
Resumos
Quatro diferentes tipos de população foram simulados com o objetivo de verificar a influência de genes de efeito principal e do tamanho da população na estimação de componentes de variância sob seleção. A estimação foi realizada por meio da utilização e comparação das metodologias clássica e Bayesiana (a Bayesiana com três níveis de informação a priori). As metodologias REML e Bayesiana com prior não-informativo, em geral, produziram resultados bastante semelhantes. Em populações cuja característica é governada por genes de efeito principal, as estimativas dos componentes de variância genética aditiva foram pouco acuradas, exceto quando se utilizou metodologia Bayesiana com prior informativo. A inclusão das informações de parentesco e dos registros de todos os indivíduos até a população-base mostrou-se necessária, exceto para populações grandes cuja característica é governada por elevado número de genes.
amostragem de Gibbs; modelo infinitesimal; modelos mistos; REML; simulação computacional; viés da seleção
This study aimed to evaluate the effects of major genes and population size on variance components estimation using four different types of selected populations. Variance components were estimated by classical and Bayesian methodologies, with three a priori information levels. In general, results from REML and Bayesian analyses with flat priors were similar. Except for Bayesian analysis with an informative prior, additive genetic variance estimates were not accurate in populations in which the trait is controlled by major genes. The use of pedigree information and records of all individuals back to the base-population was necessary to improve accuracy of variance component estimates, except for large populations in which the trait is controlled by a large number of genes.
computational simulation; Gibbs sampling; infinitesimal model; mixed models; REML; selection bias
MELHORAMENTO, GENÉTICA E REPRODUÇÃO
Estimação de componentes de variância sob influência de genes de efeito principal, comparando-se metodologias Bayesiana e clássica sob diferentes cenários
Estimation of variance components under major genes influence, comparing Bayesian and classical methodologies under different scenarios
Giselle Mariano Lessa de AssisI; José Marques Carneiro JúniorI; Ricardo Frederico EuclydesII; Robledo de Almeida TorresII; Paulo Sávio LopesII
IEmbrapa Acre, Rodovia BR 364, km 14, CP 321, CEP: 69908-970 Rio Branco - AC
IIDZO/UFV, Departamento de Zootecnia, 36571-000, Viçosa, MG
RESUMO
Quatro diferentes tipos de população foram simulados com o objetivo de verificar a influência de genes de efeito principal e do tamanho da população na estimação de componentes de variância sob seleção. A estimação foi realizada por meio da utilização e comparação das metodologias clássica e Bayesiana (a Bayesiana com três níveis de informação a priori). As metodologias REML e Bayesiana com prior não-informativo, em geral, produziram resultados bastante semelhantes. Em populações cuja característica é governada por genes de efeito principal, as estimativas dos componentes de variância genética aditiva foram pouco acuradas, exceto quando se utilizou metodologia Bayesiana com prior informativo. A inclusão das informações de parentesco e dos registros de todos os indivíduos até a população-base mostrou-se necessária, exceto para populações grandes cuja característica é governada por elevado número de genes.
Palavras-chave: amostragem de Gibbs, modelo infinitesimal, modelos mistos, REML, simulação computacional, viés da seleção
ABSTRACT
This study aimed to evaluate the effects of major genes and population size on variance components estimation using four different types of selected populations. Variance components were estimated by classical and Bayesian methodologies, with three a priori information levels. In general, results from REML and Bayesian analyses with flat priors were similar. Except for Bayesian analysis with an informative prior, additive genetic variance estimates were not accurate in populations in which the trait is controlled by major genes. The use of pedigree information and records of all individuals back to the base-population was necessary to improve accuracy of variance component estimates, except for large populations in which the trait is controlled by a large number of genes.
Key Words: computational simulation, Gibbs sampling, infinitesimal model, mixed models, REML, selection bias
Introdução
No melhoramento animal, tem-se empregado nas avaliações genéticas a Metodologia de Modelos Mistos (Henderson, 1973) para obtenção do melhor preditor linear não-viesado (BLUP), cuja predição depende normalmente da estimação dos componentes de variância, que tem sido realizada por meio do Método da Máxima Verossimilhança Restrita (REML). Ambas as metodologias se baseiam na adequabilidade do modelo infinitesimal para a característica em questão. No entanto, estudos de mapeamento e detecção de locos de características quantitativas (QTL) têm comprovado a existência de genes com efeitos expressivos (não mais infinitesimalmente pequenos) que governam características quantitativas (Barton & Keightley, 2002). Assim, as metodologias usualmente utilizadas na avaliação genética que se baseiam no modelo infinitesimal podem não ser as mais adequadas.
