ABSTRACT
CONTEXT:
Hirschsprung disease is a developmental disorder of the enteric nervous system that is characterized by absence of ganglion cells in the distal intestine, and it occurs in approximately 1 in every 500,000 live births. Hepatoblastoma is a malignant liver neoplasm that usually occurs in children aged 6 months to 3 years, with a prevalence of 0.54 cases per 100,000.
CASE REPORT:
A boy diagnosed with intestinal atresia in the first week of life progressed to a diagnosis of comorbid Hirschsprung disease. Congenital cataracts and sensorineural deafness were diagnosed. A liver mass developed and was subsequently confirmed to be a hepatoblastoma, which was treated by means of surgical resection of 70% of the liver volume and neoadjuvant chemotherapy (ifosfamide, cisplatin and doxorubicin).
CONCLUSION:
It is known that Hirschsprung disease may be associated with syndromes predisposing towards cancer, and that hepatoblastoma may also be associated with certain congenital syndromes. However, co-occurrence of hepatoblastoma and Hirschsprung disease has not been previously described. We have reported a case of a male patient born with ileal atresia, Hirschsprung disease and bilateral congenital cataract who was later diagnosed with hepatoblastoma.
KEY WORDS
Hirschsprung disease; Hepatoblastoma; Intestinal atresia; Hearing loss, sensorineural; Cataract
RESUMO
CONTEXTO:
A doença de Hirschsprung é uma desordem do desenvolvimento do sistema nervoso entérico, que é caracterizada pela ausência de células ganglionares no intestino distal, ocorrendo em cerca de 1 a cada 500.000 nascimentos. O hepatoblastoma é uma neoplasia maligna do fígado que geralmente ocorre em crianças de 6 meses a 3 anos, com prevalência de 0,54 casos por 100.000.
RELATO DE CASO:
Um menino com diagnóstico de atresia intestinal na primeira semana de vida evoluiu com diagnóstico concomitante de doença de Hirschsprung. Catarata congênita e surdez neurossensorial foram diagnosticadas. Surgiu lesão hepática com posterior confirmação de hepatoblastoma, tratado com ressecção cirúrgica de 70% do volume hepático e quimioterapia neoadjuvante (ifosfamida, cisplatina e doxorubicina).
CONCLUSÃO:
Sabe-se que a doença de Hirschsprung pode estar associada a síndromes de predisposição ao câncer, da mesma forma que o hepatoblastoma já foi correlacionado a certas síndromes congênitas malformativas. No entanto, até o momento, a associação de hepatoblastoma com a doença de Hirschsprung não foi descrita. Relatamos o caso de um menino que nasceu com atresia ileal, doença de Hirschsprung, catarata congênita bilateral e com posterior diagnóstico de hepatoblastoma.
PALAVRAS-CHAVE
Doença de Hirschsprung; Hepatoblastoma; Atresia intestinal; Perda auditiva neurossensorial; Catarata
INTRODUCTION
Hirschsprung disease is an unusual, but well-recognized cause of chronic constipation in children. It occurs in approximately 1 in every 500,000 live births, and most commonly presents as a neonatal bowel obstruction. However, in older children, it may present as chronic constipation or enterocolitis.1Haricharan RN, Georgeson KE. Hirschsprung disease. Semin Pediatr Surg. 2008;17(4):266-75. Hirschsprung disease occurs as an isolated trait in 70% of the patients, is associated with a chromosomal abnormality in 12% and occurs with additional congenital anomalies in 18%.2Amiel J, Sproat-Emison E, Garcia-Barcelo M , et al. Hirschsprung disease, associated syndromes and genetics: a review. J Med Genet. 2008;45(1):1-14.
