Abstract
Endogenous viral elements (EVEs) are the result of heritable horizontal gene transfer from viruses to hosts. In the last years, several EVE integration events were reported in plants by the exponential availability of sequenced genomes. Eucalyptus grandis is a forest tree species with a sequenced genome that is poorly studied in terms of evolution and mobile genetic elements composition. Here we report the characterization of E. grandis endogenous viral element 1 (EgEVE_1), a transcriptionally active EVE with a size of 5,664 bp. Phylogenetic analysis and genomic distribution demonstrated that EgEVE_1 is a newly described member of the Caulimoviridae family, distinct from the recently characterized plant Florendoviruses. Genomic distribution of EgEVE_1 and Florendovirus is also distinct. EgEVE_1 qPCR quantification in Eucalyptus urophylla suggests that this genome has more EgEVE_1 copies than E. grandis. EgEVE_1 transcriptional activity was demonstrated by RT-qPCR in five Eucalyptus species and one intrageneric hybrid. We also identified that Eucalyptus EVEs can generate small RNAs (sRNAs),that might be involved in de novo DNA methylation and virus resistance. Our data suggest that EVE families in Eucalyptus have distinct properties, and we provide the first comparative analysis of EVEs in Eucalyptus genomes.
Keywords:
Pararetrovirus; horizontal transfer; Eucalyptus; Caulimovirus; insertion
Introduction
In the last years, the burst of plant genome sequences has uncovered innumerable cases of horizontal gene transfer (HGT). HGT is the DNA flow between unrelated species. For many years, HGT events were considered rare and uncommon, but numerous genome analyses have since revealed the wide extent of HGT in plants (Richardson and Palmer, 2007Richardson AO and Palmer JD (2007) Horizontal gene transfer in plants. J Exp Bot 58:1-9.; Yue et al., 2012Yue J, Hu X, Sun H, Yang Y and Huang J (2012) Widespread impact of horizontal gene transfer on plant colonization of land. Nat Commun 3:1152.). Viruses play important roles in HGT, once many studies detected viral sequences integrated into several plant genomes (Bertsch et al., 2009Bertsch C, Beuve M, Dolja VV, Wirth M, Pelsy F, Herrbach E and Lemaire O (2009) Retention of the virus-derived sequences in the nuclear genome of grapevine as a potential pathway to virus resistance. Biol Direct 4:21.; Geering et al., 2014Geering ADW, Maumus F, Copetti D, Choisne N, Zwickl DJ, Zytnicki M, McTaggart AR, Scalabrin S, Vezzulli S, Wing RA, et al. (2014) Endogenous florendoviruses are major components of plant genomes and hallmarks of virus evolution. Nat Commun 5:5269.; Fonseca et al., 2016Fonseca GC, Oliveira LFV, Morais GL, Abdelnor RV, Nepomuceno AL, Waterhouse PM, Farinelli L and Margis R (2016) Unusual RNA plant virus integration in the soybean genome leads to the production of small RNAs. Plant Sci 246:62-69.). These viral DNA sequences present within the genomes of non-viral organisms are known as Endogenous Viral Elements (EVEs; Holmes, 2011Holmes EC (2011) The evolution of endogenous viral elements. Cell Host Microbe 10:368-377.). EVEs can consist of an entire viral genome or only a partial fragment (Chu et al., 2014Chu H, Jo Y and Cho WK (2014) Evolution of endogenous non-retroviral genes integrated into plant genomes. Curr Plant Biol 1:55-59.). The function of EVEs remains unclear, but some studies suggest a relationship between viral fragments in genomes and antiviral immunity (Aswad and Katzourakis 2013Aswad A and Katzourakis A (2013) Paleovirology and virally derived immunity. Trends Ecol Evolu 27:627-636.; Fonseca et al., 2016Fonseca GC, Oliveira LFV, Morais GL, Abdelnor RV, Nepomuceno AL, Waterhouse PM, Farinelli L and Margis R (2016) Unusual RNA plant virus integration in the soybean genome leads to the production of small RNAs. Plant Sci 246:62-69.). Genomic EVE regions can also act as generators of several types of virus-derived small RNAs (sRNAs; Sharma et al., 2013Sharma N, Sahu PP, Puranik S and Prasad M (2013). Recent advances in plant-virus interaction with emphasis on small interfering RNAs (siRNAs). Mol Biotechnol 55:63-77.) in some plant species (Becher et al., 2014Becher H, Ma L, Kelly LJ, Kovarik A, Leitch IJ and Leitch AR (2014) Endogenous pararetrovirus sequences associated with 24 nt small RNAs at the centromeres of Fritillaria imperialis L. (Liliaceae), a species with a giant genome. Plant J 80:823-833.; Fonseca et al., 2016Fonseca GC, Oliveira LFV, Morais GL, Abdelnor RV, Nepomuceno AL, Waterhouse PM, Farinelli L and Margis R (2016) Unusual RNA plant virus integration in the soybean genome leads to the production of small RNAs. Plant Sci 246:62-69.). The most abundant virus integrations in plants are from Caulimoviridae, a Pararetrovirus family. Using comparative genomics approaches, Caulimovirus-related sequences were identified in several angiosperms (Chabannes and Iskra-Caruana, 2013Chabannes M and Iskra-Caruana ML (2013) Endogenous pararetroviruses - a reservoir of virus infection in plants. Curr Opin Virol 3:8-13.), including Eucalyptus grandis, and they comprise a significant fraction of these plant genomes (Geering et al., 2014Geering ADW, Maumus F, Copetti D, Choisne N, Zwickl DJ, Zytnicki M, McTaggart AR, Scalabrin S, Vezzulli S, Wing RA, et al. (2014) Endogenous florendoviruses are major components of plant genomes and hallmarks of virus evolution. Nat Commun 5:5269.).