Outra pressuposição para o uso apropriado da Metodologia de Modelos Mistos é que os dados não sejam oriundos de seleção. No entanto, os dados disponíveis para predição de valores genéticos e para realização de inferências sobre os parâmetros da população são obtidos a campo a partir de rebanhos nos quais os acasalamentos não são realizados ao acaso pelos produtores, os quais selecionam os animais e determinam cruzamentos específicos. No intuito de eliminar ou minimizar o viés causado pela seleção, a análise genética deve incluir a matriz de parentesco completa até a população-base, além dos registros de todos os animais (Pollak & Quaas, 1981; Sorensen & Kennedy, 1984). No entanto, esses estudos com populações selecionadas são realizados considerando que as pressuposições implícitas ao modelo infinitesimal estão sendo atendidas.
Como alternativa às análises clássicas, metodologias baseadas na inferência Bayesiana têm sido empregadas no melhoramento animal (Gianola & Fernando, 1986; Blasco, 2001), inclusive na avaliação de dados sob seleção (Sorensen et al., 1994; Van Tassell et al., 1995), e necessitam ser avaliadas e comparadas aos métodos usuais (Shenkel, 1998). Modelos alternativos, como o poligênico finito, ou sua combinação com o modelo poligênico infinitesimal, têm sido propostos, porém, ainda são pouco empregados na estimação de componentes de variância (Bink, 2002; Gonçalves et al., 2005a; Gonçalves et al., 2005b).
Assim, é importante verificar como a estimação dos componentes de variância em populações selecionadas e não-selecionadas é afetada quando a característica é governada por poucos genes.
Objetivou-se com este trabalho verificar a influência de genes de efeito principal e do tamanho da população na estimação dos componentes de variância genética aditiva, comparando-se as metodologias Bayesiana e clássica, em populações sob seleção, por meio de simulação. Verificou-se ainda, pela metodologia Bayesiana, a influência de diferentes níveis de informação a priori nas estimativas dos componentes de variância.
Material e Métodos
Os dados utilizados na elaboração desse trabalho foram simulados em nível de gene, utilizando-se o pacote computacional Genesys (Euclydes, 1996), composto de nove programas escritos em linguagem Fortran.
Quatro diferentes tipos de população foram simulados: 1) GRAN 900: população grande, constituída de gerações compostas, cada uma de 2.400 indivíduos com registros e 1.000 pais, em que a característica sob seleção é governada por 900 pares de locos; 2) GRAN 10: população grande, constituída de gerações compostas, cada uma, de 2.400 indivíduos com registros e 1.000 pais, em que a característica sob seleção é governada por dez pares de locos; 3) PEQ 900: população pequena, constituída de gerações compostas, cada uma, de 120 indivíduos com registros e 50 pais, em que a característica sob seleção é governada por 900 pares de locos; 4) PEQ 10: população pequena, constituída de gerações compostas, cada uma, de 120 indivíduos com registros e 50 pais, em que a característica sob seleção é governada por dez pares de locos.
Inicialmente, foram simulados quatro distintos genomas para formação de cada uma das populações. Para os genomas simulados, foram estabelecidos locos bialélicos, cujas freqüências iniciais foram simuladas com base na distribuição normal, com valor médio inicial igual a 0,50. Apenas os efeitos aditivos dos locos quantitativos foram simulados conforme distribuição normal, sendo ignorados os efeitos de dominância e epistasia. As freqüências gênicas iniciais foram iguais em ambos os sexos. Foram simulados dois efeitos fixos, A e B, o primeiro com 15 e o segundo com seis subclasses. Os efeitos de ambiente não-controláveis foram simulados com base na distribuição normal. O processo de simulação empregado permitiu a geração de ligações gênicas entre determinados locos nas populações em que a característica é governada por 900 genes e, contrariamente, gerou locos não ligados nas populações em que a característica é governada por apenas dez genes (Tabela 1).
A partir da população-base, dois processos seletivos foram aplicados: seleção ao acaso (SAA), em que os indivíduos foram selecionados aleatoriamente; e seleção fenotípica (SF), em que os indivíduos foram selecionados com base no seu valor fenotípico, considerando que a seleção visa aumentar a média da população. Foram avançadas dez gerações discretas para cada um dos processos seletivos, em que o número de machos e fêmeas selecionados e o de descendentes deixados a cada geração foram iguais aos apresentados na formação da população-base (Tabela 1).
Para as populações GRAN 900 e GRAN 10, foram feitas 300 repetições e para as populações PEQ 900 e PEQ 10 foram realizadas 500 repetições para cada um dos processos seletivos na obtenção das dez gerações. Assim, os valores genéticos e ambientais para cada indivíduo a cada geração são valores médios obtidos de 300 ou 500 repetições, conforme o tamanho da população.