Primary hepatic malignancies account for approximately 1% of cancers in children, and can be divided into two major histological subgroups: hepatoblastoma and hepatocellular carcinoma.3Finegold MJ, Egler RA, Goss JA et al. Liver tumors: pediatric population . Liver Transpl.2008;14(11):1545-56. The overall prevalence of hepatoblastoma is 0.54 per 100,000 individuals, and it occurs primarily in children younger than 5 years of age.4Orphant Report Series.Rare Diseases collection. Prevalence of rare diseases: bibliographic data. Listed in alphabetical order of disease or group diseases. 2014;1. Available from: http://www.orpha.net/ orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_ alphabetical_list.pdf. Accessed in 2014 (Dec 2).
http://www.orpha.net/ orphacom/cahiers/d...
In Brazil, the median age-adjusted incidence rate (AAIR) of hepatoblastoma ranged from 0.0 to 2.8 per million in a study that included data from 13 cities; notably, the highest incidence was found in our city (Porto Alegre), with a median AAIR of 2.78.5Langer JC. Hirschsprung disease. Curr Opin Pediatr.2013;25(3):368-74. We report a case of hepatoblastoma in a child previously diagnosed with ileal atresia and Hirschsprung disease, which is an unusual association.
CASE REPORT
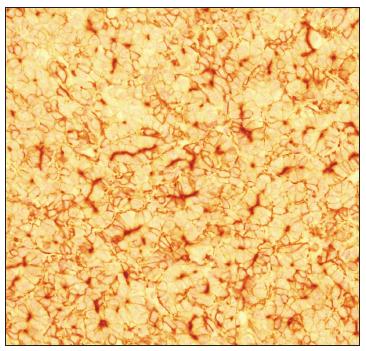
A male infant was born after 28 weeks of gestation with a birth weight of 910 grams and Apgar scores of 4 at the first minute and 7 at the fifth minute. At 2 days of age, still without bowel movements, he developed abdominal distension and vomiting. Abdominal radiography showed severe small-bowel distension and wall edema without pneumoperitoneum. Oral feeding was discontinued and antibiotics and total parenteral nutrition were started due to clinical suspicion of necrotizing enterocolitis. A barium enema revealed a state of microcolon due to disuse. On laparotomy, intestinal atresia in the terminal ileum and a disconnected cecum were identified. Ileostomy and cecostomy were performed, and a set of biopsies was obtained, going from the transverse colon to the rectum. Histopathological examination revealed absence of ganglion cells in the rectum and sigmoid colon, consistent with Hirschsprung disease (Figure 1). A Duhamel procedure was performed, with total colectomy due to absence of ganglion cells throughout the colon, which was identified during frozen section examination.
Hirschsprung disease: myenteric plexus lacking ganglion cells (hematoxylin-eosin staining, 400 x).
During a routine physical examination, the patient was diagnosed with congenital cataracts, which were surgically corrected. Genetic evaluation was normal and TORCH (Toxoplasma gondii, rubella, cytomegalovirus, herpes simples and other viruses) complex screening was negative. An echocardiogram showed a large patent ductus arteriosus with hemodynamic impairment. Indomethacin treatment was unsuccessful, and surgical closure was performed. Furthermore, the patient was evaluated by a speech therapist and deafness was detected.
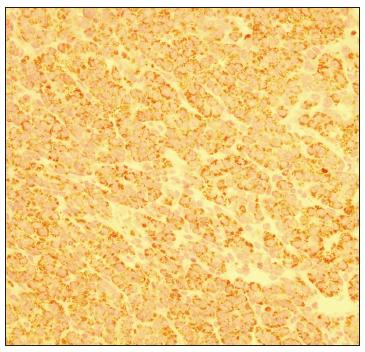
At the age of 25 months, during computed tomography (CT) on the chest to evaluate a lung malformation, a tumor in the right hepatic lobe measuring 5.6 cm x 4.3 cm, with marked contrast uptake, was incidentally observed (Figure 2). A liver biopsy was performed, and subsequent immunohistochemical examination of the biopsy specimen revealed epithelial-type hepatoblastoma (Figures 3, 4 and 5). The patient was started on neoadjuvant chemotherapy with ifosfamide, cisplatin and doxorubicin (four cycles). At that time, the alpha-fetoprotein (AFP) level was 8229 ng/ml (reference range: < 10 ng/ml). An abdominal CT scan performed after chemotherapy showed a significant reduction in tumor volume (3.2 cm x 2.6 cm). Right hepatectomy was performed, leaving a residual liver of 30%, followed by two cycles of adjuvant chemotherapy. Currently, the patient is asymptomatic, with no evidence of tumor recurrence, he has normal bowel movements and normal AFP level (2.79 ng/ml).