Previous works have already reported the serendipitous discovery of EVEs in plants during large-scale annotation of LTR retrotransposons (LTR-RTs) (Piednoël et al., 2013Piednoël M, Carrete-Veja G and Renners S (2013) Characterization of the LTR retrotransposon repertoire of a plant clade of six diploid and one tetraploid species. Plant J 75:699-709.) or during next generation sequencing analyses of genomes and transcriptomes (Villacreses et al., 2015Villacreses J, Rojas-Herrera M, Sanchez C, Hewstone N, Undurraga SF, Alzate JF, Manque P, Maracaja-Coutinho V, Polanco V (2015) Deep sequencing reveals the complete genome and evidence for transcriptional activity of the first virus-like sequences identified in Aristotelia chilensis (maqui berry). Viruses 7:1685-1699.; Fonseca et al., 2016Fonseca GC, Oliveira LFV, Morais GL, Abdelnor RV, Nepomuceno AL, Waterhouse PM, Farinelli L and Margis R (2016) Unusual RNA plant virus integration in the soybean genome leads to the production of small RNAs. Plant Sci 246:62-69.). A similar case happened during the annotation of transcriptionally active LTR-RTs in Eucalyptus (Marcon et al., 2015Marcon HS, Domingues DS, Silva JC, Borges RJ, Matioli FF, Fontes MRM and Marino CL (2015) Transcriptionally active LTR retrotransposons in Eucalyptus genus are differentially expressed and insertionally polymorphic. BMC Plant Biol 15:198.). An E. camaldulensis EST (GenBank accession FY783514), firstly identified because it contains a reverse transcriptase sequence, was in fact the fragment of a Caulimovirus. Alignment analysis of this sequence in the E. grandis genome (Myburg et al., 2014Myburg AA, Grattapaglia D, Tuskan GA, Hellsten U, Hayes RD, Grimwood J, Jenkins J, Lindquist E, Tice H, Bauer D, et al. (2014) The genome of Eucalyptus grandis. Nature 5410:356-362.) led us to the identification of a new EVE family in this genus. In this study, using publicly available genomic and transcriptomic E. grandis resources, we report the molecular characterization of this new EVE family, named E. grandis endogenous viral element 1 (EgEVE_1). We extended in silico analyses of EgEVE_1, carrying out comparative quantitative copy number analyses in two Eucalyptus species and performing transcriptional analysis in five Eucalyptus species and one intrageneric hybrid. We also compared EgEVE_1 to the Caulimoviridae genus called ‘Florendovirus’, recently identified in the E. grandis genome (Geering et al., 2014Geering ADW, Maumus F, Copetti D, Choisne N, Zwickl DJ, Zytnicki M, McTaggart AR, Scalabrin S, Vezzulli S, Wing RA, et al. (2014) Endogenous florendoviruses are major components of plant genomes and hallmarks of virus evolution. Nat Commun 5:5269.), in terms of phylogenetic position, genomic distribution and the capacity of generating sRNAs.
This study is the first fine-scale analysis of EVEs in Eucalyptus and an important step in the molecular characterization of mobile genetic elements in this woody plant genus.
Material and Methods
Virus-like sequences in Eucalyptus grandis genome
During the characterization of transcriptionally active LTR-RTs in the Eucalyptus genus (Marcon et al., 2015Marcon HS, Domingues DS, Silva JC, Borges RJ, Matioli FF, Fontes MRM and Marino CL (2015) Transcriptionally active LTR retrotransposons in Eucalyptus genus are differentially expressed and insertionally polymorphic. BMC Plant Biol 15:198.), we found a reverse transcriptase fragment in an E. camaldulensis EST sequence (GenBank accession FY783514). Similar to Piednoël et al. (2013)Piednoël M, Carrete-Veja G and Renners S (2013) Characterization of the LTR retrotransposon repertoire of a plant clade of six diploid and one tetraploid species. Plant J 75:699-709., after careful checking using CENSOR implemented in RepBase (Kohany et al., 2006Kohany O, Gentles AJ, Hankus L and Jurka J (2006) Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinformatics 7:474.), we discovered that this reverse transcriptase is in fact a partial sequence from a Caulimovirus.
Using the reverse transcriptase sequence of E. camaldulensis EST as a query, we identified a genomic region with high similarity (85% in BLASTN) in E. grandis genome scaffold 7. After manual checking of this hit using CENSOR and Repbase, we defined position 10,999,785 to 11,005,448 as a reference for further analyses. For a comparative analysis, we included four consensus Florendovirus sequences recently identified in E. grandis genome (Geering et al., 2014Geering ADW, Maumus F, Copetti D, Choisne N, Zwickl DJ, Zytnicki M, McTaggart AR, Scalabrin S, Vezzulli S, Wing RA, et al. (2014) Endogenous florendoviruses are major components of plant genomes and hallmarks of virus evolution. Nat Commun 5:5269.).
Conserved domains were identified using the CDD tool from NCBI (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) and were manually inspected.
Comparative analyses and endogenous viral family name assignment
To verify the relationships among EVEs, reverse transcriptase (RVT) regions were used to build a phylogenetic tree. RVT sequences for this analysis were the same as the ones used in a previous analysis of EVE sequences in the “Maqui Berry” genome (Villacreses et al., 2015Villacreses J, Rojas-Herrera M, Sanchez C, Hewstone N, Undurraga SF, Alzate JF, Manque P, Maracaja-Coutinho V, Polanco V (2015) Deep sequencing reveals the complete genome and evidence for transcriptional activity of the first virus-like sequences identified in Aristotelia chilensis (maqui berry). Viruses 7:1685-1699.). Nucleotide sequences were aligned using MUSCLE (Edgar, 2004Edgar RC (2004) MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792-7.) with default parameters, and the phylogenetic trees were generated using MEGA 7.0 (Kumar et al., 2016Kumar S, Stecher G and Tamura K (2016) MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870-1874.), applying the Maximum Likelihood method, with 1,000 bootstrap replicates. After performing a model test in MEGA, the General Time Reversible substitution model with Gamma distributed Invariant sites (GTR+I) was used. Gap positions were excluded when present in more than 5% of the sequences.
The new EVE found in E. grandis formed a novel lineage within the Caulimoviridae family and was, thus, named EgEVE1 (E. grandis Endogenous virus element 1). The Florendoviruses previously identified in E. grandis genome were named as EgFLOR (Florendovirus from E. grandis) 1 to 4.