Duas metodologias foram empregadas para estimar os componentes de variância: Máxima Verossimilhança Restrita (REML) e Análise Bayesiana, utilizando a Amostragem de Gibbs. Foram utilizados para obtenção das estimativas REML os programas do pacote MTDFREML Multiple Trait Drivative-Free Restricted Maximum Likelihood (Boldman et al., 1995). A análise Bayesiana foi realizada utilizando-se o programa MTGSAM Multiple Trait Gibbs Sampling in Animal Models (Van Tassell & Van Vleck, 1995), composto por um conjunto de programas escritos em FORTRAN, desenvolvido para estimação de componentes de variância por meio da Amostragem de Gibbs.
Os componentes de variância foram estimados por ambas as metodologias para as seguintes situações: 1) G0: foram utilizados os registros dos indivíduos da geração 0 para estimar os componentes de variância dessa geração; 2) G10 (SAA): foram utilizados os registros dos indivíduos da geração 10 de seleção ao acaso e informação de parentesco sem registro dos pais dos indivíduos da geração 10 para estimar os componentes de variância da geração 0; 3) G10 (SF): foram utilizados os registros dos indivíduos da geração 10 de seleção fenotípica e informação de parentesco sem registro dos pais dos indivíduos da geração 10 para estimar os componentes de variância da geração 0; 4) G0-10 (SAA): foram utilizados todos os registros e informações de parentesco completas da geração 10 até a geração 0 de seleção ao acaso para estimar os componentes de variância da geração 0; 5) G0-10 (SF): foram utilizados todos os registros e informações de parentesco completas da geração 10 até a geração 0 de seleção fenotípica para estimar os componentes de variância da geração 0.
O seguinte modelo linear misto unicaracterístico foi utilizado na análise dos dados simulados:
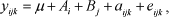
em que yijk = observação fenotípica do k-ésimo indivíduo no i-ésimo efeito fixo A e no j-ésimo efeito fixo B; µ = constante inerente a todas as observações; Ai = efeito do i-ésimo efeito fixo A; Bj = efeito do j-ésimo efeito fixo B; aijk = efeito aleatório genético aditivo do k-ésimo indivíduo no i-ésimo efeito fixo A e no j-ésimo efeito fixo B; e eijk = efeito aleatório ambiental. Matricialmente, esse modelo pode ser representado como:

em que y = vetor de observações; b = vetor de efeitos fixos desconhecidos; a = vetor de efeitos aleatórios desconhecidos que representam os valores genéticos aditivos de cada animal; e = vetor de efeitos aleatórios residuais desconhecidos; e X e Z = matrizes de incidência que relacionam os registros aos efeitos fixos e aleatórios genéticos, respectivamente.
As pressuposições acerca da distribuição de y, a e e podem ser descritas como:

em que G = matriz de variâncias e covariâncias dos efeitos aleatórios do vetor a; R = matriz de variâncias e covariâncias residuais. As matrizes G e R são descritas como:
 em que A é a matriz que indica o grau de parentesco entre os indivíduos e
em que A é a matriz que indica o grau de parentesco entre os indivíduos e  , a variância genética aditiva da característica.
, a variância genética aditiva da característica.
 em que I é a matriz identidade de ordem igual à dimensão linha de y e
em que I é a matriz identidade de ordem igual à dimensão linha de y e  , o componente de variância residual.
, o componente de variância residual.
Na análise Bayesiana, é necessário especificar também as distribuições a priori para todos os efeitos do modelo. O programa MTGSAM foi desenvolvido utilizando-se flat priors (distribuições uniformes) para os efeitos fixos, ou seja, considerando que há um vago conhecimento a priori sobre esses efeitos. Pressupõe-se que os efeitos aleatórios, inclusive os ambientais, seguem distribuição normal, de modo que, para os efeitos genéticos, existe uma pressuposição adicional sobre uma conhecida estrutura de covariâncias, representada pela matriz de parentesco. Adicionalmente, pressupõe-se que os componentes de variância genética e ambiental seguem distribuição de qui-quadrado invertida escalonada (Sorensen et al., 1994).
A derivação das distribuições condicionais completas, necessárias para a implementação da Amostragem de Gibbs, é descrita por Van Tassell & Van Vleck (1995).
