Resumos
OBJETIVOS: Estudos pós-mortem, farmacológicos, de neuroimagem e em modelos animais têm demonstrado uma possível associação de mecanismos de sinalização intracelular na fisiopatologia do transtorno afetivo bipolar (TAB). Esse trabalho tem como objetivo revisar os achados em neuropatologia e bioquímica celular. MÉTODOS: Foi realizada uma pesquisa ao MEDLINE, entre 1980 e 2003, tendo sido utilizados os unitermos: bipolar disorder, signaling, second messengers e postmortem, além de referências cruzadas dos artigos selecionados. RESULTADOS: uropatológicos demonstraram uma diminuição do número de células neuronais e gliais, principalmente no córtex pré-frontal de pacientes bipolares. Estudos neuroquímicos demonstraram alterações nas vias do AMPc, fosfatidilinositol, Wnt/GSK-3beta e Ca++ intracelular nesses pacientes. CONCLUSÃO: Os achados de alterações neuropatológicas e neuroquímicas no TAB podem estar relacionados com a fisiopatologia deste transtorno e com os efeitos dos estabilizadores de humor. No entanto, mais estudos são necessários para esclarecer o papel das cascatas de sinalização intracelular na patogênese deste transtorno.
Transtorno bipolar; Sistemas do segundo mensageiro; Química cerebral
OBJECTIVES: Postmortem, pharmacological, neuroimaging, and animal model studies have demonstrated a possible association of intracellular signaling mechanisms in the pathophysiology of bipolar disorder. The objective of this paper is to review the findings in neuropathology and cellular biochemistry. METHODS: We performed a MEDLINE research, between 1980-2003, using bipolar disorder, signaling, second messengers, and postmortem as keywords, and cross-references. RESULTS: Neuropathological studies reported a decrease in neuronal and glial cells, mainly in the prefrontal cortex of bipolar patients. Neurochemical studies reported dysfunction in cAMP, phosphoinositide, Wnt/GSK-3b, and intracellular Ca++ pathways in these patients. CONCLUSION: The neuropathological and neurochemical abnormalities demonstrated in BD may be related to the pathophysiology of this disorder and the effects of mood stabilizers. However, further studies are needed to clarify the role of the intracellular signaling cascade in the pathogenesis of this disorder.
Bipolar disorder; Second messenger systems; Brain chemistry
REVISÃO
Anormalidades neuropatológicas e neuroquímicas no transtorno afetivo bipolar
Benício Noronha FreyI, II; Manoela M Rodrigues da FonsecaI, III; Rodrigo Machado-VieiraI, IV; Jair C SoaresV; Flávio KapczinskiI, III, VI
ILaboratório de Psiquiatria Experimental do Hospital de Clínicas de Porto Alegre (HCPA)
IIDepartamento de Bioquímica Instituto de Ciências Básicas da Saúde Universidade Federal do Rio Grande do Sul (UFRGS)
IIIServiço de Psiquiatria do HCPA
IVMood Disorder Program Fundação Faculdade Federal de Ciências Médicas de Porto Alegre Hospital Materno-Infantil Presidente Vargas
VDivision of Mood and Anxiety Disorders, Department of Psychiatry, University of Texas Health Science Center at San Antonio, TX, USA
VIDepartamento de Psiquiatria e Medicina Legal da Faculdade de Medicina da Universidade Federal do Rio Grande do Sul (UFRGS)
Endereço para correspondência Endereço para correspondência Benício Noronha Frey Rua Almirante Abreu, 108/201 90420-010 Porto Alegre, RS, Brasil Tel.: (51) 3333-4821 / (51) 9916-7666 E-mail: benicio.frey@terra.com.br
RESUMO
OBJETIVOS: Estudos pós-mortem, farmacológicos, de neuroimagem e em modelos animais têm demonstrado uma possível associação de mecanismos de sinalização intracelular na fisiopatologia do transtorno afetivo bipolar (TAB). Esse trabalho tem como objetivo revisar os achados em neuropatologia e bioquímica celular.
MÉTODOS: Foi realizada uma pesquisa ao MEDLINE, entre 1980 e 2003, tendo sido utilizados os unitermos: bipolar disorder, signaling, second messengers e postmortem, além de referências cruzadas dos artigos selecionados.
RESULTADOS: uropatológicos demonstraram uma diminuição do número de células neuronais e gliais, principalmente no córtex pré-frontal de pacientes bipolares. Estudos neuroquímicos demonstraram alterações nas vias do AMPc, fosfatidilinositol, Wnt/GSK-3b e Ca++ intracelular nesses pacientes.
CONCLUSÃO: Os achados de alterações neuropatológicas e neuroquímicas no TAB podem estar relacionados com a fisiopatologia deste transtorno e com os efeitos dos estabilizadores de humor. No entanto, mais estudos são necessários para esclarecer o papel das cascatas de sinalização intracelular na patogênese deste transtorno.
Descritores: Transtorno bipolar; Sistemas do segundo mensageiro; Química cerebral
Introdução
Há décadas se sabe que o transtorno afetivo bipolar (TAB) é um transtorno mental crônico, com altos índices de recaída, na maioria das vezes incapacitante e no qual acredita-se haver um substrato neurobiológico. Entretanto, embora o entendimento da neurobiologia tenha se expandido nos últimos anos, ainda pouco se sabe sobre os verdadeiros mecanismos fisiopatológicos do TAB.1 Investigações recentes de genética no TAB têm relatado resultados que, embora conflitantes, parecem demonstrar alguma associação, pelo menos em uma percentagem dos indivíduos acometidos.2 Os estudos em neuroimagem demonstraram uma série de alterações estruturais e funcionais em determinadas regiões do cérebro de indivíduos bipolares, como o córtex préfrontal e temporal, cerebelo, gânglios da base e sistema límbico;3 no entanto, esses estudos não permitem que se alcance um substrato celular mais específico nessas regiões. Além disso, estudos neuropatológicos (pós-mortem) demonstraram diminuição da glia e da densidade e plasticidade neuronal, bem como alterações da neuroquímica intracelular.4-5
Um dos modelos que têm sido aplicados ao TAB é o modelo do kindling,6 trazido do modelo da epilepsia, no qual a repetição das crises causaria um processo de sensibilização neuronal, levando a uma diminuição progressiva do limiar, com aumento da recorrência das crises epiléticas (maníacas). Estudos em modelos animais sugerem que esse processo possa envolver uma série de alterações no que diz respeito a expressão gênica e segundos mensageiros.6 De fato, os estudos farmacológicos têm sido consistentes com esses achados, tendo sido demonstrada a ação dos antidepressivos e estabilizadores de humor em diversos mecanismos intracelulares que envolvem a regulação da expressão gênica e da plasticidade celular.7 Este trabalho tem como objetivo revisar os achados em neuropatologia e bioquímica celular, que parecem estar envolvidos na fisiopatogenia do TAB. Para tanto, foi realizada uma pesquisa ao banco de dados MEDLINE, entre 1980 e 2003, tendo sido utilizados os unitermos: bipolar disorder, signaling, second messengers e postmortem. Também foram utilizadas referências cruzadas dos artigos selecionados.