Copy number determination in E. grandis genome and diversity analysis
The copy number of EgEVE1 and of four Florendovirus families found in E. grandis genome was determined using MEGABLAST similar to Marcon et al. (2015)Marcon HS, Domingues DS, Silva JC, Borges RJ, Matioli FF, Fontes MRM and Marino CL (2015) Transcriptionally active LTR retrotransposons in Eucalyptus genus are differentially expressed and insertionally polymorphic. BMC Plant Biol 15:198., using the 2.0 genome version deposited at Phytozome (https://phytozome.jgi.doe.gov/pz/portal.html). For copy number estimation, we considered only the ones that covered over 80% of the query and with nucleotide similarity over 80% after manual inspection.
The average divergence (Pi) in RVTs among EgEVE members was calculated using DnaSp program (Librado and Rozas 2009Librado P and Rozas J (2009) DnaSP v. 5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451-2.).
Eucalyptus spp EST screening
For an initial evaluation of the transcriptional activity of EgEVE1 and EgFLOR1-4, reference sequences were used as BLASTN queries against Eucalyptus ESTs from the EUCANEXT database (Nascimento et al., 2011Nascimento LC, Lepikson JN, Salazar MM, Camargo ELO, Marques WL, Gonçalves DC, Vidal RO, Pereira GAG and Carazzolle MF (2011) An integrated database of Eucalyptus spp. genome project. BMC Proc 5:170.; Salazar et al., 2013Salazar MM, Nascimento LC, Camargo ELO, Gonçalves DC, Lepikson Neto J, Marques WL, Teixeira PJPL, Mieczkowski P, Mondego JMC, et al. (2013) Xylem transcription profiles indicate potential metabolic responses for economically relevant characteristics of Eucalyptus species. BMC Genomics 14:201.; http://bioinfo03.ibi.unicamp.br/eucalyptusdb/), in a approach similar to that of Marcon et al. (2015)Marcon HS, Domingues DS, Silva JC, Borges RJ, Matioli FF, Fontes MRM and Marino CL (2015) Transcriptionally active LTR retrotransposons in Eucalyptus genus are differentially expressed and insertionally polymorphic. BMC Plant Biol 15:198..
EgEVE1 relative quantification and transcriptional analysis
A comparative quantification by qPCR of EgEVE1 reverse transcriptase was performed between the E. grandis and E. urophylla genomes, using a single-copy gene (DUR3) as a reference. Primers for EgEVE1 quantification were: EgEVE_RVT_F 5’-CCAAGATGATAAGTTCCC TTTACC-3’ and EgEVE_RVT_R 5’-GGTGGAATTTG GAATAGATGTGG-3’. We followed the same procedures used in a previous study from our group (Marcon et al., 2015Marcon HS, Domingues DS, Silva JC, Borges RJ, Matioli FF, Fontes MRM and Marino CL (2015) Transcriptionally active LTR retrotransposons in Eucalyptus genus are differentially expressed and insertionally polymorphic. BMC Plant Biol 15:198.). We also evaluated the transcriptional activity of EgEVE1 reverse transcriptase in E. grandis, E. brassiana, E. saligna, E. tereticornis, E. urophylla and in one hybrid E. grandis x E. urophylla (termed “E. urograndis”). RT-qPCR was used to identify transcriptional activity in leaves, stalks and secondary roots, in physiological conditions and under osmotic stress. Overall procedures for the qPCR assays, including normalization, and plant harvesting, were the same as those described in Marcon et al. (2015)Marcon HS, Domingues DS, Silva JC, Borges RJ, Matioli FF, Fontes MRM and Marino CL (2015) Transcriptionally active LTR retrotransposons in Eucalyptus genus are differentially expressed and insertionally polymorphic. BMC Plant Biol 15:198..
RT-qPCR efficiency was calculated using Linreg v. 2013.0 (Ruijter et al., 2009Ruijter JM, Ramakers C, Hoogaars WMH, Karlen Y, Bakker O, Van Den Hoff MJB and Moorman AFM (2009) Amplification efficiency: Linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res 37:1-12.), and reactions with efficiency > 90% were considered for further analysis. Relative expression was calculated using the ΔΔCt method (Livak and Schmittgen, 2001Livak KJ and Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25:402-408.) with the formula (1 + E)ΔΔCt, where E represents the efficiency. Statistical analysis was performed using Assistat 7.7 beta (Silva and Azevedo, 2009Silva FAS and Azevedo CAV (2009) Principal components analysis in the software assistat-statistical attendance. In: 7 World Congress on Computers in Agriculture. American Society of Agricultural and Biological Engineers, Reno, USA.). We used one-way analysis of variance (ANOVA), and in cases where significant differences were found, the Least Square Deviation (LSD) method for multiple comparisons was applied. Results were considered significant at P < 0.05. The tissue or organ with the lowest expression (highest Ct) was used as calibrator (expression value = 1).
Small RNA mapping analysis
Public data from E. grandis small RNA sequencing (Levy et al., 2014Levy A, Szwerdszarf D, Abu-Abied M, Mordehaev I, Yaniv Y, Riov J, Arazi T and Sadot E (2014) Profiling microRNAs in Eucalyptus grandis reveals no mutual relationship between alterations in miR156 and miR172 expression and adventitious root induction during development. BMC Genomics 15:524., NCBI accession GSE58367) was used to map small RNAs against virus-like sequences to check if they may be regulated by post-transcriptional pathways. This is the only publicly available sRNA sequencing data for this genus, comprising 6,891,830 valid reads, obtained from 14-day sterile seedlings.