Computed tomography (CT) scan of the abdomen. A space-occupying lesion is visible in the liver, located between the middle and right hepatic veins (segment VIII).
Hepatoblastoma immunohistochemistry: positive for carcinoembryonic antigen (CEA), with canalicular pattern, 400 x.
Hepatoblastoma immunohistochemistry: cytoplasmic positivity for Hep Par1, with granular pattern, 400 x.
DISCUSSION
Hirschsprung disease is a developmental disorder of the enteric nervous system that is characterized by absence of ganglion cells in the myenteric (Auerbach's) and submucosal (Meissner's) plexuses of the distal intestine, which results in lack of peristalsis and functional intestinal obstruction.6Butler Tjaden NE, Trainor PA. The development etiology and pathogenesis of Hirschsprung disease.Transl Res. 2013;162(1):1-15. In 80-85% of the cases, the aganglionic region is limited to the rectum and sigmoid colon, as in our patient.7Weinberg AG, Currarino G, Besserman AM. Hirschsprung's disease and congenital deafness. Familial association. Hum Genet. 1977;38(2):157-61.
The heterogeneous nature of Hirschsprung disease seems to be supported by evidence of mutations in a variety of genes. The most commonly identified gene is the RET proto-oncogene, which is commonly found in familial and long-segment disease. It remains unclear how these mutations result in aganglionosis, but there is some evidence that early neuronal cell death may be a prominent mechanism. Hirschsprung disease is associated with a variety of other congenital abnormalities: malrotation, genitourinary abnormalities, congenital heart disease, limb abnormalities, mental retardation and dysmorphic features.6Butler Tjaden NE, Trainor PA. The development etiology and pathogenesis of Hirschsprung disease.Transl Res. 2013;162(1):1-15. Many of these patients also have other abnormalities of neural crest-derived tissues, such as pigmentation disorders and sensorineural deafness, including Waardenburg syndrome.6Butler Tjaden NE, Trainor PA. The development etiology and pathogenesis of Hirschsprung disease.Transl Res. 2013;162(1):1-15. However, the association of Hirschsprung disease with profound congenital deafness in the absence of other syndromic features, as in our patient, has been reported before.8Skinner R, Irvine D. Hirschsprung's disease and congenital deafness. J Med Genet.1973;10(4):337-9. 9Litten JB, Tomlinson GE. Liver tumors in children . Oncologist . 2008;13(7):812-20. In the present case, our patient presented with sensorineural deafness and bilateral congenital cataracts, but no pigmentation disorders.
Hirschsprung disease may also be associated with syndromes predisposing towards cancer, such as familial medullary thyroid carcinoma, multiple endocrine neoplasia type 2A and type 2B and neuroblastoma. A review of the literature was conducted through an online search for the MeSH, EMTREE and MeSH/DeCS terms "Hirschsprung disease" and "hepatoblastoma" in PubMed, Embase (via Elsevier) and LILACS (via Bireme), respectively (Table 1), but did not find any previous reports of comorbid Hirschsprung disease and hepatoblastoma. On the other hand, hepatoblastoma may also be associated with certain congenital syndromes (such as Beckwith-
Wiedemann syndrome and trisomy 18 syndrome).10Honeyman JN, La Quaglia MP. Malignant liver tumors. Semin Pediatr Surg.2012;21(3):245-54.
11Surveillance, Epidemiology, and End Results Program. Previous Version: SEER Cancer Statistics Review, 1975-2010. Available from: http://seer.cancer.gov/csr/1975_2010/sections.html. Accessed in 2014 ( Dec 2).
http://seer.cancer.gov/csr/1975_2010/sec...