Neuropatologia do TAB
Os estudos de neuroimagem estrutural demonstraram alterações significativas do volume cerebral, sugerindo atrofia e/ou perda neuronal, pelo menos em uma proporção dos indivíduos com TAB. Diversos trabalhos relataram diminuição significativa da substância cinzenta em córtex pré-frontal8-9 e temporal,10-11 além de aumento dos ventrículos laterais.12 Ainda, estudos independentes que avaliaram regiões específicas do lobo temporal demonstraram diminuição13-14 e aumento15-16 do volume da amígdala em indivíduos bipolares. Um achado consistente nos estudos em TAB é uma maior freqüência de hiperintensidades de substância branca subcortical.17 Essas lesões difusas da substância branca podem significar interrupções nos circuitos envolvidos com a regulação do humor. Embora com menos consistência, alterações em gânglios da base3 e cerebelo18 também foram observadas em pacientes bipolares. Tomados em conjunto, esses achados apontam para uma possível disfunção no circuito cortico-límbico como substrato anatômico do TAB. Os estudos de neuroimagem funcional fornecem evidências adicionais de alterações do metabolismo da glicose e diminuição do fluxo sanguíneo regional e dos fosfatos energéticos celulares nas regiões corticais e subcorticais no TAB.19 Entretanto, a resolução das técnicas de neuroimagem atuais é limitada a milímetros, sendo, portanto, fundamentais os estudos pós-mortem que possibilitam um estudo direto de resolução celular e molecular.
Em um dos estudos neuropatológicos de maior amostra de indivíduos bipolares (n=18), Öngur et al20 relataram diminuição significativa (41,2%) da densidade de células gliais no córtex pré-frontal subgenual, área 24 de Brodmann, em indivíduos bipolares e unipolares com história familiar positiva para transtornos de humor. Porém, este estudo não avaliou se tal diminuição ocorreu especificamente em astrócitos, oligodendrócitos ou micróglia. Recentemente, Rajkowska et al21 não encontraram diferenças significativas na densidade e tamanho das células gliais no córtex pré-frontal dorsolateral (DLPFC área 9 de Brodmann) de indivíduos bipolares. Entretanto, uma análise laminar mais detalhada demonstrou uma redução significativa (19%) da densidade glial na subcamada IIIc do DLPFC. Esse estudo encontrou, ainda, uma redução das células com núcleos de tamanho médio nas camadas III e V da glia, acompanhada de um aumento das células com núcleos de tamanho muito grande.21 Dois estudos independentes trouxeram dados adicionais consistentes com alterações da glia nos transtornos de humor. Cotter et al22 relataram redução da densidade glial na camada VI da área 24 de Brodmann em indivíduos unipolares e esquizofrênicos, mas não em bipolares (a maioria estava em uso de estabilizadores de humor, que parecem exercer efeito neurotrófico/neuroprotetor). Usando a mesma coorte de pacientes, Uranova et al23 investigaram a área 10 do córtex pré-frontal dorsal e demonstraram diminuição significativa da densidade glial na camada VI dos indivíduos bipolares e esquizofrênicos, e apenas nos unipolares com história familiar positiva para "transtorno mental severo". Tomados em conjunto, estes achados apontam para a hipótese de que pelo menos um subgrupo de indivíduos bipolares e unipolares, principalmente os com história familiar positiva, apresenta algum déficit da densidade glial em múltiplos sítios do córtex pré-frontal, o que pode afetar sua conexão com outras regiões cerebrais.
No sistema límbico, Benes et al24 relataram uma diminuição significativa de neurônios não-piramidais na região CA2 do hipocampo em indivíduos bipolares e esquizofrênicos. Mais recentemente, o mesmo grupo replicou o mesmo achado, porém na camada II do córtex anterior do cíngulo.25 Esses achados sugerem uma possível associação da diminuição da inibição GABAérgica (neurônios nãopiramidais) na fisiopatogenia do TAB.4 No tronco cerebral, foi demonstrado um aumento bilateral do número de neurônios pigmentados no locus ceruleus de indivíduos bipolares em comparação a unipolares.26 Esses neurônios são uma das principais fontes de noradrenalina (NA) no SNC. Nesse sentido, Young et al27 relataram um aumento do turnover da NA no córtex de indivíduos bipolares em um estudo pós-mortem.
As células gliais regulam a homeostase energética do SNC através da captação e fosforilação da glicose durante a atividade neuronal. Além disso, participam do desenvolvimento, manutenção e remodelamento das conexões sinápticas através da liberação de fatores tróficos e da regulação da concentração do glutamato na sinapse.28 Desta forma, os achados de redução da densidade glial podem resultar em uma diminuição do número de sinapses funcionais no TAB. Consistentemente com essa hipótese de disfunção sináptica, dois estudos pós-mortem que avaliaram a região hipocampal evidenciaram diminuição da expressão do mRNA de proteínas sinápticas29 e das espinhas dendríticas apicais de células piramidais30 na subregião subicular de indivíduos com TAB.
Dessa forma, os avanços da bioquímica molecular aplicada a estudos pós-mortem apontam para uma disfunção de mecanismos complexos intracelulares, que envolvem os sistemas de segundos mensageiros, regulação da expressão gênica e síntese de fatores tróficos (neuroplasticidade) como associados à fisiopatogenia do TAB.1,6-7
Sistemas de sinalização intracelular
1. Proteínas G
O mecanismo que envolve a transmissão da informação desde a sinapse até o núcleo da célula é mediado por um processo intermediário chamado sistema de segundos mensageiros, como as vias da adenosina monofosfato cíclica (AMPc) e do fosfatidilinositol (PIP2). Esse processo envolve três etapas: 1) a ligação do neurotransmissor ao receptor de membrana; 2) a ativação de proteínas que utilizam a guanosina trifosfato (GTP) como cofator, chamadas proteínas G; e 3) a ativação de sistemas efetores (via segundos mensageiros vide Figura 1). As proteínas G possuem três subunidades (a, b e g), que são firmemente ligadas à parte interna da membrana plasmática, sendo ativadas a partir da ligação do neurotransmissor ao seu receptor específico. Múltiplos sistemas de receptores do SNC são modulados pelas proteínas G, incluindo os receptores noradrenérgicos, serotoninérgicos, dopaminérgicos, colinérgicos e histaminérgicos, entre outros. As proteínas G podem exercer efeito tanto estimulatório quanto inibitório nas proteínas efetoras, sendo, portanto, classificadas como Gs (proteína G estimulatória) e Gi (inibitória). Dessa forma, as proteínas G ativadas pelos receptores modulam o fluxo de íons, através da regulação da atividade dos canais iônicos, e controlam a atividade de uma variedade de enzimas efetoras. O interesse no estudo das proteínas G no TAB surgiu dos achados em modelos animais sobre a ação regulatória do lítio em diversos subtipos de proteínas G.31-32
Young et al33-34 foram os primeiros a publicar um aumento dos níveis de Gas no córtex frontal, temporal e occipital de indivíduos com TAB. Esse achado foi replicado em um estudo que demonstrou um aumento da atividade de Gas estimulada por agonista.35 No entanto, esses resultados não descartam a possibilidade de representarem efeitos de tratamentos farmacológicos ou de amostras com n reduzido. Em um estudo que investigou uma maior amostra, Dowlatshahi et al36 não encontraram diferença nos níveis de Gas em indivíduos bipolares em relação ao grupo controle. Porém, evidenciaram um aumento significativo da Gas entre os pacientes que não estavam em uso de lítio. Desta forma, o uso do fármaco pode ter sido responsável pela falha na detecção da diferença entre o total dos pacientes e o grupo controle. Estudos com sangue periférico também têm confirmado esses achados e ampliado o entendimento da relação entre o funcionamento das proteínas G e os estados de humor. Schreiber et al37 foram os primeiros a evidenciar um aumento da atividade da proteína G em leucócitos mononucleados de pacientes em estado maníaco. Outros dois estudos observaram aumento dos níveis de Gαs em mononucleados de bipolares deprimidos sem medicação,38-39 enquanto outro estudo encontrou aumento dos níveis na mania e diminuição no estado depressivo.40 Além disso, esse aumento da expressão da Gas também foi demonstrado em plaquetas,41-42 mas não em linfoblastos.43 Esses estudos sugerem que o estado de humor e o tipo celular podem influenciar nos achados de aumento da Gas no sangue periférico de indivíduos bipolares. Tomados em conjunto, esses achados sugerem uma possível associação do funcionamento das proteínas G na fisiopatogenia do TAB. No entanto, ainda não está determinado se o TAB está associado a uma disfunção direta da atividade das proteínas G ou se esses achados representam uma manifestação secundária de uma disfunção em outras vias.