Raw read quality was assessed using FastQC version 0.10.1 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Trimmomatic version 0.35 (Bolger et al., 2014Bolger AM, Lohse M and Usadel B (2014) Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30:2114-2120.) was used to preprocess raw reads. In summary, sequencing adapters, overrepresented sequences and reads < 16nt or > 28nt were removed. Only reads with average phred quality > 30 were maintained. Using the FASTQ/A Collapser tool from FASTX-Toolkit (http://hannonlab.cshl.edu/fastx-toolkit), we obtained non-redundant small RNA sequences. This set was filtered using Bowtie 2 (Langmead and Salzberg, 2012Langmead B and Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357-359.) under stringent parameters to remove sequences from chloroplasts, mitochondria, tRNAs, rRNAs and snoRNAs. Non-reduntant sequences were mapped against E. grandis chloroplast (GenBank Accession NC_014570) and ribosomal units obtained in the SILVA ribosomal RNA gene database (Quast et al., 2013Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J and Glöckner FO (2013) The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res 41:590-596.), Gossypium barbadense, Solanum lycopersicum and Vitis vinifera mitochondria (GenBank accessions AFYB00000000, NC_028254 and NC_012119), Populus tricocharpa and Vitis vinifera tRNAs from GtRNAdb (Chan and Lowe, 2009Chan PP and Lowe TM (2009) GtRNAdb: A database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res 37:D93-D97.) and snoRNAs from the Plant snoRNA database (http://bioinf.scri.sari.ac.uk/cgi-bin/plant_snorna/home). Sequences that matched these references were discarded for further analysis. After this filtering, a total of 615,801 non-redundant sequences were mapped against EgEVE1 and EgFLOR1-4 sequences under stringent parameters, with no gaps and mismatches, using Bowtie2.
Results
Identification of a novel EVE in Eucalyptus grandis genome
We identified a sequence highly similar to an endogenous caulimovirus in the E. grandis genome using an E. camaldulensis EST as query. After manual annotation of a reference region (scaffold7: 10999785 to 11005448), we determined an EVE region of 5,664 nucleotides (nt), named EgEVE_1.
EgEVE_1 has three retroviral domains: a reverse transcriptase (RVT- cd01647) with 539 nt, a ribonuclease H (RNase H - cd09274) with 362 nt, and a pepsin-like aspartate protease (PEP - cd00303) with 245 nt (Table 1). All domains were in the same reading frame of the sense strand.
Comparative phylogenetic structure shows that EgEVE_1 is not related to Florendoviruses
We classified EgEVE_1 among viral families using the RVT sequences from previous studies on plant EVEs (Geering et al., 2014Geering ADW, Maumus F, Copetti D, Choisne N, Zwickl DJ, Zytnicki M, McTaggart AR, Scalabrin S, Vezzulli S, Wing RA, et al. (2014) Endogenous florendoviruses are major components of plant genomes and hallmarks of virus evolution. Nat Commun 5:5269.; Villacreses et al., 2015Villacreses J, Rojas-Herrera M, Sanchez C, Hewstone N, Undurraga SF, Alzate JF, Manque P, Maracaja-Coutinho V, Polanco V (2015) Deep sequencing reveals the complete genome and evidence for transcriptional activity of the first virus-like sequences identified in Aristotelia chilensis (maqui berry). Viruses 7:1685-1699.) (Figure 1). EgEVE_1 is highly related to the Caulimoviridae family, especially with Petuvirus, clustering with PVCV and AcV1 (Villacreses et al., 2015Villacreses J, Rojas-Herrera M, Sanchez C, Hewstone N, Undurraga SF, Alzate JF, Manque P, Maracaja-Coutinho V, Polanco V (2015) Deep sequencing reveals the complete genome and evidence for transcriptional activity of the first virus-like sequences identified in Aristotelia chilensis (maqui berry). Viruses 7:1685-1699.). This position suggests that EgEVE_1 may be considered part of the same genus (Figure 1), although a more comprehensive characterization using related virus sequences and experimental assays are needed to better corroborate this hypothesis. More importantly, we could demonstrate that E. grandis Florendoviruses (EgFLOR1-4, Figure 1, Figure S1), the sole Eucalyptus EVEs identified up to date, belong to another clade. In this way we confirmed that EgEVE_1 is a new family of pararetroviruses identified in the E. grandis genome.
Phylogenetic analysis of reverse transcriptase domain from the Caulimoviridae family, endogenous pararetroviruses. Ty3 retrotransposon was used as an outgroup.
To further confirm that EgEVE_1 belongs to a new family, we identified complete sequences in E. grandis genome.
EgEVE_1 distribution in the E. grandis genome: a comparative analysis with Florendoviruses
We identified six copies of EgEVE_1 in the E. grandis genome on four E. grandis chromosomes. Copy numbers for EgFLOR families ranged from 2 to 26 (Table 2), reinforcing that they belong to another group of EVEs. Among Florendoviruses, EgFLOR_1 has the highest copy number, while EgFLOR_3 has only two copies (Table 2). In Table S1 (supplementary material) we detail coordinates of each complete copy for the EgEVE and EgFLOR families.
The diversity (Pi) of EgEVE_1 complete sequences was higher than the one observed for EgFLOR families (Table 2), reinforcing that they have dissimilar genomic features.
Comparative genomic quantification between E. grandis and E. urophylla
Since we were able to find EVEs in E. grandis using a congeneric species sequence as a query, we hypothesized that a qPCR analysis in a conserved region of EgEVE_1 would allow a comparative quantification of this EVE family among Eucalyptus species. In this way, having in mind that in silico analyses were based on the E. grandis genome, we used this species to run a comparative quantification of EgEVE_1 RVT domain by qPCR in E. urophylla, similar to the one performed by Marcon et al. (2015)Marcon HS, Domingues DS, Silva JC, Borges RJ, Matioli FF, Fontes MRM and Marino CL (2015) Transcriptionally active LTR retrotransposons in Eucalyptus genus are differentially expressed and insertionally polymorphic. BMC Plant Biol 15:198., using a single-copy gene as a reference. Our analyses suggest that E. urophylla could have more EgEVE_1 copies than E. grandis (Figure 2).