Três níveis de informação a priori em relação aos componentes de variância foram considerados nessa metodologia: não-informativo (NI), pouco informativo (PI) e informativo (I). Para o nível não-informativo, esses valores resultaram em distribuição uniforme para as variâncias. Para os níveis pouco informativo e informativo, os valores do hiperparâmetro  corresponderam aos valores reais dos componentes de variância e os valores do hiperparâmetro Viforam definidos fixando-se o valor de desejado e alterando-se os de Vi, de modo que maior ou menor grau de confiança fosse atribuído às variâncias informadas. Esse procedimento foi realizado por meio da análise gráfica das distribuições de qui-quadrado invertidas escalonadas.
corresponderam aos valores reais dos componentes de variância e os valores do hiperparâmetro Viforam definidos fixando-se o valor de desejado e alterando-se os de Vi, de modo que maior ou menor grau de confiança fosse atribuído às variâncias informadas. Esse procedimento foi realizado por meio da análise gráfica das distribuições de qui-quadrado invertidas escalonadas.
Uma única cadeia de Gibbs foi gerada para cada análise. A inspeção visual das amostras geradas a cada iteração e os programas GIBBSIT (Raftery & Lewis, 1992) e GIBANAL (Van Kaam, 1998) foram utilizados para definir o tamanho da cadeia, o período de burn-in e o intervalo de amostragem (thinning interval) para obtenção das densidades marginais a posteriori. Para as populações GRAN 900 e GRAN 10, o tamanho da cadeia variou de 35.000 a 70.000 amostras e o período de burn-in de 5.000 a 10.000 iterações. Para a população PEQ 900, o tamanho da cadeia variou de 35.000 a 10.000.000 amostras e o período de burn-in de 5.000 a 5.000.000 iterações e, para a população PEQ 10, o tamanho da cadeia variou de 120.000 a 10.000.000 amostras e o período de burn-in de 20.000 a 5.000.000 iterações.
A porcentagem do erro obtida na estimação dos componentes de variância genética aditiva e ambiental foi calculada para as populações das gerações G0, G10 e G0 a G10, tanto para seleção ao acaso quanto para seleção fenotípica. Como referência, foram utilizados os valores reais dos respectivos componentes da população-base (G0). Assim:

em que  é o componente de variância estimado e , o componente de variância real da população-base.
é o componente de variância estimado e , o componente de variância real da população-base.
Resultados e Discussão
População GRAN 900
As estimativas obtidas para G0 (Tabela 2) foram relativamente próximas do valor real para ambas as metodologias. As médias da metodologia Bayesiana com prior não-informativo e prior pouco informativo foram as mais acuradas. Portanto, para população grande não-endogâmica, que não sofreu seleção e com muitos genes governando a característica, ambas as metodologias produziram resultados bastante satisfatórios.
Na prática, é comum a análise de rebanhos selecionados sem conhecimento dos registros e dos parentes (ancestrais) do indivíduo. Quando a estimação do componente de variância genética da população-base foi realizada somente com os dados da G10, sem informação de parentesco até a população-base, as estimativas obtidas pelo REML e pela metodologia Bayesiana para seleção ao acaso utilizando priors não-informativo e pouco informativo foram bastante próximas entre si, porém superestimadas. A estimativa mais acurada foi a moda obtida pela metodologia Bayesiana com prior informativo. Resultados semelhantes foram encontrados quando realizada a seleção fenotípica; as estimativas obtidas pela média e pela moda da metodologia Bayesiana com prior informativo se aproximaram mais do valor real da G0.
Ao incluir todos os registros e toda a informação de parentesco da G10 até a população-base, verificou-se que, para ambos os tipos de seleção e ambas as metodologias, as estimativas foram próximas umas das outras, porém levemente superestimadas. A semelhança encontrada para ambos os tipos de seleção, associada ao pequeno erro produzido, indica que, para população grande com muitos locos, a seleção fenotípica não causa redução na variância genética atribuída a mudanças na freqüência gênica, como ocorre quando utilizada a seleção ao acaso. Assim, as pressuposições do modelo infinitesimal estão sendo atendidas, exceto pelo fato de haver ligação entre locos na população-base, possível causa do erro encontrado. Nesse contexto, os resultados encontrados são satisfatórios. Além disso, o nível de informação a priori não influenciou os resultados, provavelmente em virtude da elevada quantidade de dados (26.400) utilizados nessas análises e, portanto, a informação contida na função de verossimilhança domina a informação a priori (Wang et al., 1994).
População GRAN 10
As estimativas obtidas para G0 por REML e pela metodologia Bayesiana foram, em geral, próximas umas das outras; as mais acuradas foram a média e a moda da metodologia Bayesiana com prior informativo. A perda da acurácia verificada nessa população, em comparação à população GRAN 900, provavelmente está associada ao desvio da normalidade dos valores genéticos da própria G0, verificado na população GRAN 10 e causado pelo pequeno número de locos.