2. Via da adenosina monofosfato cíclica (AMPc)
Uma das proteínas efetoras reguladas pelas proteínas G é a adenilato ciclase (AC), enzima que catalisa a formação do AMPc, um importante segundo mensageiro, a partir da adenosina trifosfato (ATP vide Figura 2). Uma das principais funções do AMPc é a ativação de outra enzima, uma proteína quinase dependente de AMPc (PKA), a qual integra as alterações rápidas da neurotransmissão em mudanças neurobiológicas de longa duração. Diversos estudos demonstraram um aumento significativo da atividade da AC, em estado basal e ativada, em indivíduos com TAB, alterações estas que podem estar associadas à disfunção das proteínas G descritas anteriormente.33,40,44-45 Além disso, esses trabalhos demonstraram uma relação entre a atividade da AC e o tratamento ou o estado de humor, com uma diminuição da atividade da enzima em pacientes deprimidos e em pacientes eutímicos que recorrem após tratamento com lítio.40,44-45 Um estudo pós-mortem encontrou uma diminuição da ligação do [3H]cAMP com a PKA no córtex frontal, temporal, parietal, occipital, tálamo e cerebelo de indivíduos bipolares, o que traduz uma medida indireta de aumento da atividade do AMPc nesses pacientes.46 Dois estudos pós-mortem mais recentes, que avaliaram o córtex frontal e temporal,47-48 e dois estudos em plaquetas49-50 confirmaram um aumento da atividade da PKA em pacientes bipolares. Em conjunto, esses estudos consistentemente sugerem um aumento da atividade da via do AMPc-PKA em diversas regiões cerebrais nos indivíduos com TAB.
Ainda, estudos demonstraram que o uso crônico de lítio diminui a ativação da AC e que esta ação pode ser revertida pelo aumento da concentração de GTP, sugerindo que os efeitos do tratamento com o lítio podem ser mediados no nível das proteínas G.51-52 No entanto, em estado basal o lítio aumentou a formação de AMPc no cérebro de ratos.53 Dessa forma, tem sido sugerido que a ação do lítio na atividade da AC é estado dependente: em condições basais, quando a inibição tônica da formação do AMPc mediada pela Gai é predominante, o lítio aumenta a formação do AMPc; quando da ativação da AC pelo conjunto receptor-Gas, a formação do AMPc é atenuada. Esse mecanismo de ação "bimodal" poderia ser uma das explicações do efeito terapêutico do lítio tanto na depressão como na mania. O uso crônico de valproato em concentração clinicamente relevante produziu um aumento significativo dos receptores b-adrenérgicos ligados à via do AMPc em células cultivadas in vitro.7 A carbamazepina, por sua vez, demonstrou inibir a atividade da AC tanto em estado basal54 quanto ativada,55 além de reduzir os elevados níveis de AMPc no líquor de pacientes maníacos.56
3. Via do Fosfatidilinositol (PIP2)
Diversos sistemas de neurotransmissores utilizam a via do fosfatidilinositol (Tabela 1) através da ativação das proteínas G. Nessa via, a ativação da proteína G estimula a proteína efetora fosfolipase C (PLC), a qual hidrolisa um fosfolipídio de membrana, chamado fosfatidilinositol (PIP2), formando dois importantes segundos mensageiros: o diacilglicerol (DAG) e o inositol trifosfato (IP3). O IP3 possui um receptor específico, situado no retículo endoplasmático liso, que libera os estoques de Ca+2 quando ativado. A DAG, por sua vez, tem a função de ativar a proteína quinase C (PKC Figura 3). Diversos estudos pós-mortem e com células periféricas têm demonstrado alterações desta via em indivíduos com TAB. Um estudo pós-mortem evidenciou um aumento da atividade da proteína G e da PLC no córtex occipital de indivíduos bipolares, sem diferenças nas regiões frontal e temporal.57 Outro estudo observou uma diminuição da atividade da proteína G ligada à via do PIP2, também na região occipital.58 Os autores sugeriram que esses achados controversos poderiam ser decorrentes de um processo adaptativo celular ou da ação do uso crônico de lítio. Para conservar a eficiência da transmissão desta via, a célula necessita manter uma oferta adequada de inositol para a re-síntese do PIP2. Uma vez que o inositol atravessa fracamente a barreira hemato-encefálica, o aporte do inositol se dá pela desfosforilação do IP3, através da reação catalisada pela enzima inositol monofosfatase (IMPase). Shimon et al59 compararam cérebros de indivíduos bipolares, suicidas e controles, e demonstraram uma diminuição significativa de inositol livre no córtex frontal dos indivíduos bipolares e dos suicidas em comparação ao grupo controle. Porém, não encontraram diferença na atividade da IMPase nessa região. Não houve diferenças em córtex occipital ou cerebelo. Dois estudos realizados em plaquetas relataram aumento dos níveis de PIP2 em indivíduos bipolares sem medicação, tanto na fase maníaca60 quanto depressiva,61 enquanto outros estudos encontraram níveis de PIP2 significativamente reduzidos em plaquetas de bipolares após tratamento com lítio.62-64 Esses achados suportam os estudos farmacológicos que demonstraram que o lítio, em concentrações terapêuticas (Ki=0,8mM), é um potente inibidor da IMPase.65 Dessa forma, esta ação regulatória que o lítio exerce na via do PIP2 pode ser um dos mecanismos do fármaco na regulação do humor. Mais recentemente, um estudo utilizando a espectroscopia por ressonância magnética exame capaz de mensurar substâncias neuroquímicas no cérebro in vivo , demonstrou que o lítio diminuiu significativamente o inositol no lobo frontal direito de indivíduos bipolares deprimidos.66 No entanto, embora esse efeito do lítio tenha ocorrido dentro de 5-7 dias, a melhora do humor só ocorreu após 3-4 semanas de uso do medicamento,66 sugerindo que esse efeito inicial do lítio module uma série de eventos posteriores na cascata, como a regulação da expressão gênica e da plasticidade neuronal, necessárias para que se obtenha a resposta clínica significativa.
