Abstract
Deletion-induced hemizygosity may unmask deleterious autosomal recessive variants and be a cause of the phenotypic variability observed in microdeletion syndromes. We performed complete exome sequencing (WES) analysis to examine this possibility in a patient with 1p13.2 microdeletion. Since the patient displayed clinical features suggestive of Noonan Syndrome (NS), we also used WES to rule out the presence of pathogenic variants in any of the genes associated with the different types of NS. We concluded that the clinical findings could be attributed solely to the 1p13.2 haploinsufficiency. Retrospective analysis of other nine reported patients with 1p13.2 microdeletions showed that six of them also presented some characteristics of NS. In all these cases, the deleted segment included the NRAS gene. Gain-of-function mutations of NRAS gene are causally related to NS type 6. Thus, it is conceivable that NRAS haploinsufficiency and gain-of-function mutations may have similar clinical consequences. The same phenomenon has been described for two other genes belonging to the Ras/MAPK pathway: MAP2K2 and SHOC2. In conclusion, we here report genotype-phenotype correlations in patients with chromosome 1p13.2 microdeletions and we propose that NRAS may be a critical gene for the NS characteristics in the patients.
Keywords
1p13.2 deletion; Noonan syndrome type 6; NRAS gene; RASopathy; unmasking heterozygosity
Introduction
Patients with chromosomal deletions may present with variable clinical phenotypes for different reasons. First, the deletions frequently vary in size and the phenotypic alterations may depend on which loci are deleted in the specific case, as exemplified by the contiguous gene syndromes such as, for instance, Langer-Giedion syndrome (OMIM #150230) and Williams-Beuren syndrome (OMIM #194050). Second, the phenotypic effect of the deleted segment may be modulated by some specific regions of the rest of the genome, in analogy with autosomal dominant diseases with variable expressivity and as, for instance, is the case in digenic inheritance (Schaffer, 2013Schaffer AA (2013) Digenic inheritance in medical genetics. J Med Genet 50:641-652.). Third, early postzygotic events, and/or environmental factors may influence phenotypic discordances, such as in cases of monozygotic twins with discordant phenotype and chromosome 22q11.2 microdeletion (Yamagishi et al., 1998Yamagishi H, Ishii C, Maeda J, Kojima Y, Matsuoka R, Kimura M, Takao A, Momma K and Matsuo N (1998) Phenotypic discordance in monozygotic twins with 22q11.2 deletion. Am J Med Genet 78:319-321.). Fourth, the clinical phenotype may be influenced by the presence of a pathogenic variant in a gene located in the region homologous to the deleted segment, in the intact member of the chromosome pair. This phenomenon has been termed "unmasking heterozygosity" (UH) and when it occurs the phenotype of a recessive disease might emerge from the microdeletion due to hemizygosity (Coman and Gardner, 2007Coman DJ and Gardner RJ (2007) Deletions that reveal recessive genes. Eur J Hum Genet 15:1103-1104.; Poot, 2012Poot M (2012) Beware of hemizygous deletions that may unmask deleterious variants. Mol Syndromol 3:45-46.). To our knowledge, the earliest example of this phenomenon was described in retinoblastoma (OMIM #180200), which may occur because a chromosomal deletion may cause hemizygosity for an RB1 (OMIM *614041) pathogenic variant on chromosome 13q14.
Next generation sequencing (NGS) techniques, such as whole genome sequencing (WGS) and whole exome sequencing (WES) allow the screening of the genome including the non-deleted allele for variants that may contribute to variable phenotypic expression in deletion syndromes. Accordingly, NGS has been reported to reveal deleterious variants unmasked by hemizygous deletions in individuals with 22q11.2 deletion syndrome (OMIM #192430; #188400) (McDonald-McGinn et al., 2013McDonald-McGinn DM, Fahiminiya S, Revil T, Nowakowska BA, Suhl J, Bailey A, Mlynarski E, Lynch DR, Yan AC, Bilaniuk LT, et al. (2013) Hemizygous mutations in SNAP29 unmask autosomal recessive conditions and contribute to atypical findings in patients with 22q11.2DS. J Med Genet 50:80-90.), with mental retardation or multiple congenital abnormalities and hemizygous deletions (Hochstenbach et al., 2012Hochstenbach R, Poot M, Nijman IJ, Renkens I, Duran KJ, Van't Slot R, van Binsbergen E, van der Zwaag B, Vogel MJ, Terhal PA, et al. (2012) Discovery of variants unmasked by hemizygous deletions. Eur J Hum Genet 20:748-753.), and with thrombocytopenia absent radius syndrome (OMIM #274000) (Albers et al., 2012Albers CA, Paul DS, Schulze H, Freson K, Stephens JC, Smethurst PA, Jolley JD, Cvejic A, Kostadima M, Bertone P, et al. (2012) Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nat Genet 44:435-439.).