Em análise apenas dos dados da G10, verificaram-se resultados bastante distintos dos obtidos para GRAN 900. Houve grande diferença entre as estimativas obtidas para seleção ao acaso e para seleção fenotípica. Para o primeiro caso, as estimativas foram levemente superestimadas para ambas as metodologias empregadas, com exceção da moda da análise Bayesiana com prior informativo, que gerou resultado bastante acurado. As estimativas obtidas pela seleção fenotípica, porém, foram bastante distintas do valor real para todas as análises, inclusive pela metodologia Bayesiana com prior informativo.
Como para esta população a característica é governada por poucos genes, o decréscimo na variância aditiva é notável quando realizada seleção fenotípica, o que não ocorre quando a seleção é ao acaso. Portanto, ao realizar a análise de dados em que a seleção atuou de forma expressiva sob característica governada por poucos genes, sem incluir registros e informação de parentesco, obtiveram-se estimativas com erros expressivos.
Quando incluídos todos os registros e toda a informação de parentesco da G10 até a população-base, obtiveram-se para seleção ao acaso estimativas muito próximas umas das outras, as quais foram levemente subestimadas. Contudo, quando os dados sofreram seleção fenotípica, as estimativas foram bem menos acuradas em todas as análises, as quais produziram resultados muito próximos entre si. Esse resultado é bastante interessante, pois comprova que, mesmo em populações grandes, com todos os registros e informações de parentesco até a população-base, a violação da pressuposição do modelo infinitesimal, em que a característica não é mais governada por um infinito número de locos de pequeno efeito (mesmo considerando que esses locos têm somente efeito aditivo e que não estão ligados), é suficiente para produzir estimativas bem distantes do valor real. Novamente, percebe-se que, para análises com grande quantidade de dados, a informação a priori com alto grau de confiança não é capaz de produzir estimativas não-viesadas, pois a informação dos dados proporcionada pela função de verossimilhança se sobrepõe à distribuição a priori do componente de variância gené-tica aditiva. Resultados de experimentos com camundongos também mostraram a não-adequabilidade do modelo infinitesimal na análise de dados que sofreram seleção (Meyer & Hill, 1991; Beniwal et al., 1992).
População PEQ 900
Para G0, a estimativa REML foi levemente superestimada, enquanto, para a metodologia Bayesiana, foram encontrados resultados bastante distintos, que variaram conforme o nível de informação considerado. Utilizando prior não-informativo, a metodologia gerou estimativas bastante distintas entre média e moda, no entanto, a média foi bem mais próxima do valor real. A associação da pequena quantidade de dados à falta de informação a priori é a possível causa da falha nessa estimação, uma vez que a informação a priori tem maior peso no processo quando há poucos dados disponíveis. Por outro lado, as médias dos priors pouco informativo e informativo e a moda do informativo foram bastante acuradas, o que confirma a importância do nível de informação a priori nessa população.
Pela análise dos resultados da G10, as estimativas obtidas pela seleção ao acaso foram superestimadas, com exceção da metodologia Bayesiana com prior informativo, a qual produziu estimativas acuradas. Quando realizada seleção fenotípica, foram obtidas novamente estimativas pouco acuradas, com exceção da metodologia Bayesiana com prior informativo. Verificou-se, inclusive, grande diferença entre média e moda da análise Bayesiana para priors não-informativo e pouco informativo, comprovando a grande falta de simetria das distribuições a posteriori geradas.
As estimativas obtidas ao serem incluídos todos os registros e o parentesco completo dos indivíduos de G10 até G0 para ambos os tipos de seleção e para todas as análises foram, em geral, levemente superestimadas e bastante próximas entre si, resultado muito semelhante ao encontrado para a mesma situação na população GRAN 900. A seleção fenotípica não está causando decréscimo substancial na variância aditiva em virtude da mudança na freqüência gênica, portanto, as metodologias foram adequadas para estimar o componente de variância na população-base, uma vez que corrigiram a queda na variância causada pela endogamia e pelo desequilíbrio gamético. Novamente, a possível causa do erro encontrado pode estar relacionada à ocorrência de genes ligados na população-base, não satisfazendo assim todas as pressuposições do modelo infinitesimal. Vale ressaltar que, incluindo todos os registros até G0, obteve-se maior quantidade de dados (1.320 registros) em relação às situações em que havia somente 120 registros, portanto, o nível de informação a priori deixa de ser tão relevante.
Pelos resultados encontrados, a metodologia Bayesiana com prior informativo fornece estimativas acuradas dos componentes de variância genética quando há pequena quantidade de dados. O mesmo não se aplica a grande volume de dados.