A PKC é uma importante enzima da via do PIP2, tendo ação na regulação da excitabilidade neuronal, liberação de neurotransmissores, expressão gênica e plasticidade sináptica.1 Um estudo pós-mortem demonstrou um aumento significativo da atividade da PKC no córtex frontal de pacientes bipolares,67 achado também demonstrado em estudos com plaquetas.68-69 Ainda, Soares et al62 não encontraram diferenças dos níveis da PKC em plaquetas de bipolares eutímicos tratados com lítio. De fato, o lítio demonstrou efeito na inibição da atividade da PKC também em estudos animais.70-71 Dessa forma, os achados de aumento da atividade da PKC no TAB e de sua diminuição com o uso do lítio podem ser clinicamente relevantes. Neste sentido, Bebchuk et al,72 recentemente, publicaram um estudo piloto demonstrando possíveis efeitos antimaníacos com o uso do tamoxifeno, um antiestrogênico inibidor da PKC.
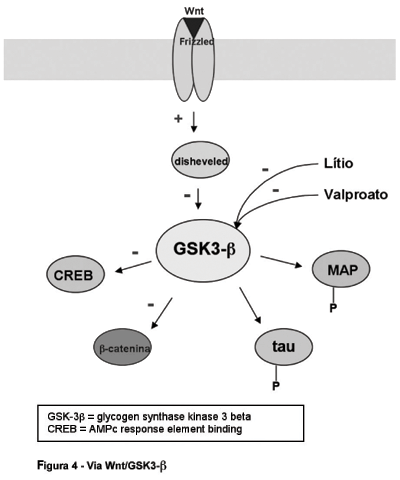
4. Via Wnt (wingless)/Glicogen synthase kinase 3b (GSK3-b)
As proteínas Wnt ligam-se aos receptores (frizzled) de membrana ligados à proteína G, ativando a proteína quinase disheveled, esta que inibe a atividade da glicogen synthase kinase 3ß (GSK3-b) (Figura 4). O interesse do estudo do papel da GSK3-b no TAB surgiu a partir das observações de que o lítio e, mais recentemente, o valproato diminuem a atividade desta proteína em concentrações terapêuticas.73 A GSK3-b é capaz de fosforilar uma extensa gama de proteínas metabólicas, sinalizadoras e estruturais, além de fatores de transcrição gênica.74 Dentre estas atividades, destacam-se a modulação de proteínas associadas aos microtúbulos do citoesqueleto, como a tau, a MAP-1B e a MAP-2, e a regulação da morte celular programada (apoptose).73 A fosforilação da tau e da MAP-1B pela GSK3-b está associada à perda ou desestabilização da conformação dos microtúbulos75-76 e o uso de lítio demonstrou diminuir a fosforilação da tau em cultura de neurônios humanos.77 A GSK3-b está associada diretamente ao aumento da apoptose neuronal,74 diminuindo as atividades de proteínas que promovem a sobrevivência neuronal, como a AMPc response element binding protein (CREB) e a heat shock factor-1 (HSF-1).78-79 Além disso, o lítio, o valproato e a lamotrigina protegem as células SH-SY5Y da apoptose facilitada pela GSK3-b.80
A atividade da GSK3-b pode ser modulada por uma série de cascatas de sinalização intracelular. Mais especificamente, a fosforilação da GSK3-b pela PKA, PKC e Akt diminui, enquanto o Ca++ intracelular pode aumentar sua atividade.73-74 Dessa forma, tem sido sugerido que os efeitos neuroprotetores das neurotrofinas (NGF, BDNF) e do lítio podem decorrer, ao menos em parte, da inibição da GSK3-b, através da via PI3K/Akt.81 Dois estudos pós-mortem não encontraram diferenças nos níveis da GSK3-b no córtex pré-frontal de pacientes bipolares.82-83 Entretanto, embora não haja evidências diretas de anormalidades da via Wnt/GSK3-b no TAB, evidências robustas salientam a importância da regulação desta via no tratamento deste transtorno.73-80
5. Cálcio (Ca++) intracelular
A variação dos níveis de cálcio intracelular (Ca++i) modula a plasticidade sináptica, a sobrevivência e a morte celular. De fato, a sinalização do Ca++i interage com diversas outras cascatas de sinalização, incluindo as vias do AMPc e do PIP2.84 Além disso, o cálcio pode interagir com outras proteínas regulatórias, como a calmodulina, formando complexos que modulam a atividade de outras enzimas importantes, incluindo as proteínas quinases dependentes de cálcio-calmodulina (CaMKs). A PKC ativada pelo Ca++i demonstrou diminuir a atividade da PKA em fibroblastos,85 enquanto o AMPc causou dessensibilização dos receptores para IP3 e diminuiu o influxo de Ca++, desta forma reduzindo os níveis de Ca++i.86 Dubovsky et al87 foram os primeiros a descrever um aumento dos níveis de Ca++i em leucócitos e plaquetas de bipolares maníacos e deprimidos sem medicação. Além disso, utilizando estímulos que aumentam as concentrações de Ca++i, verificou-se uma resposta significativamente aumentada em bipolares maníacos e não-medicados em relação a pacientes eutímicos e em uso de estabilizadores de humor.88-89 Estes estudos sugerem que as variações do estado de humor podem estar relacionadas com alterações dos níveis de Ca++i e que estas alterações podem ser revertidas com a remissão das crises,87 embora outros estudos não tenham replicado estes achados.90-91 Recentemente, Emagoreishi et al92 demonstraram que indivíduos bipolares com altos níveis de Ca++i basal apresentaram uma menor produção de AMPc após estímulo dos receptores b-adrenérgicos com isoproterenol e uma maior atividade basal da AC ligada à proteína G, corroborando os achados de aumento da atividade das proteínas G descritos anteriormente e sugerindo que alterações em uma via podem descompensar ou induzir mudanças adaptativas em outras vias de sinalização.
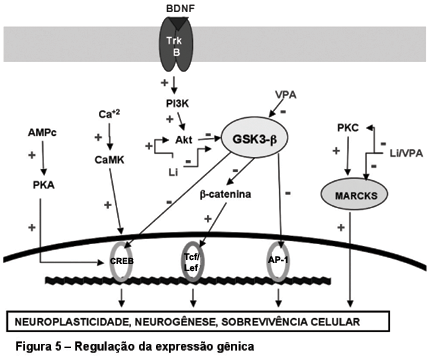
Regulação da expressão gênica e neuroplasticidade
A atividade das vias de segundos mensageiros tem como um importante alvo final a modulação de uma família de proteínas que agem como fatores de transcrição gênica. Essas proteínas ligam-se a sítios específicos do DNA e regulam a expressão de uma ampla variedade de genes capazes de regular funções celulares como a proliferação e a apoptose celular (Figura 5). Um fator de transcrição que tem sido estudado no TAB é a CREB, uma proteína que se localiza no núcleo da célula, usualmente na forma inativa, sendo ativada por uma série de proteínas quinases, como a PKA, a MAPK ou a CaMK. Após ativada, a CREB promove a produção de RNAm ao ligar-se a um determinado sítio na região promotora de genes alvo, com a formação de proteínas que podem alterar permanentemente a estrutura ou a função de regiões específicas do cérebro. Dowlatshahi et al36 não encontraram alterações significativas dos níveis de CREB em indivíduos bipolares em um estudo pós-mortem; entretanto, encontraram níveis diminuídos da proteína nos que cometeram suicídio e naqueles tratados com anticonvulsivantes no momento da morte. Estudos farmacológicos que examinaram os efeitos do lítio na atividade da CREB encontraram achados conflitantes,68-78 enquanto a carbamazepina demonstrou diminuir a fosforilação da proteína em gliomas.54 Além disso, o uso crônico de ECT (eletroconvulsoterapia) e de antidepressivos aumentou a expressão do BDNF (brain-derived neurotrophic factor) e de seu receptor TrkB através do aumento da atividade da CREB,93-94 sendo que a GSK3-b diminuiu a fosforilação da CREB induzida pelo BDNF.95
A activator protein-1 (AP-1) constitui uma família de fatores de transcrição gênica, que incluem os membros Fos, Jun e ATF, também chamados immediate early genes, que regulam genes envolvidos com crescimento, proliferação e morte celular. O aumento da atividade da GSK3-b em culturas de células diminuiu significativamente a atividade da AP-1,96 enquanto o uso de lítio demonstrou reverter esta ação.97 Outro fator de transcrição passível de modulação pela via Wnt/GSK3-b é a b-catenina. A ativação da b-catenina leva à sua ligação com os fatores de transcrição Tcf/Lef, regulando a expressão de genes moduladores do ritmo circadiano, da adesão e do desenvolvimento celular,98 sendo sua atividade também diminuída pela ação da GSK3-b.