Since UH can be revealed using exome sequencing, we used it in a genotype-phenotype correlation study of chromosome 1p13.2 microdeletions. We here report the case of a 21-year-old man with a multisystem phenotype containing some features of Noonan syndrome (OMIM #613224) and 1p13.2 deletion extending from 1:112,096,417 to 1:115,805,157 (hg19) diagnosed by aCGH. Exome sequencing analysis examined the possibility of a recessive pathogenic variant unmasked by his hemizygous deletion and also verified if he had pathogenic variants in any of the Noonan Syndrome genes. We concluded that the clinical characteristics of the patient could be attributed to the 1p13.2 haploinsufficiency. Additionally, we compared his clinical characteristics with the phenotype of six patients with similar deletions overlapping the one of our patient reported previously in DECIPHER databaseDECIPHER database, https://decipher.sanger.ac.uk (accessed June 2015).
https://decipher.sanger.ac.uk...
and three individuals reported in the literature (Mattia et al., 1992Mattia FR, Wardinsky TD, Tuttle DJ, Grix Jr. A, Smith KA and Walling P (1992) Interstitial deletion of the short arm of chromosome 1 (46XY, del(1)(p13p22.3)). Am J Med Genet 44:551-554.; Bisgaard et al., 2007Bisgaard AM, Rasmussen LN, Moller HU, Kirchhoff M and Bryndorf T (2007) Interstitial deletion of the short arm of chromosome 1 (1p13.1p21.1) in a girl with mental retardation, short stature and colobomata. Clin Dysmorphol 16:109-112.; Fitzgibbon et al., 2009Fitzgibbon GJ, Kingston H, Needham M and Gaunt L (2009) Haploinsufficiency of the nerve growth factor beta gene in a 1p13 deleted female child with an insensitivity to pain. Dev Med Child Neurol 51:833-837.). On these bases we propose that haploinsufficiency of NRAS gene (OMIM *164790) may be important in determining the clinical phenotype of this microdeletion.
Clinical report
Our patient was a 21-year-old man born of healthy unrelated parents (DECIPHER patient no. 274660). He was born at 37 weeks of pregnancy by cesarean delivery for breech presentation. Birth weight was 2,720 g (3rd-10th centile), with length of 47 cm (10th centile) and occipital frontal circumference (OFC) 34.5 cm (25-50th centile). He presented with hypotonia and feeding difficulties in infancy. His motor development was delayed, having walked independently at 22 months of age. At 3 years-old he spoke single words and at 4 years-old he spoke sentences. He never learned to write or to read. Since infancy he has had recurring bouts of vomiting. Colonoscopy revealed a polypoid mucosa in the terminal ileum and chronic enteritis. Ophthalmologic exams showed myopia, with normal fundus. Echocardiography and nuclear magnetic resonance of the brain were normal.
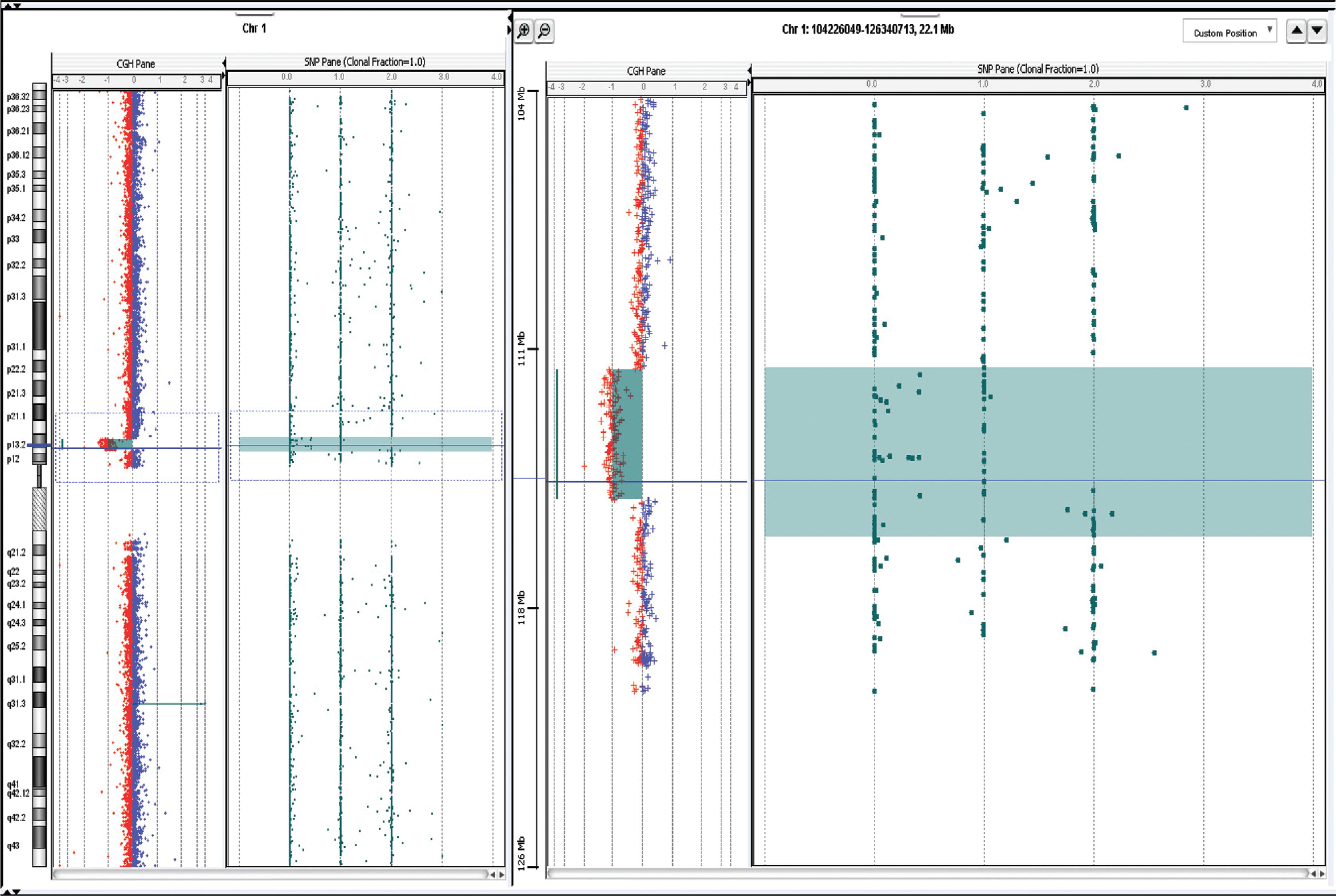
He was evaluated by a geneticist at 20 years of age, when his weight was 68.5 kg (25-50th centile), height 161 cm (< 3rd percentile) and OFC 53 cm (< 3rd percentile). Besides the short stature, his phenotype had several features reminiscent of Noonan Syndrome, including intellectual disability, ptosis, low hairline at the nape, broad neck, excess of pigmented nevi resembling lentigines, pectus excavatum and scoliosis (see Figure 1). Complementary examinations showed patellar chondromalacia, osteoporosis, asymmetry of the lower limbs, nephrolithiasis with increased loss of K+ and Ca+ in urine, hypercholesterolemia and primary hypothyroidism. The conventional karyotype of the patient and his parents were normal. However, subsequent aCGH analysis (Agilent SurePrint G3 4x180K) revealed a 3.71 Mb microdeletion on chromosome 1p13.2 (chr1:112,096,417–115,805,157; UCSC Genome Browser hg19) (Figure 2).