EgEVE_1 transcriptional activity in Eucalyptus species and in different organs
This is the first report on transcriptional activity of EVEs in forest trees. We BLAST searched the transcriptome of six Eucalyptus species deposited in the EUCANEXT database (Nascimento et al., 2011Nascimento LC, Lepikson JN, Salazar MM, Camargo ELO, Marques WL, Gonçalves DC, Vidal RO, Pereira GAG and Carazzolle MF (2011) An integrated database of Eucalyptus spp. genome project. BMC Proc 5:170.; Salazar et al., 2013Salazar MM, Nascimento LC, Camargo ELO, Gonçalves DC, Lepikson Neto J, Marques WL, Teixeira PJPL, Mieczkowski P, Mondego JMC, et al. (2013) Xylem transcription profiles indicate potential metabolic responses for economically relevant characteristics of Eucalyptus species. BMC Genomics 14:201.), using EgEVE_1 and EgFLOR1-4 as queries. We did not find any hit for EgFLOR1-4, indicating that Florendoviruses are not transcriptionally active in Eucalyptus genomes. EgEVE_1 only showed similarity with ESTs from E. calmadulensis (Supplementary material Table S2).
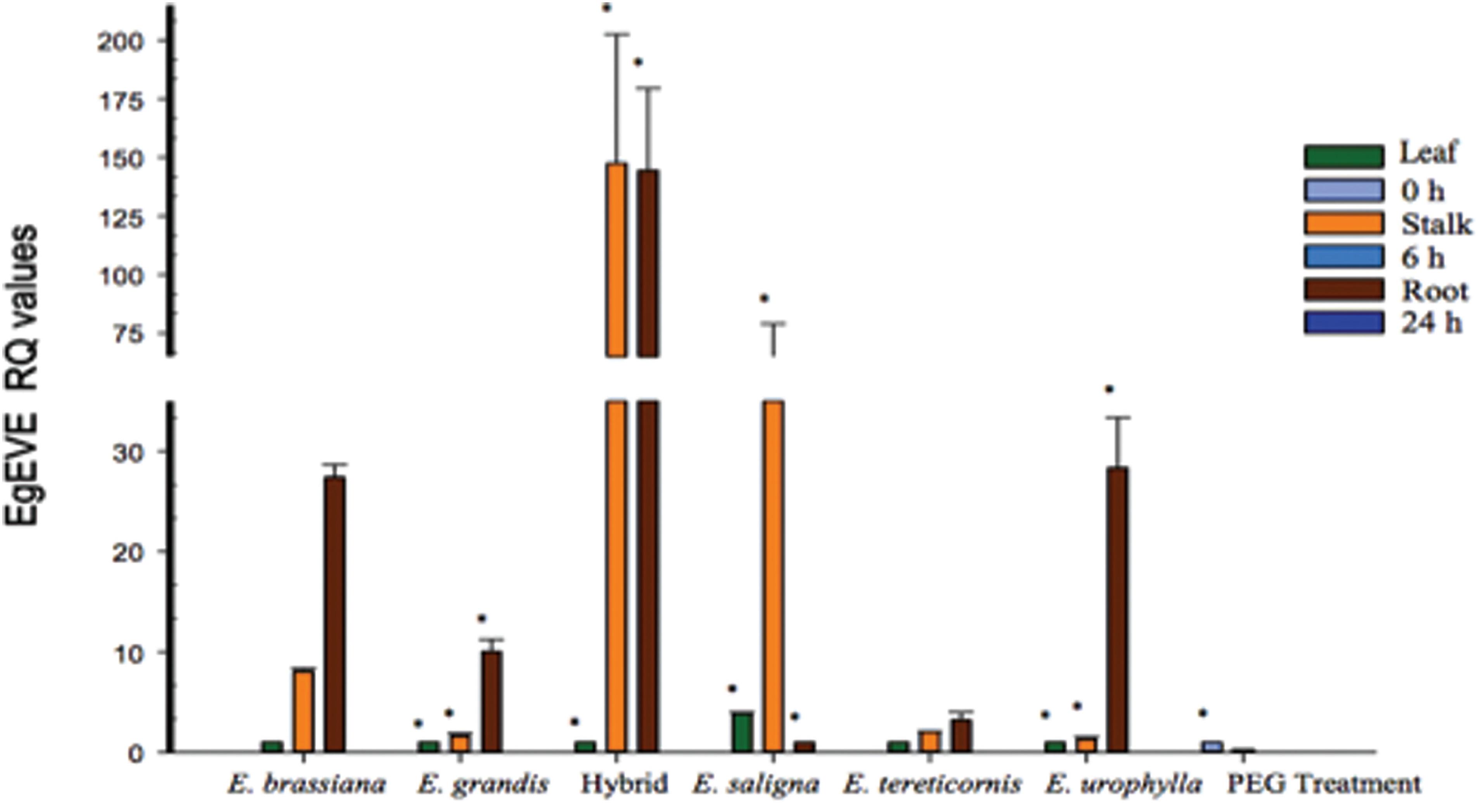
We also analyzed EgEVE_1 transcriptional levels using RT-qPCR for three tissues (leaves, stalk and secondary roots) from five Eucalyptus species (E. brassiana, E. grandis, E. saligna, E. tereticornis and E. urophylla) and one intrageneric hybrid (E. grandis x E. urophylla – termed “E. urograndis” to facilitate discussion). We also evaluated secondary roots from E. grandis under osmotic stress imposed by PEG treatment (Rodrigues et al., 2013Rodrigues MR, Bravo JP, Sassaki FT, Severino FE and Maia IG (2013) The tonoplast intrinsic aquaporin (TIP) subfamily of Eucalyptus grandis: Characterization of EgTIP2, a root-specific and osmoticstress-responsive gene. Plant Sci 213:106-113.) (Figure 3).
Transcriptional profile of EgEVE in three tissues from five Eucalyptus species and one interspecific hybrid using RT-qPCR. Asterisk indicates differential expression (*p ≤ 0.05, ANOVA followed by LSD test).
The highest transcriptional levels for EgEVE_1 were found in stalks and roots from E. urograndis and E. saligna (Figure 3). Interestingly, EgEVE_1 displayed low transcriptional activity in leaves (Figure 3). Considering that most transcriptome analyses use leaves, this may explain the lack of EgFLOR in expressed sequences. EgEVE_1 transcriptional levels were repressed in roots submitted to osmotic stress by PEG treatment (Figure 3).