População PEQ 10
Para G10, a estimativa obtida por REML foi altamente subestimada, assim como as obtidas pela Bayesiana com prior não-informativo, cuja densidade a posteriori mostrou-se bastante assimétrica. Com prior pouco informativo, a densidade a posteriori também foi assimétrica, porém, sua média mostrou-se levemente subestimada. Ao utilizar prior informativo, as estimativas também foram bastante acuradas. A pequena quantidade de dados e o maior desvio dos valores genéticos da normalidade desta população em comparação a PEQ 900 podem ser os responsáveis pelo erro encontrado pela metodologia REML. Para a metodologia Bayesiana, esses fatores não prejudicam a análise quando se têm priors informativos.
Ao analisar os dados da G10, considerando seleção ao acaso, foram encontradas estimativas pouco acuradas pela metodologia REML, pela Bayesiana com prior não-informativo (média e moda bastante divergentes) e pela moda Bayesiana com prior pouco informativo. A média com prior pouco informativo e a média e a moda com prior informativo foram as que mais se aproximaram do valor real. Ao realizar seleção fenotípica, houve falha na estimação pela metodologia REML, assim como pela Bayesiana com prior não-informativo. Novamente, o desvio da normalidade associado ao tamanho pequeno da população prejudica substancialmente a estimação pela metodologia REML.
As estimativas obtidas pela Bayesiana com prior pouco informativo foram subestimadas, enquanto as mais próximas do valor real foram geradas pela Bayesiana com prior informativo.
Incluindo todos os registros e o parentesco completo dos indivíduos da G10 até G0, para seleção ao acaso, as estimativas obtidas foram superestimadas para todas as metodologias e o maior volume de dados ao considerar todas as gerações diminuiu relativamente a importância da informação a priori. Ao realizar seleção fenotípica, encontraram-se, de modo geral, estimativas acuradas, inclusive a obtida pelo REML, que foi altamente acurada. Esse resultado não era esperado, uma vez que, apesar de se terem incluído todo o parentesco e todos os registros, a característica é governada por apenas dez genes. Possivelmente, a própria estrutura dos dados (relacionada aos valores reais dos componentes de variância) está favorecendo a ocorrência desses valores.
As porcentagens de erro das estimativas dos componentes de variância genética aditiva e ambiental obtidas pela metodologia de máxima verossimilhança restrita (REML) e pela metodologia Bayesiana, considerando G0, G10, G0 a G10, provenientes de seleção fenotípica encontram-se na Figura 1. Foram utilizadas as modas a posteriori geradas pela metodologia Bayesiana na elaboração das Figuras.
Na análise dos resultados produzidos na G0 (Figura 1 a e b), verificou-se que as estimativas obtidas para população GRAN 900 foram bastante acuradas e muito próximas entre si para todas as análises realizadas, tanto para a variância aditiva (cuja maior PE foi de 4,7% para metodologia Bayesiana com prior não-informativo e a menor de 1,8% para metodologia Bayesiana com prior informativo) quanto para a ambiental (em que a maior PE foi de 6,6% para metodologia Bayesiana com prior não-informativo e a menor, de 2,5% para metodologia Bayesiana com prior informativo). Para GRAN 10, as estimativas da variância ambiental foram também próximas entre si para todas as análises e bastante acuradas (o pior resultado foi gerado pelo método REML, pelo qual a PE foi igual a -4,5%), enquanto as estimativas da variância genética foram relativamente menos acuradas (a PE variou de 8,0%, para metodologia Bayesiana com prior informativo, a 16,1%, para metodologia Bayesiana com prior pouco informativo). As diferenças nos valores das estimativas obtidas para a população PEQ 900 se acentuaram mais conforme a metodologia utilizada: a mais acurada foi gerada pela metodologia Bayesiana com prior informativo (PE igual a -0,4%) e a menos acurada, pela metodologia Bayesiana com prior não-informativo (PE igual a -58,2%). As diferenças na variância ambiental entre as metodologias foram de menor magnitude; a menos acurada foi a estimativa gerada pela análise Bayesiana com prior não-informativo (PE igual a 16,5%). Para PEQ 10, as estimativas foram as menos acuradas para ambos os componentes de variância quando comparadas às demais populações, com exceção das obtidas pela metodologia Bayesiana com prior informativo (em que PE foi igual a -2,0% para variância aditiva e igual a 2,2% para variância ambiental). As estimativas menos acuradas foram geradas pela metodologia Bayesiana com prior não-informativo (PE igual a -99,4%, para variância aditiva e PE igual a 52,6%, para variância ambiental). Portanto, mesmo na situação em que a população analisada é não-endogâmica e não sofreu seleção, as metodologias utilizadas (com exceção da Bayesiana com prior informativo) não forneceram estimativas satisfatórias quando utilizada a característica governada por poucos genes, principalmente em populações pequenas. Além disso, para as populações pequenas, para ambos os componentes de variância, as estimativas REML foram relativamente mais acuradas que as obtidas pela moda da análise Bayesiana com prior não-informativo.