Frontal view of patients with deleted NRAS. Patients present features of Noonan Syndrome 6, including macrocephaly, short/webbed neck, low hairline, skin abnormalities, triangular face with age, low-set ears, arched eyebrows, hypertelorism, ptosis, downslating palpebral fissures and epicanthal folds. Considering the patients with NRAS deletion, we did not have a picture of patient 253793 and 258063.
Copy number profile of chromosome 1 of our patient obtained by aCGH. The chromosome 1 copy number imbalances are indicated on the left panel and shown in detail on the right panel: the alterations marked by the square show a ~3.71 Mb 1p13.2 deletion.
Material and Methods
Samples and DNA isolation
This study was approved by the Research Ethics Committee of Universidade Federal de Minas Gerais and by the Brazilian National Committee of Ethics in Research (CONEP – Brazil) with number 854.709. Written informed consent including permission for publication was provided by the parents. Genomic DNA was isolated from peripheral blood of the patient using a modified salting out procedure (Miller et al., 1988Miller SA, Dykes DD and Polesky HF (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16:1215.).
Whole exome sequencing and analysis
Whole exome sequencing was performed in the patient sample by The Centre for Applied Genomics, The Hospital for Sick Children (Toronto, Canada) using the Agilent SureSelect Human All Exon V4 Kit (Agilent Technologies) and SOLiD 5500xl platform (Applied Biosystems). All data were aligned to the hg19/GRCh37 reference genome build via BFAST and BWA aligner. Variants were quality trimmed using the Genome Analysis ToolkitGenome Analysis Toolkit (GATK), http://www.broadinstitute.org/gatk/ (accessed June 2015).
http://www.broadinstitute.org/gatk/...
(GATK 1.1.28) and they were annotated for functional effect by SnpEffSnpEff, http://snpeff.sourceforge.net/ (accessed June 2015).
http://snpeff.sourceforge.net/...
2.0.5. SnpEff also provided a simple assessment of the putative impact of the variant (e.g. High, Moderate, or Low impact). Alignment, calling and annotation of the variants against databases such as 1000 Genomes (April 2012 release), NHLBI Exome Sequencing ProjectNHLBI Exome Sequencing Project (ESP), http://evs.gs.washington.edu/EVS/ (Accessed June 2015).
http://evs.gs.washington.edu/EVS/...
(ESP6500) and Single Nucleotide Polymorphism database (dbSNP137) were done using a software developed in-house called Mendel,MD (Cardenas et al., 2015Cardenas RGCCL, Linhares ND and Pena SDJ (2015) Mendel, MD: A user-friendly online program for clinical exome analysis. BMC Bioinform 16 (Suppl 8):A2.). The prediction software SIFTSIFT, http://sift.jcvi.org/ (accessed June 2015).
http://sift.jcvi.org/...
(Sorting Intolerant From Tolerant) was used in order to estimate whether a given amino acid substitution affected protein function. SIFT prediction is based on the degree of conservation of amino acid residues in sequence alignments derived from closely related sequences and on the physical properties of amino acids.
Results
Whole exome sequencing of the patient sample identified 76,822 variants. Approximately 26,863 of these variants passed in the GATK quality filters and had a Minor Allele Frequency (MAF) ≤ 0.01 in the 1000 Genomes, ESP6500 and dbSNP137. In the deleted region, which extended from 1:112,096,417 to 1:115,805,157 (hg19), exome analysis revealed 35 hemizygous variants that passed in GATK quality filters. Only 10 of these variants were considered to have moderate or high impact by SnpEff, which means that they are assumed to have high disruptive impact in the protein or might change protein effectiveness (examples: stop gained, frameshift, missense and splice donor variants). Based on the assumption that the potential damaging variants are rare, the variants were then filtered considering an allele frequency < 0.01 in the 1000 Genomes1000 Genomes, http://browser.1000genomes.org (accessed June 2015).
http://browser.1000genomes.org...