Como vimos, estudos recentes demonstraram que o lítio, o valproato e a carbamazepina podem exercer efeitos terapêuticos através da regulação da expressão gênica via fatores de transcrição. O lítio age em diversos níveis da cascata de segundos mensageiros, bem como em proteínas quinases. Além disso, existem evidências da ação do lítio em um dos principais substratos da PKC no cérebro, a proteína MARCKS (miristoylated alanine-rich C kinase substrate), que também é uma proteína implicada na regulação de eventos neuroplásticos, alterando a conformação do citoesqueleto através dos filamentos de actina. O uso de lítio por quatro semanas demonstrou reduzir dramaticamente a expressão da proteína MARCKS em células do hipocampo.99 Estudos posteriores demonstraram que essa ação do lítio ocorria através da regulação da transcrição gênica da proteína100 e era dependente da concentração do inositol e da ativação do receptor ligado à cascata do PIP2.101 Outro estudo demonstrou que o valproato também é capaz de reduzir a expressão da MARCKS nas células hipocampais, através de mecanismos distintos aos do lítio,102 o que corrobora as observações clínicas do efeito terapêutico sinérgico dessas duas drogas na regulação do humor.
Conclusões
Os estudos pós-mortem e os achados de neuroimagem têm revelado com consistência uma diminuição significativa do volume de determinadas regiões do SNC, acompanhada de perda ou atrofia das células nervosas, principalmente das células gliais. No entanto, ainda não está determinado se esses achados representam alterações precoces na migração neuronal, perdas celulares decorrentes da própria progressão da doença, alterações bioquímicas que acompanham as crises de humor ou a ação dos diversos medicamentos utilizados. Em contrapartida, o possível papel da morte/atrofia celular no declínio progressivo do funcionamento encontrado em muitos pacientes permanece a ser elucidado. Estudos comparando pacientes medicados com aqueles que nunca receberam medicação podem auxiliar no entendimento dos efeitos dos psicofármacos na morfologia e funcionamento celular.
Evidências crescentes apontam para a associação de eventos intracelulares, envolvendo o sistema de segundos mensageiros como parte das alterações neurobiológicas do TAB. Foi demonstrado um aumento da atividade das proteínas G e das vias do AMPc e do PIP2 que, através da regulação da síntese de DNA, modificam proteínas envolvidas com a plasticidade sináptica, neurogênese e conformação do citoesqueleto. Entretanto, permanece incerto se essas alterações refletem um aumento da vulnerabilidade do indivíduo (como resultado de fatores genéticos/eventos de vida precoces), efeitos dos tratamentos instituídos ou o próprio processo etiológico central da doença. Estudos prospectivos precoces, que avaliem as alterações da expressão gênica durante o curso e o tratamento do transtorno, são um campo promissor de pesquisa na área. Além disso, o aperfeiçoamento dos modelos animais, como o uso de ratos mutantes, se faz necessário para que se possa testar se as alterações em determinadas cascatas de sinalização intracelular são suficientes para promover as alterações comportamentais e se o bloqueio dessas vias são capazes de inibir a ação dos psicofármacos.
Finalmente, os estudos farmacológicos têm revelado ação dos principais estabilizadores de humor em diversos destes mecanismos intracelulares. Desta forma, é possível que os efeitos agudos desses fármacos desencadeiem uma cascata de eventos intracelulares capazes de alterar a síntese protéica, produzindo efeitos reparatórios na plasticidade sináptica e restaurando a transmissão nervosa. A que ponto essas drogas podem interferir nas alterações fisiopatológicas do TAB e estabilizar o curso e a progressão da doença ainda está para ser definido. Esses avanços na neurobiologia do TAB devem ser interpretados com cautela e sem generalizações, por serem estudos iniciais, que necessitam ser replicados e conduzidos com amostras maiores e menos heterogêneas. Além disso, também têm sido relatadas anormalidades envolvendo outras vias, como o sistema purinérgico.103 Esperamos que esse novo e promissor campo de pesquisa possa auxiliar no desenvolvimento de tratamentos mais eficazes para um transtorno tão incapacitante como o TAB.
Agradecimento
Os autores agradecem à Dra. Betina Kruter pelo auxílio no preparo do manuscrito.
Financiamento e Conflito de Interesses: Inexistente
Recebido em 05.01.2004
Aceito em 27.04.2004
- 1. Manji HK, Lenox RH. Signaling: cellular insights into the pathophysiology of bipolar disorder. Biol Psychiatry. 2000;48(6):518-30.
- 2. Zandi PP, Willour VL, Huo Y, Chellis J, Potash JB, MacKinnon DF, et al. Genome scan of a second wave of NIMH genetics initiative bipolar pedigrees: chromosomes 2, 11, 13, 14, and X. Am J Med Genet. 2003;119B(1):69-76.
- 3. Soares JC, Mann JJ. The anatomy of mood disorders - review of structural neuroimaging studies. Biol Psychiatry. 1997;41(1):86-106.
- 4. Rajkowska G. Cell pathology in bipolar disorder. Bipolar Disord. 2002;4(2):105-16.
- 5. Vawter MP, Freed WJ, Kleinman JE. Neuropathology of bipolar disorder. Biol Psychiatry. 2000;48(6):486-504.
- 6. Ghaemi SN, Boiman EE, Goodwin FK. Kindling and second messengers: an approach to the neurobiology of recurrence in bipolar disorder. Biol Psychiatry. 1999;45(2):137-44.
- 7. Lenox RH, Frazer A. Mechanism of action of antidepressants and mood stablizers. In: Davis KL, Charney D, Coyle JT, Nemeroff C, editors. Neuropsychopharmacology: the fifth generation of progress. Philadelphia: Lippincott Williams & Wilkins; 2002. p. 1139-63.
- 8. Blumberg HP, Stern E, Martinez D, Ricketts S, de Assis J, White T, et al. Rostral and orbital prefrontal cortex dysfunction in the manic state of bipolar disorder. Am J Psychiatry. 1999;156(12):1986-8.
- 9. Drevets WC, Price JL, Simpson Jr JR, Todd RD, Reich T, Vannier M, et al. Subgenual prefrontal cortex abnormalities in mood disorders. Nature. 1997;386(6627):824-7.
