Resumos
OBJETIVO: As mudanças no eixo hipotálamo-pituitária-adrenal (HPA) são características da depressão. Devido aos efeitos dos glicocorticóides serem mediados por receptores intracelulares, como os receptores de glicocorticóides (RGs), inúmeros estudos examinaram o número e/ou função dos RGs em pacientes com depressão. MÉTODOS: Os autores fazem uma revisão das evidências científicas dos estudos que têm consistentemente demonstrado que a função dos RGs está prejudicada na depressão maior, em conseqüência da redução da resposta do eixo HPA ao feedback negativo mediado pelos RGs e a um aumento na produção e secreção de HLC em várias regiões cerebrais, sugerindo que esses mecanismos estão envolvidos na etiologia da depressão e no tratamento antidepressivo. RESULTADOS: Esta revisão faz um resumo da literatura atual sobre RG na depressão e sobre o impacto dos antidepressivos nos RGs em estudos clínicos e pré-clínicos, e dá suporte ao conceito de que a sinalização deficiente dos RGs é parte fundamental na fisiopatogenia da depressão, na ausência de evidências claras de redução na expressão dos RGs. Embora os efeitos dos antidepressivos nos hormônios glicocorticóides e seus receptores sejam relevantes para a ação terapêutica dessas drogas, os mecanismos moleculares subjacentes a esses efeitos ainda não estão esclarecidos. Estudos indicam que os antidepressivos têm efeitos diretos nos RGs, levando a uma melhora da função e a um aumento da expressão dos RGs. Nós propomos que, em humanos, os antidepressivos podem inibir os transportadores de esteróides localizados na barreira hemato-liquórica e nos neurônios, como o complexo de resistência a múltiplas drogas glicoproteína-p ("multidrug resistance p-glycoprotein"), e podem aumentar o acesso do cortisol ao cérebro e o feedback negativo mediado por glicocorticoides no eixo HPA. CONCLUSÃO: O aumento da ação do cortisol no cérebro pode ser uma abordagem eficaz para maximizar os efeitos terapêuticos dos antidepressivos. Hipóteses referentes aos mecanismos destes receptores envolvem compostos não esteróides que regulam a função dos RGs via segundos mensageiros. A pesquisa nesta área trará novos entendimentos à fisiopatologia e ao tratamento dos transtornos afetivos, em especial na depressão.
Estresse; Depressão; Hipotálamo; Hidrocortisona; Antidepressivos; Neuroendocrinologia; Citocinas; Pseudoneuroimunologia
OBJECTIVES: Changes in the hypothalamic-pituitary-adrenocortical (HPA) system are characteristic of depression. Because the effects of glucocorticoids are mediated by intracellular receptors including, most notably, the glucocorticoid receptor (GR), several studies have examined the number and/or function of GRs in depressed patients. METHODS: Review scientific evidences have consistently demonstrated that GR function is impaired in major depression, resulting in reduced GR-mediated negative feedback on the HPA axis and increased production and secretion of CRF in various brain regions postulated to be involved in the causality of depression. RESULTS: This article summarizes the literature on GR in depression and on the impact of antidepressants on the GR in clinical and preclinical studies, and supports the concept that impaired GR signalling is a key mechanism in the pathogenesis of depression, in the absence of clear evidence of decreased GR expression. The data also indicate that antidepressants have direct effects on the GR, leading to enhanced GR function and increased GR expression. Although the effects of antidepressants on glucocorticoid hormones and their receptors are relevant for the therapeutic action of these drugs, the molecular mechanisms underlying these effects are unclear. We propose that antidepressants in humans could inhibit steroid transporters localised on the blood-brain barrier and in neurones, like the multidrug resistance p-glycoprotein, and thus increase the access of cortisol to the brain and the glucocorticoid-mediated negative feedback on the HPA axis. CONCLUSION: Enhanced cortisol action in the brain might prove to be a successful approach to maximise therapeutic antidepressant effects. Hypotheses regarding the mechanism of these receptor changes involve non-steroid compounds that regulate GR function via second messenger pathways. Research in this field will lead to new insights into the pathophysiology and treatment of affective disorders.