database, ESP6500 and dbSNP137, however no plausibly pathogenic variant was retained. Besides, all of these 10 variants were predicted as tolerated by SIFT (score ≥ 0.05) (Table S1). In addition, we verified by exome analysis that the patient did not have any pathogenic variants in the genes associated with Noonan Syndrome and other rasopathies (PTPN11, SHOC2, KRAS, CBL, SOS1, RAF1, HRAS, NRAS, RASA1, SPRED1, BRAF, MAP2K1, MAP2K2, RIT1 and RRAS) (Rauen, 2013Rauen KA (2013) The RASopathies. Annu Rev Genomics Hum Genet 14:355-369.; Ekvall et al., 2015Ekvall S, Wilbe M, Dahlgren J, Legius E, van Haeringen A, Westphal O, Anneren G and Bondeson ML (2015) Mutation in NRAS in familial Noonan syndrome—case report and review of the literature. BMC Med Genet 16:e95.).
The deleted 1p13.2 region of our patient contained 69 genes according to the NCBI Map Viewer Annotation Release 105, of which 28 were listed in OMIM (Table S2). However, only eight of these genes were known to be associated with Mendelian disease phenotypes (Table S3). Among the recessive disease genes there was the TSHB gene (OMIM *188540), in which homozygous mutations had been reported in patients with congenital nongoitrous hypothyroidism-4 (OMIM #275100). Our patient had hypothyroidism, but it was not congenital.
In order to perform a genotype-phenotype correlation study of chromosome 1p13.2 microdeletions, we compared the clinical characteristics of our patient to those of previously described cases (Table 1 and Figure 1). Isolated chromosome 1p13.2 microdeletions are not common; we could find only three individuals reported in the literature (Mattia et al., 1992Mattia FR, Wardinsky TD, Tuttle DJ, Grix Jr. A, Smith KA and Walling P (1992) Interstitial deletion of the short arm of chromosome 1 (46XY, del(1)(p13p22.3)). Am J Med Genet 44:551-554.; Bisgaard et al., 2007Bisgaard AM, Rasmussen LN, Moller HU, Kirchhoff M and Bryndorf T (2007) Interstitial deletion of the short arm of chromosome 1 (1p13.1p21.1) in a girl with mental retardation, short stature and colobomata. Clin Dysmorphol 16:109-112.; Fitzgibbon et al., 2009Fitzgibbon GJ, Kingston H, Needham M and Gaunt L (2009) Haploinsufficiency of the nerve growth factor beta gene in a 1p13 deleted female child with an insensitivity to pain. Dev Med Child Neurol 51:833-837.). Additionally, six patients with isolated deletions partially overlapping the one of our patient had been reported previously in DECIPHER database. All these nine patients were included in our analysis.
The deletions in the several patients ranged from 2.19 Mb to 17.93 Mb in size and eight of them included a region extending from 115,018,964 to 115,713,872 (hg19). The region of overlap of all patients contained 10 genes: TRIM33, BCAS2, DENND2C, AMPD1, NRAS, CSDE1, SIKE1, SYCP1, TSHB, TSPAN2 (Figure 3). The Database of Genomic Variants (DGVbeta) lists numerous copy-number variations (CNVs) in individuals with normal phenotypes within this region. The segment with the least number of CNVs was the one containing the genes AMPD1, NRAS and CSDE1 (Figure 4).
Schematic representation of the deleted segments in our patient and those previously reported with isolated 1p13.2 microdeletions. Except for the patient described by Mattia et al. (1992)Mattia FR, Wardinsky TD, Tuttle DJ, Grix Jr. A, Smith KA and Walling P (1992) Interstitial deletion of the short arm of chromosome 1 (46XY, del(1)(p13p22.3)). Am J Med Genet 44:551-554., who has a microdeletion from 1p13 to 1p22.3, all other patients have their breakpoints defined by molecular methods. Ideogram of chromosome 1, physical map and deleted segments are indicated according to their placement on the Ensembl Genome BrowserEnsembl Genome Browser, http://www.ensembl.org/index.html (accessed June 2015).
http://www.ensembl.org/index.html... .
Genomic variants listed in the Database of Genomic Variants (DGVbeta)Database of Genomic Variants (DGVbeta), http://dgvbeta.tcag.ca/dgv/app/home (accessed June 2015).
http://dgvbeta.tcag.ca/dgv/app/home... in the smallest region of overlap between the patients (chr1:115,018,964–115,713,872) (hg19). The black arrow indicate the region less populated with CNVs.
The patient reported by Fitzgibbon et al. (2009)Fitzgibbon GJ, Kingston H, Needham M and Gaunt L (2009) Haploinsufficiency of the nerve growth factor beta gene in a 1p13 deleted female child with an insensitivity to pain. Dev Med Child Neurol 51:833-837. presented congenital insensitivity to pain and temperature, which led the authors to propose that this might be a consequence of haploinsufficiency of the NGF gene (OMIM *162030), which is related to hereditary sensory and autonomic neuropathy type V, an autosomal recessive disease (sequencing of the undeleted homologous chromosome had not shown any pathogenic variants). However, in disagreement with this suggestion, NGF was also deleted in three patients described in DECIPHER, none of whom presented congenital insensitivity to pain and temperature (patients no. 250335, 253793 and 260230). In relation to coloboma, Bisgaard et al. (2007)Bisgaard AM, Rasmussen LN, Moller HU, Kirchhoff M and Bryndorf T (2007) Interstitial deletion of the short arm of chromosome 1 (1p13.1p21.1) in a girl with mental retardation, short stature and colobomata. Clin Dysmorphol 16:109-112. hypothesized that haploinsufficiency of the WNT2B gene (OMIM *601968) might have caused it in their patient. Indeed, this gene was deleted in two other patients with ocular coloboma (Patients no. 250335 and 257066), but it was also deleted in our patient, who did not have this malformation.