- 10. Altshuler LL, Conrad A, Hauser P, Li XM, Guze BH, Denikoff K, et al. Reduction of temporal lobe volume in bipolar disorder: a preliminary report of magnetic resonance imaging. Arch Gen Psychiatry. 1991;48(5):482-3.
- 11. Hauser P, Altshuler LL, Berrettini W, Dauphinais ID, Gelernter J, Post RM. Temporal lobe measurement in primary affective disorder by magnetic resonance imaging. J Neuropsychiatry Clin Neurosci. 1989;1(2):128-34.
- 12. Elkis H, Friedman L, Wise A, Meltzer HY. Meta-analyses of studies of ventricular enlargement and cortical sulcal prominence in mood disorders. Arch Gen Psychiatry. 1995;52(9):735-46.
- 13. Sheline Yi, Gado MH, Price JL. Amygdala core nuclei volumes are decreased in recurrent major depression. Neuroreport. 1998;9(9):2023-8.
- 14. Pearlson GD, Barta PE, Powers RE, Menon RR, Richards SS, Aylward EH, et al. Medial and superior temporal gyral volumes and cerebral asymmetry in schizophrenia versus bipolar disorder. Biol Psychiatry. 1997;41(1):1-4.
- 15. Strakowski SM, DelBello MP, Sax KW, Zimmerman ME, Shear PK, Hawkins JM, et al. Brain magnetic resonance imaging of structural abnormalities in bipolar disorder: a pilot study. Arch Gen Psychiatry. 1999;56(3):254-60.
- 16. Altshuler LL, Bartzokis G, Grieder T, Curran J, Mintz J. Amygdala enlargement in bipolar disorder and hippocampal reduction in schizophrenia: an MRI study demonstrating neuroanatomic specificity. Arch Gen Psychiatry. 1998;55(7):663-4.
- 17. McDonald WM, Tupler LA, Marsteller FA, Figiel GS, DiSouza S, Nemeroff CB, et al. Hyperintense lesions on magnetic resonance images in bipolar disorder. Biol Psychiatry. 1999;45(8):965-71.
- 18. Loeber RT, Sherwood AR, Renshaw PF, Cohen BM, Yurgelun-Todd DA. Differences in cerebellar blood volume in schizophrenia and bipolar disorder. Schizophr Res. 1999;37(1):81-9.
- 19. Stoll AL, Renshaw PF, Yurgelun-Todd D, Cohen BM. Neuroimaging in bipolar disorder: what have we learned? Biol Psychiatry. 2000;48(6):505-17.
- 20. Öngur D, Drevets WC, Price JL. Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc Natl Acad Sci USA. 1998;95(22):13290-5.
- 21. Rajkowska G, Halaris A, Selemon LD. Reductions in neuronal and glial density characterize the dorsolateral prefrontal cortex in bipolar disorder. Biol Psychiatry. 2001;49(9):741-52.
- 22. Cotter D, Mackay D, Landau S, Kerwin R, Everall I. Reduced glial cell density and neuronal size in the anterior cingulate cortex in major depressive disorder. Arch Gen Psychiatry. 2001;58(6):545-53.
- 23. Uranova N, Orlovskaya D, Vikhreva O, Zimina I, Kolomeets N, Vorstrikov V, et al. Electron microscopy of oligodendroglia in severe mental illness. Brain Res Bull. 2001;55(5):597-610.
- 24. Benes FM, Kwok EW, Vincent SL, Todtenkopf MS. A reduction of nonpyramidal cells in sector CA2 of schizophrenics and manic-depressives. Biol Psychiatry. 1998;44(2):88-97.
- 25. Benes FM, Vincent SL, Todtenkopf MS. The density of pyramidal and nonpyramidal neurons in anterior cingulate cortex of schizophrenic and bipolar subjects. Biol Psychiatry. 2001;50(6):395-406.
- 26. Baumann B, Danos P, Krell D, Diekmann S, Wurthmann C, Bielau H, et al. Unipolar-bipolar dichotomy of mood disorder in supported by noradrenergic brainstem system morphology. J Affect Disord. 1999;54(1-2):217-24.
- 27. Young LT, Warsh JJ, Kish SJ, Shannak K, Hornykeiwicz O. Reduced brain 5-HT and elevated NE turnover and metabolites in bipolar affective disorder. Biol Psychiatry. 1994;35(2):121-7.
- 28. Magistretti PJ, Ransom BR. Astrocytes. In: Davis KL, Charney D, Coyle JT, Nemeroff C, editors. Neuropsychopharmacology: the fifth generation of progress. Philadelphia: Lippincott Williams & Wilkins; 2002. p. 133-45.
- 29. Eastwood SL, Harrison PJ. Hippocampal synaptic pathology in schizophrenia, bipolar disorder, and major depression: a study of complexin mRNAs. Mol Psychiatry. 2000;5(4):425-32.
- 30. Rosoklija G, Toomayan G, Ellis SP, Keilp J, Mann JJ, Latov N, et al. Structural abnormalities of subicular dendrites in subjects with schizophrenia and mood disorders. Arch Gen Psychiatry. 2000;57(4):349-56.
- 31. Avissar S, Schreiber G, Danon A, Belmaker RH. Lithium inhibits adrenergic and cholinergic increases in GTP binding in rat cortex. Nature. 1988;331(6155):440-2.
- 32. Mork A, Geisler A. The effects of lithium in vitro and ex vivo on adenylate ciclase in brain are exerted by distinct mechanisms. Neuropharmacology. 1989;28(3):307-11.
- 33. Young LT, Li PP, Kish SJ, Siu KP, Warsh JJ. Postmortem cerebral cortex Gs alpha-subunit levels are elevated in bipolar in bipolar affective disorder. Brain Res. 1991;553(2):323-6.
- 34. Young LT, Li PP, Kish SJ, Siu KP, Kamble A, Hornykiewicz O, et al. Cerebral cortex Gs alpha protein levels and forskolin-stimulated cyclic AMP formation are increased in bipolar affective disorder. J Neurochem. 1993;61(3):890-8.
- 35. Friedman E, Wang HY. Receptor-mediated activation of G proteins is increased in postmortem brains of bipolar affective disorder subjects. J Neurochem. 1996;67(3):1145-52.
- 36. Dowlatshahi D, MacQueen GM, Wang JF, Reiach JS, Young LT. Proteincoupled cyclic AMP signaling in post mortem brain of subjects with mood disorders: effects of diagnosis, suicide, and treatment at time of death. J Neurochem. 1999;73(3):1121-6.
- 37. Schreiber G, Avissar S, Danon A, Belmaker RH. Hyperfunctional G proteins in mononuclear leukocytes in patients with mania. Biol Psychiatry. 1991;29(3):273-80.
- 38. Spleiss O, Van Calker D, Scharer L, Adamovic K, Berger M, Gebicke-Haerter PJ. Abnormal G protein alpha(s)- and alpha(i2)- subunit mRNA expression in bipolar affective disorder. Mol Psychiatry. 1998;3(6):512-20.
- 39. Young LT, Li PP, Kamble A, Siu KP, Warsh JJ. Mononuclear leukocyte levels of G proteins in depressed patients with bipolar disorder and major depressive disorder. Am J Psychiatry. 1994;151(4):594-6.
- 40. Avissar S, Nechamkin Y, Barki-Harrington L, Roitman G, Schreiber G. Differential G protein measures in mononuclear leukocytes of patients with bipolar mood disorder are state dependent. J Affect Disord. 1997;43(2):85-93.
