyme activity measurements (expressed in μM SH/3 min/mL of plasma) of the patient, her parents and her siblings, are presented in Table 1.
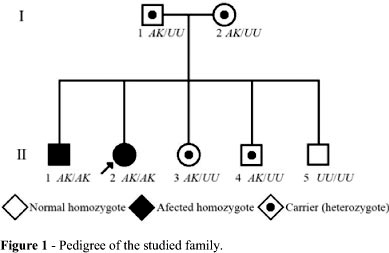
The molecular analyses revealed that the proband (II.2) and her brother (II.1), who has low enzyme activity too, present two point mutations in homozygosis: nt1615 GCA > ACA (Ala539Thr), responsible for the K variant, and nt209 GAT > GGT (Asp70Gly), which produces the atypical variant. Her parents (I.1 e I.2), as well as two of her siblings (II.3 e II.4), are heterozygotes for these two sites of the BCHE gene, and her other brother (II.5), who presented the highest enzyme activity, is homozygous for the usual (normal) variants at both examined sites (Figures 1 and 2). The analysis of exon 1 including 5'UTR showed that the proband and her brother with low enzyme activity (II.1) are both homozygous for -116G.
Prolonged neuromuscular blockade after the use of succinylcholine occurs in patients with a significant BChE deficiency, compromising more than 70% of the enzyme activity, and is caused mostly by genetic abnormalities. Hypocholinesterasemia may also occur during pregnancy, liver diseases, malnutrition and chronic diseases, but in these cases the enzyme deficiency is usually low, and there is rarely a prolonged neuromuscular blockade (about 6% of the cases) (Viby-Mogensen and Hanel, 1978).
Variant K presents a linkage disequilibrium regarding the point mutation responsible for the atypical variant in 89% of its carriers (Bartels et al., 1992). This association contributes to the additional 33% reduction in the BChE enzyme activity Vmax in atypical (A) serum. Patients with genotype AK/AK have reduced enzyme activity, and this is the most common genotype responsible for hereditary hypocholinesterasemia (44%) (Yen et al., 2003). Furtado-Alle et al. (2008) found that point mutation -116G > A in exon 1 of the BCHE gene in cis with the K variant is also responsible for the decrease in BChE activity.
In the case reported here, the prolonged neuromuscular blockade after use of succinylcholine was due to the association of variants A and K in cis (genotype AK/AK), as described by Yen et al (2003), and not to the association of variants K and -116A in cis. We also showed the importance of knowing the BChE phenotype and genotype of patients who will undergo surgical procedures when the use of succinylcholine or mivacurium is indicated as a neuromuscular blocker drug. The risk of prolonged neuromuscular blockade in these cases is estimated at 2.18% (Vaisi-Raygani et al., 2007).
This study makes it clear that, whenever a subject with butyrylcholinesterase deficiency is detected, the patient's family, especially his/her siblings, should be investigated, because, although the enzyme activity is determined in a codominant manner, the clinical phenotype usually displays a recessive pattern.
Acknowledgments
The authors are grateful to Fernanda Udinal, for her valuable help with the English version.
Received: March 18, 2010; Accepted: October 22, 2010.
Associate Editor: Paulo A. Otto
License information: This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
- Bartels CF, Jensen FS, Lockridge O, Van der Spek AFL, Rubinstein HM, Lubrano T and La Du BN (1992) DNA mutation associated with the human butyrylcholinesterase K-variant and its linkage to the atypical variant mutation and other polymorphic sites. Am J Hum Genet 50:1086-1103.
- Caldwell JE (2004) The continuing search for a succinylcholine replacement. Anesthesiology 100:763-764.
- Evans FT, Gray PW, Lehmann H and Silk E (1952) Sensitivity to succinylcholine in relation to serum-cholinesterase. Lancet 1:1229-1230.
- Furtado-Alle L, Andrade FA, Nunes K, Mikami LR, Souza RL and Chautard-Freire-Maia EA (2008) Association of variants of the -116 site of the butyrylcholinesterase BCHE gene to enzyme activity and body mass index. Chem Biol Interact 175:115-118.
- Garry PJ and Routh JI (1965) A micro method for serum cholinesterase. Clin Chem 11:91-96.
- Goodall R (2004) Cholinesterase: Phenotyping and genotyping. Ann Clin Biochem 41:98-110.
- Kalow W and Genest K (1957) A method for the detection of atypical forms of human serum cholinesterase; determination of dibucaine numbers. Can J Biochem Physiol 35:339-346.
- La Du BN, Bartels CF, Nogueira CP, Hajra A, Lightstone H, Van der Spek AFL and Lockridge O (1990) Phenotypic and molecular biological analysis of human butyrylcholinesterase variants. Clin Biochem 23:423-431.
- La Du BN, Bartels CF, Nogueira CP, Arpagaus M and Lockridge O (1991) Proposed nomenclature for human butyrylcholinesterase genetic variants identified by DNA sequencing. Cell Mol Neurobiol 11:79-89.
- Levano S, Ginz H, Siegemund M, Filipovic M, Voronkov E, Urwyler A and Girard T (2005) Genotyping the butyrylcholinesterase in patients with prolonged neuromuscular block after succinylcholine. Anesthesiology 102:531-535.
- Lockridge O and Masson P (2000) Pesticides and susceptible populations: People with butyrylcholinesterase genetic variants may be at risk. Neurotoxicology 21:113-126.
- Miller R (2004) Will succinylcholine ever disappear? Anesth Analg 98:1674-1675.
- Parmo-Folloni F, Nunes K, Lepienski LM, Mikami LR, Souza RL, Tsuneto LT, Petzl-Erler ML and Chautard-Freire-Maia EA (2008) Two new mutations of the human BCHE gene (IVS3-14T > C and L574fsX576). Chem Biol Interact 175:135-137.
- Rubinstein HM, Dietz AA and Lubrano T (1978) E1k; another quantitative variant at cholinesterase locus 1. J Med Genet 15:27-29.
- Vaisi-Raygani A, Rahimi Z, Kharazi H, Tavilan H, Aminiani M, Kiani A, Vaisi-Raygani A and Pourmotabbed T (2007) Determination of butyrylcholinesterase (BChE) phenotypes to predict the risk of prolonged apnea in persons receiving succinylcholine in the healthy population of western Iran. Clin Biochem 40:629-633.
- Viby-Mogensen J and Hanel HK (1978) Prolonged apnea after suxamethonium: An analysis of the first 225 cases reported to the Danish Cholinesterase Research Unit. Acta Anaesthesiol Scand 22:371-380.
- Yen T, Nightingale BN, Burns JC, Sullivan DR and Stewart PM (2003) Butyrylcholinesterase (BCHE) genotyping for post-succinylcholine apnea in an Australian population. Clin Chem 49:1297-1308.
Publication Dates
-
Publication in this collection
28 Jan 2011 -
Date of issue
2011
History
-
Accepted
22 Oct 2010 -
Received
18 Mar 2010