Eucalyptus EVEs as sources of small RNAs
There is evidence that EVEs might act as sources of sRNAs, probably shaping epigenic features and/or having a role on antiviral defenses (Becher et al., 2014Becher H, Ma L, Kelly LJ, Kovarik A, Leitch IJ and Leitch AR (2014) Endogenous pararetrovirus sequences associated with 24 nt small RNAs at the centromeres of Fritillaria imperialis L. (Liliaceae), a species with a giant genome. Plant J 80:823-833.; Geering et al., 2014Geering ADW, Maumus F, Copetti D, Choisne N, Zwickl DJ, Zytnicki M, McTaggart AR, Scalabrin S, Vezzulli S, Wing RA, et al. (2014) Endogenous florendoviruses are major components of plant genomes and hallmarks of virus evolution. Nat Commun 5:5269.; Fonseca et al., 2016Fonseca GC, Oliveira LFV, Morais GL, Abdelnor RV, Nepomuceno AL, Waterhouse PM, Farinelli L and Margis R (2016) Unusual RNA plant virus integration in the soybean genome leads to the production of small RNAs. Plant Sci 246:62-69.). To check if Eucalyptus EVEs could be involved in sRNA production, we mapped filtered non-redundant sRNAs ranging from 16 to 26 nt with zero mismatches to consensus EVE sequences (EgEVE_1 and EgFLOR1-4). Although the numbers of sRNAs matches are probably underestimated due to polymorphisms between reference copies and genomic sequences, this analysis can provide an initial overview of sRNA production in Eucalyptus EVEs.
We mapped a total of 727 sRNA reads (Figure 4; Table S3). EgEVE_1 was the element with most mapped reads (434) and EgFLOR2 had the lowest number of mapped reads (16), and the most abundant class for all EVEs was 24-nt sRNAs (Supplementary material Table S3; Figure 4), a class usually associated to transposable elements and repetitive sequences and involved in RNA-directed DNA methylation (Zhang and Zhu, 2011Zhang H and Zhu JK (2011) RNA-directed DNA methylation. Curr Opin Plant Biol 14:142-147.; Parent et al., 2012Parent J-S, Martínez de Alba AE and Vaucheret H (2012) The origin and effect of small RNA signaling in plants. Frontiers Plant Sci 3:179.). 24 nt sRNAs have already been associated to plant-integrated pararetroviruses (Becher et al., 2014Becher H, Ma L, Kelly LJ, Kovarik A, Leitch IJ and Leitch AR (2014) Endogenous pararetrovirus sequences associated with 24 nt small RNAs at the centromeres of Fritillaria imperialis L. (Liliaceae), a species with a giant genome. Plant J 80:823-833.).
Size variation of sRNAs (16 to 26 nucleotiotides) according to EVE. (A) EgEVE_1, (B) EgFLOR_1, (C) EgFLOR_2, (D) EgFLOR_3, (E) EgFLOR_4.
In all EVEs, most sRNAs were mapped in the 3’ region. In the case EgEVE_1, we observed a clustered mapping of sRNAs in RVT and RNAseH regions (Figure 5), suggesting a prominent role of these regions in sRNA regulation.
Eucalyptus grandis small RNA reads distribution along EVE reference sequences. (A) EgEVE_1, (B) EgFLOR_1, (C) EgFLOR_2, (D) EgFLOR_3, (D) EgFLOR_4.
Discussion
Our data report the first transcriptionally active EVE in the E. grandis genome, EgEVE_1, using bioinformatics and experimental approaches. Similar to EVEs that were recently described in several plant genomes (Chabannes and Iskra-Caruana, 2013Chabannes M and Iskra-Caruana ML (2013) Endogenous pararetroviruses - a reservoir of virus infection in plants. Curr Opin Virol 3:8-13.; Becher et al., 2014Becher H, Ma L, Kelly LJ, Kovarik A, Leitch IJ and Leitch AR (2014) Endogenous pararetrovirus sequences associated with 24 nt small RNAs at the centromeres of Fritillaria imperialis L. (Liliaceae), a species with a giant genome. Plant J 80:823-833.; Villacreses et al., 2015Villacreses J, Rojas-Herrera M, Sanchez C, Hewstone N, Undurraga SF, Alzate JF, Manque P, Maracaja-Coutinho V, Polanco V (2015) Deep sequencing reveals the complete genome and evidence for transcriptional activity of the first virus-like sequences identified in Aristotelia chilensis (maqui berry). Viruses 7:1685-1699.), EgEVE_1 is also classified as being close to the genus Petuvirus within the Caulimoviridae.
We could not recover the Gag domain of EgEVE_1 in any of its genomic copies. Such arrangements of fragmented copies dispersed at several genomic loci have been also described in EVEs from Musa and Nicotiana species (Chabannes and Iskra-Caruana, 2013Chabannes M and Iskra-Caruana ML (2013) Endogenous pararetroviruses - a reservoir of virus infection in plants. Curr Opin Virol 3:8-13.). On the other hand, Eucalyptus Florendoviruses (EgFLOR families) are bigger than EgEVE_1 (7731 to 7854 bp), with similar size when compared to other pararetroviruses (Calvert et al., 1995Calvert LA, Ospina MD and Shepherd RJ (1995) Characterization of cassava vein mosaic virus: A distinct plant pararetrovirus. J Genet 76:1271-1276.; Villacreses et al., 2015Villacreses J, Rojas-Herrera M, Sanchez C, Hewstone N, Undurraga SF, Alzate JF, Manque P, Maracaja-Coutinho V, Polanco V (2015) Deep sequencing reveals the complete genome and evidence for transcriptional activity of the first virus-like sequences identified in Aristotelia chilensis (maqui berry). Viruses 7:1685-1699.).
EgEVE_1 is clearly distinct from the EgFLOR families by phylogenetic (Figure 1) and genomic (Table 1) analysis. Floredoviruses also contain a domain that encodes a putative protein of unknown function (Figure 5).