- 41. Mitchell PB, Manji HK, Chen G, Jolkovsky L, Smith-Jackson E, Denicoff K, et al. High levels of Gs alpha in platelets of euthymic patients with bipolar affective disorder. Am J Psychiatry. 1997;154(2):218-23.
- 42. Manji HK, Chen G, Shimon H, Hsiao JK, Potter WZ, Belmaker RH. Guanine nucleotide-binding proteins in bipolar affective disorder: effects of longterm lithium treatment. Arch Gen Psychiatry. 1995;52(2):135-44.
- 43. Alda M, Keller D, Grof E, Turecki G, Cavazzoni P, Duffy A, et al. Is lithium response related to G(s) alpha levels in transformed lymphoblasts from subjects with bipolar disorder? J Affect Disord. 2001;65(2):117-22.
- 44. Avissar S, Barki-Harrington L, Nechamkin Y, Roitman G, Schreiber G. Reduced beta-adrenergic receptor-coupled Gs protein function and Gs alpha immunoreactivity in mononuclear leukocytes of patients with depression. Biol Psychiatry. 1996;39(9):755-60.
- 45. Ebstein RP, Lerer B, Shapira B, Shemesh Z, Moscovich DG, Kindler S. Cyclic AMP second-messenger signal amplification in depression. Br J Psychiatry. 1988;152:665-9.
- 46. Rahman S, Li PP, Young LT, Kofman O, Kish SJ, Warsh JJ. Reduced [3H]cyclic AMP binding in postmortem brain from subjects with bipolar affective disorder. J Neurochem. 1997;68(1):297-304.
- 47. Chang A, Li PP, Warsh JJ. Altered cAMP-dependent protein kinase subunit immunolabeling in postmortem brain from patients with bipolar affective disorder. J Neurochem. 2003;84(4):781-91.
- 48. Fields A, Li PP, Kish SJ, Warsh JJ. Increased cyclic AMP-dependent protein kinase activity in postmortem brain from patients with bipolar affective disorder. J Neurochem. 1999;73(4):1704-10.
- 49. Perez J, Tardito D, Mori S, Racagni G, Smeraldi E, Zanardi R. Altered Rap1 endogenous phosphorylation and levels in platelets from patients with bipolar disorder. J Psychiatr Res. 2000;34(2):99-104.
- 50. Perez J, Zanardi R, Mori S, Gasperini M, Smeraldi E, Racagni G. Abnormalities of cAMP-dependent endogenous phosphorylation in platelets from patients with bipolar disorder. Am J Psychiatry. 1995;152(8):1204-6.
- 51. Newman ME, Belmaker RH. Effects of lithium in vitro and ex vivo on components of the adenylate ciclase system in membranes from the cerebral cortex of the rat. Neuropharmacology. 1987;26(2-3):211-7.
- 52. Newman M, Klein E, Birmaher B, Feinsod M, Belmaker RH. Lithium at therapeutic concentrations inhibits human brain noradrenaline-sensitive cyclic AMP accumulation. Brain Res. 1983;278(1-2):380-1.
- 53. Masana MI, Bitran JA, Hsiao JK, Potter WZ. In vivo evidence that lithium inactivates G[I] modulation of adenylate cyclase in brain. J Neurochem. 1992;59(1):200-5.
- 54. Chen G, Pan B, Hawver D, Wright C, Potter WZ, Manji HK. Attenuation of cyclic AMP production by carbamazepine. J Neurochem. 1996;67(5):2079-86.
- 55. Elphick M, Anderson SM, Hallis KF, Grahame-Smith DG. Effects of carbamazepine on 5-hydroxytryptamine function in rodents. Psychopharmacology. (Berl.) 1990;100(1):49-53.
- 56. Post RM, Ballenger JC, Uhde TW, Smith C, Rubinow DR, Bunney WE Jr. Effect of carbamazepine on cyclic nucleotides in CSF of patients with affective illness. Biol Psychiatry. 1982;17(9):1037-45.
- 57. Mathews R, Li PP, Young LT, Kish SJ, Warsh JJ. Increased G alpha /11 immunoreactivity in postmortem occipital cortex from patients with bipolar affective disorder. Biol Psychiatry. 1997;41(6):649-56.
- 58. Jope RS, Song L, Li PP, Young LT, Kish SJ, Pacheco MA, et al. The phosphoinositide signal transduction system is impaired in bipolar affective disorder brain. J Neurochem. 1996;66(6):2402-9.
- 59. Shimon H, Agam G, Belmaker RH, Hyde TM, Kleinman JE. Reduced frontal cortex inositol levels in postmortem brain of suicide victims and patients with bipolar disorder. Am J Psychiatry. 1997;154(8):1148-50.
- 60. Brown AS, Mallinger AG, Renbaum LC. Elevated platelet membrane phophatidylinositol-4,5-biphosphate in bipolar mania. Am J Psychiatry. 1993;150(8):1252-4.
- 61. Soares JC, Dippold CS, Wells KF, Frank E, Kupfer DJ, Mallinger AG. Increased platelet membrane phophatidylinositol-4,5-biphosphate in drugfree bipolar patients. Neurosci Lett. 2001;299(1-2):150-2.
- 62. Soares JC, Chen G, Dippold CS, Wells KF, Frank E, Kupfer DJ, et al. Concurrent measures of protein kinase C and phosphoinositides in lithium-treated bipolar patients and healthy individuals: a preliminary study. Psychiatry Res. 2000;95(2):109-18.
- 63. Soares JC, Mallinger AG, Dippold CS, Frank E, Kupfer DJ. Platelet membrane phospholipids in euthymic bipolar disorder patients: are they affected by lithium treatment? Biol Psychiatry. 1999;45(4):453-7.
- 64. Soares JC, Dippold CS, Mallinger AG. Platelet membrane phophatidylinositol-4,5-biphosphate alterations in bipolar disorder - evidence from a single case study. Psychiatry Res. 1997;69(2-3):197-202.
- 65. Sherman WR, Gish BG, Honchar MP, Munsell LY. Effects of lithium on phosphoinositide metabolism in vivo. Fed Proc. 1986;45(11):2639-46.
- 66. Moore GJ, Bebchuk JM, Parrish JK, Faulk MW, Arfken CL, Strahl-Bevacqua J, et al. Temporal dissociation between lithium-induced changes in frontal lobe myo-inositol and clinical response in manic-depressive illness. Am J Psychiatry. 1999;156(12):1902-8.
- 67. Wang HY, Friedman E. Enhanced protein kinase C activity and translocation in bipolar affective disorders brains. Biol Psychiatry. 1996;40(7):568-75.
- 68. Wang HY, Markowitz P, Levinson D, Undie AS, Friedman E. Increased membrane-associated protein kinase C activity and translocation in blood platelets from bipolar affective disorder patients. J Psychiatr Res. 1999;33(2):171-9.
- 69. Friedman E, Hoau YW, Levinson D, Connell TA, Singh H. Altered platelet protein kinase C activity in bipolar affective disorder, manic episode. Biol Psychiatry. 1993;33(7):520-5.
- 70. Manji HK, Etcheberrigaray R, Chen G, Olds JL. Lithium dramatically decreases membrane-associated protein kinase C in the hippocampus: selectivity for the alpha isozyme. J Neurochem. 1993;61(6):2303-10.