Discussion
We here report a patient with isolated 1p13.2 microdeletion diagnosed by aCGH analysis. We performed exome sequencing in the patient in order to check for possible unmasking heterozygosity and to exclude variants in any of the genes related to Noonan Syndrome or other RASopathies. The analysis of complete exome sequencing (WES) identified 35 hemizygous variants in the deleted region. However, evaluation of frequency in the population and bioinformatics protein predictions did not indicate pathogenicity of these variants. Moreover, we did not find pathogenic variants in any of the genes known to be associated with Noonan Syndrome. Consequently, we conclude that the phenotype of our patient is exclusively related to his hemizygous 1p13.2 state.
We then tried to identify in the deletion region, genes possibly related with the phenotype of our patient and others. Our attention was drawn to the presence of deletion of NRAS for four reasons:
-
The phenotype of our patient and also of other patients with the 1p13.2 deletion includes several features of Noonan Syndrome (Table 1 and Figure 1);
-
Variants of NRAS have been described as causing Noonan Syndrome, Type 6 (OMIM #613224), an autosomal dominant disorder (Cirstea et al., 2010Cirstea IC, Kutsche K, Dvorsky R, Gremer L, Carta C, Horn D, Roberts AE, Lepri F, Merbitz-Zahradnik T, Konig R, et al. (2010) A restricted spectrum of NRAS mutations causes Noonan syndrome. Nat Genet 42:27-29.). However, these have been shown to be gain-of-function mutations;
-
Haploinsufficiency Index predictions (HI) shown in the DECIPHER database for all the genes deleted in our patient, revealed that the NRAS gene was the one with higher rank of HI (which suggested that this gene is more likely to exhibit haploinsufficiency) (Huang et al., 2010Huang N, Lee I, Marcotte EM and Hurles ME (2010) Characterising and predicting haploinsufficiency in the human genome. PLoS Genet 6:e1001154.);
-
The region surrounding NRAS is rarely seen in polymorphic CNVs (Figure 4).
NRAS is a member of the Ras subfamily, which includes 36 members; KRAS, HRAS and NRAS have been the most studied because of their critical roles in human oncogenesis (Wennerberg et al., 2005Wennerberg K, Rossman KL and Der CJ (2005) The Ras superfamily at a glance. J Cell Sci 118:843-846.). Ras proteins belong to a class of signal-transducing GTPases, which cycle between an active GTP-bound and an inactive GDP-bound conformation. Alterations of conditions, including gene variants, may result in stabilization of Ras proteins in their active state leading to malignant transformation (Scheffzek et al., 1997Scheffzek K, Ahmadian MR, Kabsch W, Wiesmuller L, Lautwein A, Schmitz F and Wittinghofer A (1997) The Ras-RasGAP complex: Structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science 277:333-338.). Ras proteins activate several pathways, including the Ras/mitogen activated protein kinase (MAPK) pathway (Wennerberg et al., 2005Wennerberg K, Rossman KL and Der CJ (2005) The Ras superfamily at a glance. J Cell Sci 118:843-846.). This pathway plays an essential role in regulating the cell survival, differentiation, proliferation and apoptosis, among many others (Pearson et al., 2001Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K and Cobb MH (2001) Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr Rev 22:153-183.).
Germline variants in genes that encode components or regulators of the Ras/MAPK pathway have been shown to cause developmental syndromes collectively referred to as RASopathies. These disorders include several forms of Noonan syndrome, including Costello syndrome (OMIM #218040), cardio-facio-cutaneous syndrome (OMIM #115150), and capillary malformation–arteriovenous malformation syndrome (OMIM #608354) (Rauen, 2013Rauen KA (2013) The RASopathies. Annu Rev Genomics Hum Genet 14:355-369.). In all of these cases the mutations have been shown to involve gain-of-function, increasing activity in the Ras/MAPK pathway. It is believed that this might be the reason why they are clinically similar, with overlapping phenotypic features including characteristic facial features, cardiac defects, cutaneous abnormalities, neurocognitive delay and an increased cancer risk (Tidyman and Rauen, 2009Tidyman WE and Rauen KA (2009) The RASopathies: Developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev 19:230-236.; Rauen, 2013Rauen KA (2013) The RASopathies. Annu Rev Genomics Hum Genet 14:355-369.).