Ao observar os resultados representados na Figura 1 (c e d), em que foi considerada somente a G10 para se estimar os componentes de variância da população-base, sendo realizada seleção fenotípica durante dez gerações, verifica-se que, para a variância genética aditiva, as PE foram bastante expressivas para todas as populações, com exceção da GRAN 900 e das obtidas pela metodologia Bayesiana com prior informativo. Para GRAN 900, a menor e a maior PE foram, respectivamente, de 3,0% para metodologia Bayesiana com prior informativo e de 14,1% para metodologia Bayesiana com prior pouco informativo. Para GRAN 10, a estimativa menos acurada foi gerada pelo REML (-64,7%) e a mais acurada pela metodologia Bayesiana com prior informativo (-29,9%). Na população PEQ 900, o melhor resultado foi obtido para metodologia Bayesiana com prior informativo (PE igual a 3,8%) e o pior para metodologia Bayesiana com prior não-informativo (PE igual a -79,9%). Para a população PEQ 10, as estimativas menos acuradas foram produzidas pelo REML e pela metodologia Bayesiana com prior não-informativo (PE iguais a -100,0 e -99,9%, respectivamente) e a mais acurada, pela metodologia Bayesiana com prior informativo (PE igual a -8,4%). As estimativas da variância ambiental foram mais acuradas que as da variância aditiva para todas as populações. Para GRAN 900 e GRAN 10, as menos acuradas foram obtidas pela metodologia Bayesiana com prior pouco informativo (PE igual a -9,5%) e pela metodologia Bayesiana com prior informativo (PE igual a -13,6%), respectivamente. Para as populações PEQ 900 e PEQ 10, as menos acuradas foram geradas pela metodologia Bayesiana com prior não-informativo (PE igual a 41,1%) e pelo REML (PE igual a 20,0%).
Realizando seleção fenotípica e considerando toda a informação de parentesco e todos os registros da G0 até G10 (Figura 1 e e f), verificou-se que os componentes de variância genética aditiva foram estimados satisfatoriamente por todas as metodologias e para todas as populações, com exceção da GRAN 10. Nesta população, todas as metodologias produziram porcentagens de erro semelhantes, que variaram de -24,8 a -26,3% para metodologia Bayesiana com prior informativo e para o método REML, respectivamente. As porcentagens de erro para variância ambiental foram insignificantes para todas as populações e metodologias empregadas e variaram de 0,0%, para população GRAN 10, a 1,6%, para população PEQ 10. Neste caso, o tipo de seleção, o número de pares de locos envolvidos na expressão da característica, o tamanho da população, assim como a metodologia utilizada, tiveram influência desprezível na estimação do componente de variância ambiental quando todos os registros e toda a informação de parentesco foram incluídos na análise. O mesmo não ocorreu para a variância genética aditiva.
Conclusões
Quando a característica é governada por elevado número de genes, os componentes de variância genética aditiva e ambiental são satisfatoriamente estimados em populações selecionadas grandes ou pequenas pelas metodologias usuais, desde que os registros de todos os indivíduos e a matriz completa de parentesco sejam conhecidos. Em populações grandes, no entanto, a exclusão dessas informações não altera substancialmente a acurácia das estimativas.
Quando a característica é governada por reduzido número de genes, estimativas menos acuradas do componente de variância genética aditiva são obtidas em populações grandes, mesmo quando os registros de todos os indivíduos e a matriz completa de parentesco são incluídos nas análises. Caso essas informações sejam desconhecidas, o erro na estimação desse componente aumenta consideravelmente em populações grandes ou pequenas.
As metodologias REML e Bayesiana, em geral, produzem resultados bastante semelhantes na estimação dos componentes de variância. Para análises com menor quantidade de dados, no entanto, estimativas mais acuradas são obtidas se utilizados priors informativos por meio da análise Bayesiana.
A inclusão de informações externas altamente confiáveis por meio da análise Bayesiana possibilita gerar estimativas acuradas dos componentes de variância em situações que o método REML não tem sua convergência garantida.
Agradecimento
À CAPES, pela bolsa de estudos concedida à primeira e ao segundo autores durante seus respectivos programas de doutoramento.
Literatura Citada
Recebido: 22/2/2006
Aprovado: 28/3/2007
Correspondências devem ser enviadas para: giselle@cpafac.embrapa.br
- BARTON, N.H.; KEIGHTLEY, P.D. Understanding quantitative genetic variation. Nature Reviews Genetics, v.3, p.11-21, 2002.