- 71. Chen G, Rajkowska G, Du F, Seraji-Bozorgzad N, Manji HK. Enhancement of hipocampal neurogenesis by lithium. J Neurochem. 2000;75(4):1729-34.
- 72. Bebchuk JM, Arfken CL, Dolan-Manji S, Murphy J, Hasanat K, Manji HK. A preliminary investigation of a protein kinase C inhibitor in the treatment of acute mania. Arch Gen Psychiatry. 2000;57(1):95-7.
- 73. Gould TD, Manji HK. The Wnt signaling pathway in bipolar disorder. Neuroscientist. 2002;8(5):497-511.
- 74. Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3b in cellular signaling. Prog Neurobiol. 2001;65(4):391-426.
- 75. Johnson GV, Hartigan JA. Tau protein in normal in Alzheimer's disease brain: an update. J Alzheimers Dis. 1(4-5):329-41.
- 76. Lucas FR, Goold RG, Gordon-Weeks PR, Salinas PC. Inhibition of GSK-3b leading to the loss of phosphorylated MAP-1B is an early event in axonal remodeling induced by WNT-7a or lithium. J Cell Sci. 1998;111(Pt 10):1351-61.
- 77. Hong M, Chen DC, Klein PS, Lee VM. Lithium reduces tau phosphorylation by inhibition of glycogen synthase kinase-3. J Biol Chem. 1997;272(40):25326-32.
- 78. Grimes CA, Jope RS. CREB DNA binding activity is inhibited by glycogen synthase kinase-3 beta and facilitated by lithium. J Neurochem. 2001;78(6):1219-32.
- 79. He B, Meng YH, Mivechi NF. Glycogen synthase kinase 3beta and extracellular signal-regulated kinase inactivate heat shock transcription factor 1 by facilitating the disappearance of trancriptionally active granules after heat shock. Mol Cell Biol. 1998;18(11):6624-33.
- 80. Li X, Bijur GN, Jope RS. Glycogen synthase kinase 3-beta, mood stabilizers, and neuroprotection. Bipolar Disord. 2002;4(2):137-44.
- 81. Chuang DM, Chen RW, Chalecka-Franaszek E, Ren M, Hashimoto R, Senatorov V, et al. Neuroprotective effects of lithium in cultured cells and animal models of disease. Bipolar Disord. 2002;4(2):129-36.
- 82. Kozlovsky N, Belmaker RH, Agam G. Low GSK-3beta immunoreactivity in postmortem frontal cortex of schizophrenic patients. Am J Psychiatry 2000;157:831-3.
- 83. Lesort M, Greendorfer A, Stockmeier C, Johnson GV, Jope RS. Glycogen synthase kinase 3-beta, beta-catenin, and tau in postmortem bipolar brain. J Neural Transm. 1999;106(11-12):1217-22.
- 84. Liu M, Simon MI. Regulation by cAMP-dependent protein kinase of a G protein-mediated phospholipase C. Nature. 1996;382(6586):83-7.
- 85. Dobbeling U, Berchtold MW. Down-regulation of the protein kinase A pathway by activators of protein kinase C and intracellular Ca2+ in fibroblasts cells. FEBS Lett. 1996;391(1-2):131-3.
- 86. Supattapone S, Danoff SK, Theibert A, Joseph SK, Steiner J, Snyder SH. Cyclic AMP-dependent phosphorilation of a brain inositol triphosphate receptor decreases its release of calcium. Proc Natl Acad Sci USA. 1988;85(22):8747-50.
- 87. Dubovsky SL, Murphy J, Thomas M, Rademacher J. Abnormal intracellular calcium ion concentration in platelets and lymphocytes of bipolar patients. Am J Psychiatry. 1992;149(1):118-20.
- 88. Okamoto Y, Kagaya A, Shinno H, Motohashi N, Yamawaki S. Serotonininduced platelet calcium mobilization is enhanced in mania. Life Sci. 1995;56(5):327-32.
- 89. Kusumi I, Koyama T, Yamashita I. Trombin-induced platelet calcium mobilization is enhanced in bipolar disorder. Biol Psychiatry. 1992;32(8):731-4.
- 90. Suzuki K, Kusumi I, Sasaki Y, Koyama T. Serotonin-induced platelet intracellular calcium mobilization in various psychiatric disorders: is it specific to bipolar disorder? J Affect Disord. 2001;64(2-3):291-6.
- 91. El Khoury AE, Petterson U, Kallner G, Aberg-Wistedt A, Stain-Malmgren R. Calcium homeostasis in long-term lithium-treated women with bipolar affective disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2002;26(6):1063-9.
- 92. Emamghoreishi M, Li PP, Shlichter L, Parikh SV, Cooke R, Warsh JJ. Associated disturbances in calcium homeostasis and G protein-mediated camp signaling in bipolar I disorder. Biol Psychiatry. 2000;48(7):665-73.
- 93. Chen B, Dowlatshahi D, McQueen GM, Wang GF, Young LT. Increased hippocampal BDNF immunoreactivity in subjects treated with antidepressant medication. Biol Psychiatry. 2001;50(4):260-5.
- 94. Thome J, Sakai N, Shin KH, Steffen C, Zhang YJ, Impey S, et al. cAMP response element-mediated gene transcription is up-regulated by chronic antidepressant treatment. J Neurosci. 2000;20(11):4030-6.
- 95. Mai L, Jope RS, Li X. BDNF-mediated signal transduction is modulated by GSK3beta and mood stabilizing agents. J Neurochem. 2002;82(1):75-83.
- 96. Nikolakaki E, Coffer PJ, Hemelsoet R, Woodgett JR, Defize LH. Glycogen synthase kinase phosphorilates Jun family members in vitro and negatively regulates their transactivating potential in intact cells. Oncogene. 1993;8(4):833-40.
- 97. Hedgepeth CM, Conrad LJ, Zhang J, Huang HC, Lee VM, Klein PS. Activation of the Wnt pathway: a molecular mechanism of lithium action. Dev Biol. 1997;185(1):82-91.
- 98. Novak A, Dedhar S. Signaling through b-catenin and Lef/Tcf. Cell Mol Life Sci. 1999;56(5-6):532-7.
- 99. Lenox RH, Watson DG, Patel J, Ellis J. Chronic lithium administration alters a prominent PKC substrate in rat hippocampus. Brain Res. 1992;570(1-2):333-40.
- 100. Wang L, Liu X, Lenox RH. Transcriptional down-regulation of MARCKS gene expression in immortalized hipocampal cells by lithium. J Neurochem. 2001;79(4):816-25.
- 101. Manji HK, Bersudsky Y, Chen G, Belmaker RH, Potter WZ. Modulation of protein kinase C isozymes and substrates by lithium: the role of myo-inositol. Neuropsychopharmacology. 1996;15(4):370-81.
- 102. Watson DG, Watterson JM, Lenox RH. Sodium valproate down-regulates the miristoylated alanine-rich C kinase substrate (MARCKS) in immortalized hippocampal cells: a property unique to PKC-mediated mood stabilizers. J Pharmacol Exp Ther. 1998;285(1):307-16.
- 103. Machado-Vieira R, Lara DR, Souza DO, Kapczinski F. Purinergic dysfunction in mania: an integrative model. Med Hypotheses. 2002;58(4):297-304.
Datas de Publicação
-
Publicação nesta coleção
23 Fev 2005 -
Data do Fascículo
Set 2004
Histórico
-
Recebido
05 Jan 2004 -
Aceito
27 Abr 2004