Few heterozygous mutations have been described as cause of the Noonan type 6 (NS6) and they have all been associated with gain-of-function in NRAS (De Filippi et al., 2009De Filippi P, Zecca M, Lisini D, Rosti V, Cagioni C, Carlo-Stella C, Radi O, Veggiotti P, Mastronuzzi A, Acquaviva A, et al. (2009) Germ-line mutation of the NRAS gene may be responsible for the development of juvenile myelomonocytic leukaemia. Br J Haematol 147:706-709.; Cirstea et al., 2010Cirstea IC, Kutsche K, Dvorsky R, Gremer L, Carta C, Horn D, Roberts AE, Lepri F, Merbitz-Zahradnik T, Konig R, et al. (2010) A restricted spectrum of NRAS mutations causes Noonan syndrome. Nat Genet 42:27-29.; Runtuwene et al., 2011Runtuwene V, van Eekelen M, Overvoorde J, Rehmann H, Yntema HG, Nillesen WM, van Haeringen A, van der Burgt I, Burgering B and den Hertog J (2011) Noonan syndrome gain-of-function mutations in NRAS cause zebrafish gastrulation defects. Dis Model Mech 4:393-399.; Denayer et al., 2012Denayer E, Peeters H, Sevenants L, Derbent M, Fryns JP and Legius E (2012) NRAS Mutations in Noonan Syndrome. Mol Syndromol 3:34-38.; Kraoua et al., 2012Kraoua L, Journel H, Bonnet P, Amiel J, Pouvreau N, Baumann C, Verloes A and Cave H (2012) Constitutional NRAS mutations are rare among patients with Noonan syndrome or juvenile myelomonocytic leukemia. Am J Med Genet A 158A:2407-2411.; Ekvall et al., 2015Ekvall S, Wilbe M, Dahlgren J, Legius E, van Haeringen A, Westphal O, Anneren G and Bondeson ML (2015) Mutation in NRAS in familial Noonan syndrome—case report and review of the literature. BMC Med Genet 16:e95.). Cirstea et al. (2010)Cirstea IC, Kutsche K, Dvorsky R, Gremer L, Carta C, Horn D, Roberts AE, Lepri F, Merbitz-Zahradnik T, Konig R, et al. (2010) A restricted spectrum of NRAS mutations causes Noonan syndrome. Nat Genet 42:27-29. expressed each mutant as yellow fluorescent protein–NRAS fusion proteins in cells from the COS-7 line and showed for example that NRAS substitutions p.Thr50Ile or p.Gly60Glu resulted in enhanced phosphorylation of MEK and ERK in the presence of serum or after epidermal growth factor (EGF) stimulation. In addition, the authors observed that the p.Gly60Glu NRAS mutant accumulated constitutively in the active, GTP-bound form (Cirstea et al., 2010Cirstea IC, Kutsche K, Dvorsky R, Gremer L, Carta C, Horn D, Roberts AE, Lepri F, Merbitz-Zahradnik T, Konig R, et al. (2010) A restricted spectrum of NRAS mutations causes Noonan syndrome. Nat Genet 42:27-29.). Runtuwene et al. (2011)Runtuwene V, van Eekelen M, Overvoorde J, Rehmann H, Yntema HG, Nillesen WM, van Haeringen A, van der Burgt I, Burgering B and den Hertog J (2011) Noonan syndrome gain-of-function mutations in NRAS cause zebrafish gastrulation defects. Dis Model Mech 4:393-399. showed that similar effects occurs in GTP-bound in p.Ile24Asn mutants and they revealed that p.Gly60Glu, p.Ile24Asn and the positive control (p.Gly12Val) mutants also enhanced MAPK activation. These patients with NRAS mutations often show a relatively mild phenotype of typical Noonan features, including hypertelorism, low-set ears, short stature, webbed neck, low hairline, thorax deformities, motor delay/muscular hypotonia and lentigenes/cafe-au-lait spots (reviewed by Kraoua et al., 2012Kraoua L, Journel H, Bonnet P, Amiel J, Pouvreau N, Baumann C, Verloes A and Cave H (2012) Constitutional NRAS mutations are rare among patients with Noonan syndrome or juvenile myelomonocytic leukemia. Am J Med Genet A 158A:2407-2411.; Ekvall et al., 2015Ekvall S, Wilbe M, Dahlgren J, Legius E, van Haeringen A, Westphal O, Anneren G and Bondeson ML (2015) Mutation in NRAS in familial Noonan syndrome—case report and review of the literature. BMC Med Genet 16:e95.).
On the other hand, our patient and other cases with pure chromosome 1p13.2 microdeletions and hemizygosity for NRAS reviewed by us from the literature or from the DECIPHER database all seemed to present several clinical features of Noonan Syndrome (Table 1 and Figure 1). The presence of such features related to Noonan Syndrome in patients with deletion of NRAS and presumably haploinsufficiency of the gene product appears prima facie paradoxical.