The incidence of hepatoblastoma in the United States (2.2 cases per 1 million children aged 0-14 years, over the period 2006201012Bulterys M, Goodman MT, Smith MA, Buckley JD. Hepatic tumors. In: Ries LA, Smith MA, Gurney JG, et al., eds. Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. National Cancer Institute, SEER Program, 1999. NIH Pub. No. 99-4649. p. 91-8. Available from: http://seer.cancer.gov/archive/publications/childhood/childhood-monograph.pdf. Accessed in 2014 (Dec 2).
http://seer.cancer.gov/archive/publicati...
) appears to have doubled over recent decades.13Ikeda H, Hachitanda Y, Tanimura M , et al. Development of unfavorable hepatoblastoma in children of very low birth weight: results of a surgical and pathologic review. Cancer. 1998;82(9):1789-96. The cause of this increase in incidence is unknown, but it may be related to increasing survival of very low birth weight premature infants.14de Camargo B, de Oliveira Ferreira JM, de Souza Reis R, et al. Socioeconomic status and the incidence of non-central nervous system childhood embryonic tumours in Brazil. BMC. Cancer 2011;11:160. In Brazil, there have been very few cases, and they are recorded in only 8 of the 14 population-based cancer registries. The incidence appears to be highest in the central-western region of the country.14de Camargo B, de Oliveira Ferreira JM, de Souza Reis R, et al. Socioeconomic status and the incidence of non-central nervous system childhood embryonic tumours in Brazil. BMC. Cancer 2011;11:160.
15de Camargo B , de Oliveira Santos M, Rebelo MS, et al. Cancerincidence among children and adolescents in Brazil: first report of 14 population-based cancer registries. Int J. Cancer 2010;126(3):715-20. The patient in this case report met the criteria for the highest risk of hepatoblastoma (male, white and extremely premature, with birth weight < 1 kg).16Tanimura M, Matsui I, Abe J , et al. Increased risk of hepatoblastoma among immature children with a lower birth weight. Cancer Res. 1998;58(14):3032-5.
17Schnater JM, Köhler SE, Lamers WH, et al. Where do we stand with hepatoblastoma? A review.. Cancer 2003;98(4):668-78.
One sensitive but nonspecific biomarker for the presence of hepatoblastoma is AFP. This is a useful clinical marker for monitoring treatment effectiveness and tumor recurrence, since 90% of the patients at diagnosis have highly elevated serum levels of AFP.11Surveillance, Epidemiology, and End Results Program. Previous Version: SEER Cancer Statistics Review, 1975-2010. Available from: http://seer.cancer.gov/csr/1975_2010/sections.html. Accessed in 2014 ( Dec 2).
http://seer.cancer.gov/csr/1975_2010/sec...
Because the liver has excellent regeneration capacity, up to 80% of this organ can be resected.3Finegold MJ, Egler RA, Goss JA et al. Liver tumors: pediatric population . Liver Transpl.2008;14(11):1545-56. The goal of therapy for hepatoblastoma is complete surgical resection3Finegold MJ, Egler RA, Goss JA et al. Liver tumors: pediatric population . Liver Transpl.2008;14(11):1545-56. (which was the result achieved in the case reported here), because the majority of patients survive if a hepatoblastoma is removed completely. The overall 5-year survival rate for children with hepatoblastoma is 70%.12Bulterys M, Goodman MT, Smith MA, Buckley JD. Hepatic tumors. In: Ries LA, Smith MA, Gurney JG, et al., eds. Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. National Cancer Institute, SEER Program, 1999. NIH Pub. No. 99-4649. p. 91-8. Available from: http://seer.cancer.gov/archive/publications/childhood/childhood-monograph.pdf. Accessed in 2014 (Dec 2).
http://seer.cancer.gov/archive/publicati...
Metastases are found in approximately 20% of patients at diagnosis (usually in the lungs, central nervous system (CNS) and eyes).15de Camargo B , de Oliveira Santos M, Rebelo MS, et al. Cancerincidence among children and adolescents in Brazil: first report of 14 population-based cancer registries. Int J. Cancer 2010;126(3):715-20.