The quantification of genomic repetitive units by comparative qPCR has been performed in several species (Baruch and Kashkush, 2012Baruch O and Kashkush K (2012) Analysis of copy-number variation, insertional polymorphism, and methylation status of the tiniest class I (TRIM) and class II (MITE) transposable element families in various rice strains. Plant Cell Rep 31:885-893.; Yaakov et al., 2013Yaakov B, Ben-David S and Kashkush K (2013) Genome-wide analysis of stowaway-like MITEs in wheat reveals high sequence conservation, gene association, and genomic diversification. Plant Physiol 161:486-496.; Marcon et al., 2015Marcon HS, Domingues DS, Silva JC, Borges RJ, Matioli FF, Fontes MRM and Marino CL (2015) Transcriptionally active LTR retrotransposons in Eucalyptus genus are differentially expressed and insertionally polymorphic. BMC Plant Biol 15:198.). The genomes of E. grandis (1C = 630 Mb) and E. urophylla (1C = 640 Mb) are of similar size and diverged < 20 Mya (Myburg et al., 2014Myburg AA, Grattapaglia D, Tuskan GA, Hellsten U, Hayes RD, Grimwood J, Jenkins J, Lindquist E, Tice H, Bauer D, et al. (2014) The genome of Eucalyptus grandis. Nature 5410:356-362.), making them a good congeneric pair for comparative analyses of EgEVE_1 distribution in the two genomes. EgEVE_1 showed more copies (approximately four times more) in the E. urophylla genome than in E. grandis, suggesting recombination and/or recurrent invasion of this EVE family.
The transcriptomic data associated with EgEVE_1 and EgFLOR families gave an initial overall picture of transcriptional activity of these elements in Eucalyptus genomes. EgFLOR families seem to have a very low transcriptional activity, since we could find transcripts for only one family. Further experimental analyses using other organs should better address the question of whether these elements are in fact “silent” components of Eucalyptus genomes.
To our knowledge, EgEVE_1 is the transcriptionally most active EVE found in a Eucalyptus genome up to date. Furthermore, RT-qPCR analyses also showed that EgEVE_1 has transcriptional activity differences among Eucalyptus spp. tissues and species (Figure 3).
The transcriptional activity of EgEVE_1 suggests that this family can act in small interference RNA (siRNA) pathways mediated by virus infections. This feature has also been observed in RT-qPCR analyses of other pararetroviruses in different plants, which revealed a low level of transcription associated to asymptomatic plants under normal growth conditions (Noreen et al., 2007Noreen F, Akbergenov R, Hohn T and Richert-Pöggeler KR (2007) Distinct expression of endogenous Petunia vein clearing virus and the DNA transposon dTph1 in two Petunia hybrida lines is correlated with differences in histone modification and siRNA production. Plant J 50:219-229.; Villacreses et al., 2015Villacreses J, Rojas-Herrera M, Sanchez C, Hewstone N, Undurraga SF, Alzate JF, Manque P, Maracaja-Coutinho V, Polanco V (2015) Deep sequencing reveals the complete genome and evidence for transcriptional activity of the first virus-like sequences identified in Aristotelia chilensis (maqui berry). Viruses 7:1685-1699.). In support of this hypothesis, our sRNA analysis showed that the “24 nt pattern” is prevalent in all analyzed of EVEs, which are known to be associated to viral siRNA pathways (Sharma et al., 2013Sharma N, Sahu PP, Puranik S and Prasad M (2013). Recent advances in plant-virus interaction with emphasis on small interfering RNAs (siRNAs). Mol Biotechnol 55:63-77.) and de novo DNA methylation (Blevins et al., 2015Blevins T, Podicheti R, Mishra V, Marasco M, Tang H and Pikaard CS (2015) Identification of Pol IV and RDR2-dependent precursors of 24 nt siRNAs guiding de novo DNA methylation in Arabidopsis. eLife 4:e09591.), thus also explaining the observed low transcriptional activity of EgFLOR families due to methylation. In this way, EVE sequences integrated in the Eucalyptus genome may have roles in both DNA methylation patterns, as well as virus-plant interactions, warranting further studies on the impact of EVEs under biotic stress conditions.
In summary, this first fine-scale analysis of EVE integration in Eucalyptus species highlighted the importance of mobile elements in reshaping genomes and providing molecular tools to confer viral resistances in a tree genome.
Acknowledgments
We thank Drs. Ivan de Godoy Maia and Juliana Pereira Bravo for their assistance in transcriptional analyses and for providing plant material; to Izabel Gava and Shinitiro Oda who kindly provided access to plant material at Suzano Papel e Celulose. This work was supported by a CNPq grant (474123/2010-3). HSM was also supported by fellowships from CAPES and CNPq. CLM and DSD are research fellows of CNPq.
References
- Aswad A and Katzourakis A (2013) Paleovirology and virally derived immunity. Trends Ecol Evolu 27:627-636.
- Baruch O and Kashkush K (2012) Analysis of copy-number variation, insertional polymorphism, and methylation status of the tiniest class I (TRIM) and class II (MITE) transposable element families in various rice strains. Plant Cell Rep 31:885-893.
- Becher H, Ma L, Kelly LJ, Kovarik A, Leitch IJ and Leitch AR (2014) Endogenous pararetrovirus sequences associated with 24 nt small RNAs at the centromeres of Fritillaria imperialis L. (Liliaceae), a species with a giant genome. Plant J 80:823-833.
- Bertsch C, Beuve M, Dolja VV, Wirth M, Pelsy F, Herrbach E and Lemaire O (2009) Retention of the virus-derived sequences in the nuclear genome of grapevine as a potential pathway to virus resistance. Biol Direct 4:21.
- Blevins T, Podicheti R, Mishra V, Marasco M, Tang H and Pikaard CS (2015) Identification of Pol IV and RDR2-dependent precursors of 24 nt siRNAs guiding de novo DNA methylation in Arabidopsis. eLife 4:e09591.
- Bolger AM, Lohse M and Usadel B (2014) Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30:2114-2120.
- Calvert LA, Ospina MD and Shepherd RJ (1995) Characterization of cassava vein mosaic virus: A distinct plant pararetrovirus. J Genet 76:1271-1276.
- Chabannes M and Iskra-Caruana ML (2013) Endogenous pararetroviruses - a reservoir of virus infection in plants. Curr Opin Virol 3:8-13.