We searched the literature and we have found three similar examples. In Noonan syndrome, Edwards et al. (2014)Edwards JJ, Martinelli S, Pannone L, Lo IF, Shi L, Edelmann L, Tartaglia M, Luk HM and Gelb BD (2014) A PTPN11 allele encoding a catalytically impaired SHP2 protein in a patient with a Noonan syndrome phenotype. Am J Med Genet A 164A:2351-2355. described a 5-year-old male with two de novo pathogenic PTPN11 variants in cis (OMIM *176876). The double-mutant gene product (SHP-2) was found to be catalytically impaired. More recently, Chen et al. (2014)Chen JL, Zhu X, Zhao TL, Wang J, Yang YF and Tan ZP (2014) Rare copy number variations containing genes involved in RASopathies: Deletion of SHOC2 and duplication of PTPN11. Mol Cytogenet 7:28. described a patient with a Noonan-like phenotype associated with a deletion of the 10q25.2 chromosomal region that included the SHOC2 gene (OMIM *602775). Gain-of-function mutations in this gene have been shown to cause a Noonan Syndrome-like syndrome. In the same vein, Nowaczyk et al. (2014)Nowaczyk MJ, Thompson BA, Zeesman S, Moog U, Sanchez-Lara PA, Magoulas PL, Falk RE, Hoover-Fong JE, Batista DA, Amudhavalli SM, et al. (2014) Deletion of MAP2K2/MEK2: A novel mechanism for a RASopathy? Clin Genet 85:138-146. reported seven patients with deletions of chromosome 19p13.3 including the MAP2K2 gene (OMIM *601263) and phenotype features of Cardio-facio-cutaneous syndrome, which is a RASopathy known to be caused by activating mutations of the BRAF, MAP2K1, MAP2K2, or KRAS genes. On these bases, these authors proposed that haploinsufficiency of MAP2K2/MEK2 "appears to be a new model of a RASopathy, where a deletion of one of the components of the pathway, MEK2, results in a RASopathy-like phenotype" (Nowaczyk et al., 2014Nowaczyk MJ, Thompson BA, Zeesman S, Moog U, Sanchez-Lara PA, Magoulas PL, Falk RE, Hoover-Fong JE, Batista DA, Amudhavalli SM, et al. (2014) Deletion of MAP2K2/MEK2: A novel mechanism for a RASopathy? Clin Genet 85:138-146.). We believe that the association here described of 1p13.2 microdeletion involving hemizygosity for NRAS and clinical features of Noonan syndrome, represent a further example of the model proposed by Nowaczyk et al. (2014)Nowaczyk MJ, Thompson BA, Zeesman S, Moog U, Sanchez-Lara PA, Magoulas PL, Falk RE, Hoover-Fong JE, Batista DA, Amudhavalli SM, et al. (2014) Deletion of MAP2K2/MEK2: A novel mechanism for a RASopathy? Clin Genet 85:138-146., according to which, haploinsufficiency of a gene in the Ras/MAPK pathway may cause dysregulation of the pathway and produce a RASopathy phenotype.
Acknowledgments
The authors are grateful to the patients and their family for their precious cooperation in this study. We thank Dr. Sérgio Pereira of The Centre for Applied Genomics, The Hospital for Sick Children (Toronto, Canada) for assistance with exome data interpretation. This work was supported by FAPEMIG grant to SDJP (process CDS-30/11). MCMF and RGCCLC were supported by a fellowship from CAPES and NDL was supported by a fellowship from CNPq. This study makes use of data generated by the DECIPHER Consortium. A full list of centres that contributed to the generation of the data is available from the website and via email from decipher@sanger.ac.uk. Funding for the DECIPHER project was provided by the Wellcome Trust.
References
- Albers CA, Paul DS, Schulze H, Freson K, Stephens JC, Smethurst PA, Jolley JD, Cvejic A, Kostadima M, Bertone P, et al (2012) Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nat Genet 44:435-439.
- Bisgaard AM, Rasmussen LN, Moller HU, Kirchhoff M and Bryndorf T (2007) Interstitial deletion of the short arm of chromosome 1 (1p13.1p21.1) in a girl with mental retardation, short stature and colobomata. Clin Dysmorphol 16:109-112.
- Cardenas RGCCL, Linhares ND and Pena SDJ (2015) Mendel, MD: A user-friendly online program for clinical exome analysis. BMC Bioinform 16 (Suppl 8):A2.
- Chen JL, Zhu X, Zhao TL, Wang J, Yang YF and Tan ZP (2014) Rare copy number variations containing genes involved in RASopathies: Deletion of SHOC2 and duplication of PTPN11. Mol Cytogenet 7:28.
- Cirstea IC, Kutsche K, Dvorsky R, Gremer L, Carta C, Horn D, Roberts AE, Lepri F, Merbitz-Zahradnik T, Konig R, et al (2010) A restricted spectrum of NRAS mutations causes Noonan syndrome. Nat Genet 42:27-29.
- Coman DJ and Gardner RJ (2007) Deletions that reveal recessive genes. Eur J Hum Genet 15:1103-1104.
- De Filippi P, Zecca M, Lisini D, Rosti V, Cagioni C, Carlo-Stella C, Radi O, Veggiotti P, Mastronuzzi A, Acquaviva A, et al (2009) Germ-line mutation of the NRAS gene may be responsible for the development of juvenile myelomonocytic leukaemia. Br J Haematol 147:706-709.
- Denayer E, Peeters H, Sevenants L, Derbent M, Fryns JP and Legius E (2012) NRAS Mutations in Noonan Syndrome. Mol Syndromol 3:34-38.
- Edwards JJ, Martinelli S, Pannone L, Lo IF, Shi L, Edelmann L, Tartaglia M, Luk HM and Gelb BD (2014) A PTPN11 allele encoding a catalytically impaired SHP2 protein in a patient with a Noonan syndrome phenotype. Am J Med Genet A 164A:2351-2355.
- Ekvall S, Wilbe M, Dahlgren J, Legius E, van Haeringen A, Westphal O, Anneren G and Bondeson ML (2015) Mutation in NRAS in familial Noonan syndrome—case report and review of the literature. BMC Med Genet 16:e95.
- Fitzgibbon GJ, Kingston H, Needham M and Gaunt L (2009) Haploinsufficiency of the nerve growth factor beta gene in a 1p13 deleted female child with an insensitivity to pain. Dev Med Child Neurol 51:833-837.
- Hochstenbach R, Poot M, Nijman IJ, Renkens I, Duran KJ, Van't Slot R, van Binsbergen E, van der Zwaag B, Vogel MJ, Terhal PA, et al (2012) Discovery of variants unmasked by hemizygous deletions. Eur J Hum Genet 20:748-753.