CONCLUSION
We have reported a case of an unusual association of hepatoblastoma in a child with previous diagnoses of Hirschsprung disease, ileal atresia, deafness and cataracts. Complete resection of the tumor was achieved, with favorable clinical evolution. We emphasize the importance of comprehensive assessment of patients with Hirschsprung disease, due to the possibility of several chromosomal abnormalities and associated congenital anomalies.
REFERENCES
- Haricharan RN, Georgeson KE. Hirschsprung disease. Semin Pediatr Surg. 2008;17(4):266-75.
- Amiel J, Sproat-Emison E, Garcia-Barcelo M , et al. Hirschsprung disease, associated syndromes and genetics: a review. J Med Genet. 2008;45(1):1-14.
- Finegold MJ, Egler RA, Goss JA et al. Liver tumors: pediatric population . Liver Transpl.2008;14(11):1545-56.
- Orphant Report Series.Rare Diseases collection. Prevalence of rare diseases: bibliographic data. Listed in alphabetical order of disease or group diseases. 2014;1. Available from: http://www.orpha.net/ orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_ alphabetical_list.pdf Accessed in 2014 (Dec 2).
» http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf - Langer JC. Hirschsprung disease. Curr Opin Pediatr.2013;25(3):368-74.
- Butler Tjaden NE, Trainor PA. The development etiology and pathogenesis of Hirschsprung disease.Transl Res. 2013;162(1):1-15.
- Weinberg AG, Currarino G, Besserman AM. Hirschsprung's disease and congenital deafness. Familial association. Hum Genet. 1977;38(2):157-61.
- Skinner R, Irvine D. Hirschsprung's disease and congenital deafness. J Med Genet.1973;10(4):337-9.
- Litten JB, Tomlinson GE. Liver tumors in children . Oncologist . 2008;13(7):812-20.
- Honeyman JN, La Quaglia MP. Malignant liver tumors. Semin Pediatr Surg.2012;21(3):245-54.
- Surveillance, Epidemiology, and End Results Program. Previous Version: SEER Cancer Statistics Review, 1975-2010. Available from: http://seer.cancer.gov/csr/1975_2010/sections.html Accessed in 2014 ( Dec 2).
» http://seer.cancer.gov/csr/1975_2010/sections.html - Bulterys M, Goodman MT, Smith MA, Buckley JD. Hepatic tumors. In: Ries LA, Smith MA, Gurney JG, et al., eds. Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. National Cancer Institute, SEER Program, 1999. NIH Pub. No. 99-4649. p. 91-8. Available from: http://seer.cancer.gov/archive/publications/childhood/childhood-monograph.pdf Accessed in 2014 (Dec 2).
» http://seer.cancer.gov/archive/publications/childhood/childhood-monograph.pdf - Ikeda H, Hachitanda Y, Tanimura M , et al. Development of unfavorable hepatoblastoma in children of very low birth weight: results of a surgical and pathologic review. Cancer. 1998;82(9):1789-96.
- de Camargo B, de Oliveira Ferreira JM, de Souza Reis R, et al. Socioeconomic status and the incidence of non-central nervous system childhood embryonic tumours in Brazil. BMC. Cancer 2011;11:160.
- de Camargo B , de Oliveira Santos M, Rebelo MS, et al. Cancerincidence among children and adolescents in Brazil: first report of 14 population-based cancer registries. Int J. Cancer 2010;126(3):715-20.
- Tanimura M, Matsui I, Abe J , et al. Increased risk of hepatoblastoma among immature children with a lower birth weight. Cancer Res. 1998;58(14):3032-5.
- Schnater JM, Köhler SE, Lamers WH, et al. Where do we stand with hepatoblastoma? A review.. Cancer 2003;98(4):668-78.
-
Sources of funding: None
Publication Dates
-
Publication in this collection
09 Oct 2015 -
Date of issue
Mar-Apr 2016
History
-
Received
07 July 2014 -
Accepted
03 Nov 2014