- Chan PP and Lowe TM (2009) GtRNAdb: A database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res 37:D93-D97.
- Chu H, Jo Y and Cho WK (2014) Evolution of endogenous non-retroviral genes integrated into plant genomes. Curr Plant Biol 1:55-59.
- Edgar RC (2004) MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792-7.
- Fonseca GC, Oliveira LFV, Morais GL, Abdelnor RV, Nepomuceno AL, Waterhouse PM, Farinelli L and Margis R (2016) Unusual RNA plant virus integration in the soybean genome leads to the production of small RNAs. Plant Sci 246:62-69.
- Geering ADW, Maumus F, Copetti D, Choisne N, Zwickl DJ, Zytnicki M, McTaggart AR, Scalabrin S, Vezzulli S, Wing RA, et al. (2014) Endogenous florendoviruses are major components of plant genomes and hallmarks of virus evolution. Nat Commun 5:5269.
- Holmes EC (2011) The evolution of endogenous viral elements. Cell Host Microbe 10:368-377.
- Kohany O, Gentles AJ, Hankus L and Jurka J (2006) Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinformatics 7:474.
- Kumar S, Stecher G and Tamura K (2016) MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870-1874.
- Langmead B and Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357-359.
- Levy A, Szwerdszarf D, Abu-Abied M, Mordehaev I, Yaniv Y, Riov J, Arazi T and Sadot E (2014) Profiling microRNAs in Eucalyptus grandis reveals no mutual relationship between alterations in miR156 and miR172 expression and adventitious root induction during development. BMC Genomics 15:524.
- Librado P and Rozas J (2009) DnaSP v. 5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451-2.
- Livak KJ and Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25:402-408.
- Marcon HS, Domingues DS, Silva JC, Borges RJ, Matioli FF, Fontes MRM and Marino CL (2015) Transcriptionally active LTR retrotransposons in Eucalyptus genus are differentially expressed and insertionally polymorphic. BMC Plant Biol 15:198.
- Myburg AA, Grattapaglia D, Tuskan GA, Hellsten U, Hayes RD, Grimwood J, Jenkins J, Lindquist E, Tice H, Bauer D, et al. (2014) The genome of Eucalyptus grandis Nature 5410:356-362.
- Nascimento LC, Lepikson JN, Salazar MM, Camargo ELO, Marques WL, Gonçalves DC, Vidal RO, Pereira GAG and Carazzolle MF (2011) An integrated database of Eucalyptus spp. genome project. BMC Proc 5:170.
- Noreen F, Akbergenov R, Hohn T and Richert-Pöggeler KR (2007) Distinct expression of endogenous Petunia vein clearing virus and the DNA transposon dTph1 in two Petunia hybrida lines is correlated with differences in histone modification and siRNA production. Plant J 50:219-229.
- Parent J-S, Martínez de Alba AE and Vaucheret H (2012) The origin and effect of small RNA signaling in plants. Frontiers Plant Sci 3:179.
- Piednoël M, Carrete-Veja G and Renners S (2013) Characterization of the LTR retrotransposon repertoire of a plant clade of six diploid and one tetraploid species. Plant J 75:699-709.
- Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J and Glöckner FO (2013) The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res 41:590-596.
- Richardson AO and Palmer JD (2007) Horizontal gene transfer in plants. J Exp Bot 58:1-9.
- Rodrigues MR, Bravo JP, Sassaki FT, Severino FE and Maia IG (2013) The tonoplast intrinsic aquaporin (TIP) subfamily of Eucalyptus grandis: Characterization of EgTIP2, a root-specific and osmoticstress-responsive gene. Plant Sci 213:106-113.
- Ruijter JM, Ramakers C, Hoogaars WMH, Karlen Y, Bakker O, Van Den Hoff MJB and Moorman AFM (2009) Amplification efficiency: Linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res 37:1-12.
- Salazar MM, Nascimento LC, Camargo ELO, Gonçalves DC, Lepikson Neto J, Marques WL, Teixeira PJPL, Mieczkowski P, Mondego JMC, et al. (2013) Xylem transcription profiles indicate potential metabolic responses for economically relevant characteristics of Eucalyptus species. BMC Genomics 14:201.
- Sharma N, Sahu PP, Puranik S and Prasad M (2013). Recent advances in plant-virus interaction with emphasis on small interfering RNAs (siRNAs). Mol Biotechnol 55:63-77.
- Silva FAS and Azevedo CAV (2009) Principal components analysis in the software assistat-statistical attendance. In: 7 World Congress on Computers in Agriculture. American Society of Agricultural and Biological Engineers, Reno, USA.
- Villacreses J, Rojas-Herrera M, Sanchez C, Hewstone N, Undurraga SF, Alzate JF, Manque P, Maracaja-Coutinho V, Polanco V (2015) Deep sequencing reveals the complete genome and evidence for transcriptional activity of the first virus-like sequences identified in Aristotelia chilensis (maqui berry). Viruses 7:1685-1699.
- Yaakov B, Ben-David S and Kashkush K (2013) Genome-wide analysis of stowaway-like MITEs in wheat reveals high sequence conservation, gene association, and genomic diversification. Plant Physiol 161:486-496.
- Yue J, Hu X, Sun H, Yang Y and Huang J (2012) Widespread impact of horizontal gene transfer on plant colonization of land. Nat Commun 3:1152.
- Zhang H and Zhu JK (2011) RNA-directed DNA methylation. Curr Opin Plant Biol 14:142-147.
-
Associate Editor: Marcia Pinheiro Margis
Supplementary material
The following online material is available for this article:
Table S1 - Distribution of EVE elements in Eucalyptus grandis genome.
Table S2 - Expressed sequence tags (ESTs) matching to EVE elements.
Table S3 - sRNAs mapped to Eucalyptus EVEs.
Figure S1 - Phylogenetic analysis of reverse transcriptase domain from EgEVE copies.
Publication Dates
-
Publication in this collection
23 Feb 2017 -
Date of issue
2017
History
-
Received
25 Apr 2016 -
Accepted
10 Oct 2016