- BENIWAL, B.K.; HASTINGS, I.M.; THOMPSON, R. et al. Estimation of changes in genetic parameters in selected lines of mice using REML with an animal model. 1. Lean mass. Heredity, v.69, p.352-360, 1992.
- BINK, M.C.A.M. On flexible finite polygenic models for multiple-trait evaluation. Genetical Research, v.80, p.245-256, 2002.
- BLASCO, A. The Bayesian controversy in animal breeding. Journal of Animal Science, v.79, p.2023-2046, 2001.
- BOLDMAN, K.G; KRIESE, L.A.; Van VLECK, L.D. et al. A manual for use of MTDFREML. A set of programs to obtain estimates of variances and covariances United States Department of Agriculture, Agriculture Research Service, 1995. 115p.
- EUCLYDES, R.F. Uso do sistema para simulação Genesys na avaliação de métodos de seleção clássicos e associados a marcadores moleculares Viçosa, MG: Universidade Federal de Viçosa, 1996. 150p. Tese (Doutorado em Genética e Melhoramento) - Universidade Federal de Viçosa, 1996.
- GIANOLA, D.; FERNANDO, R.L. Bayesian methods in animal breeding theory. Journal of Animal Science, v.63, p.217-244, 1986.
- GONÇALVES, T.M.; OLIVEIRA, H.N.; BOVENHUIS, H. et al. Comparação de diferentes estratégias para a análise de características de crescimento e de carcaça de suínos cruzados: modelos finito e infinitesimal poligênico. Revista Brasileira de Zootecnia, v.34, n.5, p.1531-1539, 2005a.
- GONÇALVES, T.M.; OLIVEIRA, H.N.; BOVENHUIS, H. et al. Modelos alternativos para detecção de locos de características quantitativas (QTL) de carcaça e crescimento nos cromossomos 4, 5 e 7 de suínos. Revista Brasileira de Zootecnia, v.34, n.5, p.1540-1552, 2005b.
- HENDERSON, C.R. Sire evaluation and genetic trends. In: ANIMAL BREEDING GENETIC SYMPOSIUM IN HONOR OF DR. J.L. LUSH, 1973, Champaign. Proceedings Champaign: ASAS/ADSA, 1973. p.10-28.
- MEYER, K.; HILL, W.G. Mixed model analysis of a selection experiment for food intake in mice. Genetical Research Cambridge, v.57, p.71-81, 1991.
- POLLAK, E.J.; QUAAS, R.L. Monte Carlo study of within-herd multiple trait evaluation of beef cattle growth traits. Journal of Animal Science, v.52, n.2, p.248-256, 1981.
- RAFTERY, A.E.; LEWIS, S.M. [1992]. How many iterations in the Gibbs sampler? Disponível em: <http://www.stat.washington.edu/www/research/online]> Acesso em: 31/1/2006.
- SCHENKEL, F.S. Studies on effects of parental selection on estimation of genetic parameters and breeding values of metric traits Guelph: University of Guelph, 1998. 191p. Ph.D. (Thesis in Animal Production) - University of Guelph, 1998.
- SORENSEN, D.A.; KENNEDY, B.W. Estimation of genetic variances from unselected and selected populations. Journal of Animal Science, v.59, n.5, p.1213-1223, 1984.
- SORENSEN, D.A.; WANG, C.S.; JENSEN, J. Bayesian analysis of genetic change due to selection using Gibbs sampling. Genetics, Selection, Evolution, v.26, p.333-360, 1994.
- Van KAAM, J.B.C.H.M. "GIBANAL" Analyzing program for Markov Chain Monte Carlo Sequences (Version 2.10). Wageningen: Department of Animal Sciences, Wageningen Agricultural University, 1998. 4p. (Manual).
- Van TASSEL, C.P.; CASELLA, G.; POLLAK, E.J. Effects of selection on estimates of variance components using Gibbs Sampling and Restricted Maximum Likelihood. Journal of Dairy Science, v.78, p.678-692, 1995.
- Van TASSEL, C.P.; Van VLECK, L.D. A manual for use of MTGSAM. A set of Fortran programs to apply Gibbs Sampling to animal models for variance components estimation Lincoln: United States Department of Agriculture, Agriculture Research Service, 1995. 85p.
- WANG, C.S.; GIANOLA, D.; SORENSEN, D.A. et al. Response to selection for litter size in Danish Landrace pigs: a Bayesian analysis. Theoretical and Applied Genetics, v.88, p.220-230, 1994.
Datas de Publicação
-
Publicação nesta coleção
30 Nov 2007 -
Data do Fascículo
Out 2007
Histórico
-
Aceito
28 Mar 2007 -
Recebido
22 Fev 2006