- Huang N, Lee I, Marcotte EM and Hurles ME (2010) Characterising and predicting haploinsufficiency in the human genome. PLoS Genet 6:e1001154.
- Kraoua L, Journel H, Bonnet P, Amiel J, Pouvreau N, Baumann C, Verloes A and Cave H (2012) Constitutional NRAS mutations are rare among patients with Noonan syndrome or juvenile myelomonocytic leukemia. Am J Med Genet A 158A:2407-2411.
- Mattia FR, Wardinsky TD, Tuttle DJ, Grix Jr. A, Smith KA and Walling P (1992) Interstitial deletion of the short arm of chromosome 1 (46XY, del(1)(p13p22.3)). Am J Med Genet 44:551-554.
- McDonald-McGinn DM, Fahiminiya S, Revil T, Nowakowska BA, Suhl J, Bailey A, Mlynarski E, Lynch DR, Yan AC, Bilaniuk LT, et al (2013) Hemizygous mutations in SNAP29 unmask autosomal recessive conditions and contribute to atypical findings in patients with 22q11.2DS. J Med Genet 50:80-90.
- Miller SA, Dykes DD and Polesky HF (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16:1215.
- Nowaczyk MJ, Thompson BA, Zeesman S, Moog U, Sanchez-Lara PA, Magoulas PL, Falk RE, Hoover-Fong JE, Batista DA, Amudhavalli SM, et al (2014) Deletion of MAP2K2/MEK2: A novel mechanism for a RASopathy? Clin Genet 85:138-146.
- Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K and Cobb MH (2001) Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr Rev 22:153-183.
- Poot M (2012) Beware of hemizygous deletions that may unmask deleterious variants. Mol Syndromol 3:45-46.
- Rauen KA (2013) The RASopathies. Annu Rev Genomics Hum Genet 14:355-369.
- Runtuwene V, van Eekelen M, Overvoorde J, Rehmann H, Yntema HG, Nillesen WM, van Haeringen A, van der Burgt I, Burgering B and den Hertog J (2011) Noonan syndrome gain-of-function mutations in NRAS cause zebrafish gastrulation defects. Dis Model Mech 4:393-399.
- Schaffer AA (2013) Digenic inheritance in medical genetics. J Med Genet 50:641-652.
- Scheffzek K, Ahmadian MR, Kabsch W, Wiesmuller L, Lautwein A, Schmitz F and Wittinghofer A (1997) The Ras-RasGAP complex: Structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science 277:333-338.
- Tidyman WE and Rauen KA (2009) The RASopathies: Developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev 19:230-236.
- Wennerberg K, Rossman KL and Der CJ (2005) The Ras superfamily at a glance. J Cell Sci 118:843-846.
- Yamagishi H, Ishii C, Maeda J, Kojima Y, Matsuoka R, Kimura M, Takao A, Momma K and Matsuo N (1998) Phenotypic discordance in monozygotic twins with 22q11.2 deletion. Am J Med Genet 78:319-321.
Internet Resources
- 1000 Genomes, http://browser.1000genomes.org (accessed June 2015).
» http://browser.1000genomes.org - Database of Genomic Variants (DGVbeta), http://dgvbeta.tcag.ca/dgv/app/home (accessed June 2015).
» http://dgvbeta.tcag.ca/dgv/app/home - DECIPHER database, https://decipher.sanger.ac.uk (accessed June 2015).
» https://decipher.sanger.ac.uk - Ensembl Genome Browser, http://www.ensembl.org/index.html (accessed June 2015).
» http://www.ensembl.org/index.html - Genome Analysis Toolkit (GATK), http://www.broadinstitute.org/gatk/ (accessed June 2015).
» http://www.broadinstitute.org/gatk/ - NCBI dnSNP, http://www.ncbi.nlm.nih.gov/projects/SNP/ (Accessed June 2015).
» http://www.ncbi.nlm.nih.gov/projects/SNP/ - NHLBI Exome Sequencing Project (ESP), http://evs.gs.washington.edu/EVS/ (Accessed June 2015).
» http://evs.gs.washington.edu/EVS/ - Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/ (accessed June 2015).
» http://www.omim.org/ - SIFT, http://sift.jcvi.org/ (accessed June 2015).
» http://sift.jcvi.org/ - SnpEff, http://snpeff.sourceforge.net/ (accessed June 2015).
» http://snpeff.sourceforge.net/
Supplementary material
The following online material is available for this article:
Table S1 - Hemizygous variants within the deleted region that passed in GATK quality filters.
Table S2 - Genes deleted in the patient according to the NCBI Map Viewer Annotation Release 105.
Table S3 - Deleted genes in our patient associated with Mendelian disease phenotypes in OMIM.
-
Associate Editor: Francisco Mauro Salzano
Data availability
Data citations
1000 Genomes, http://browser.1000genomes.org (accessed June 2015).
Database of Genomic Variants (DGVbeta), http://dgvbeta.tcag.ca/dgv/app/home (accessed June 2015).
DECIPHER database, https://decipher.sanger.ac.uk (accessed June 2015).
NCBI dnSNP, http://www.ncbi.nlm.nih.gov/projects/SNP/ (Accessed June 2015).
NHLBI Exome Sequencing Project (ESP), http://evs.gs.washington.edu/EVS/ (Accessed June 2015).
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/ (accessed June 2015).
Publication Dates
-
Publication in this collection
04 Aug 2016 -
Date of issue
Jul-Sep 2016
History
-
Received
29 Feb 2016 -
Accepted
09 May 2016