Stress; Depression; Hypothalamus; Hidrocortisone; Antidepressive agents; Neuroendocrinology; Cytokines; Psychoneuroimmunology
REVISÃO
O eixo hipotálamo-pituitária-adrenal, a função dos receptores de glicocorticóides e sua importância na depressão
Mario F Juruena; Anthony J Cleare; Carmine M Pariante
Division of Psychological Medicine, Section of Neurobiology of Mood Disorders, Institute of Psychiatry, University of London e Affective Disorders Unit, South London Maudsley Trust, London, UK
Endereço para correspondência Endereço para correspondência Mario Francisco Juruena 103 Denmark Hill Division of Psychological Medicine, Institute of Psychiatry University of London SE5 8AZ London - UK Tel.: 44 (20) 7848 5305 Fax: 44 (20) 7848 5408 E-mail: m.juruena@iop.kcl.ac.uk
RESUMO
OBJETIVO: As mudanças no eixo hipotálamo-pituitária-adrenal (HPA) são características da depressão. Devido aos efeitos dos glicocorticóides serem mediados por receptores intracelulares, como os receptores de glicocorticóides (RGs), inúmeros estudos examinaram o número e/ou função dos RGs em pacientes com depressão.
MÉTODOS: Os autores fazem uma revisão das evidências científicas dos estudos que têm consistentemente demonstrado que a função dos RGs está prejudicada na depressão maior, em conseqüência da redução da resposta do eixo HPA ao feedback negativo mediado pelos RGs e a um aumento na produção e secreção de HLC em várias regiões cerebrais, sugerindo que esses mecanismos estão envolvidos na etiologia da depressão e no tratamento antidepressivo.
RESULTADOS: Esta revisão faz um resumo da literatura atual sobre RG na depressão e sobre o impacto dos antidepressivos nos RGs em estudos clínicos e pré-clínicos, e dá suporte ao conceito de que a sinalização deficiente dos RGs é parte fundamental na fisiopatogenia da depressão, na ausência de evidências claras de redução na expressão dos RGs. Embora os efeitos dos antidepressivos nos hormônios glicocorticóides e seus receptores sejam relevantes para a ação terapêutica dessas drogas, os mecanismos moleculares subjacentes a esses efeitos ainda não estão esclarecidos. Estudos indicam que os antidepressivos têm efeitos diretos nos RGs, levando a uma melhora da função e a um aumento da expressão dos RGs. Nós propomos que, em humanos, os antidepressivos podem inibir os transportadores de esteróides localizados na barreira hemato-liquórica e nos neurônios, como o complexo de resistência a múltiplas drogas glicoproteína-p ("multidrug resistance p-glycoprotein"), e podem aumentar o acesso do cortisol ao cérebro e o feedback negativo mediado por glicocorticoides no eixo HPA.
CONCLUSÃO: O aumento da ação do cortisol no cérebro pode ser uma abordagem eficaz para maximizar os efeitos terapêuticos dos antidepressivos. Hipóteses referentes aos mecanismos destes receptores envolvem compostos não esteróides que regulam a função dos RGs via segundos mensageiros. A pesquisa nesta área trará novos entendimentos à fisiopatologia e ao tratamento dos transtornos afetivos, em especial na depressão.
Descritores: Estresse; Depressão; Hipotálamo; Hidrocortisona; Antidepressivos; Neuroendocrinologia; Citocinas; Pseudoneuroimunologia
Introdução
Os hormônios desempenham um papel crítico no desenvolvimento e expressão de uma ampla gama de comportamentos. Um aspecto da influência dos hormônios no comportamento é a sua potencial contribuição para a fisiopatologia dos transtornos psiquiátricos e para o mecanismo de ação dos psicotrópicos, particularmente na depressão maior. De todos os eixos endócrinos, o eixo hipotálamo-pituitária-adrenal (HPA) tem sido o mais amplamente estudado.1-2 Este eixo exerce um papel fundamental na resposta aos estímulos externos e internos, incluindo os estressores psicológicos. Anormalidades na função do eixo HPA têm sido descritas em pessoas que experimentam transtornos psiquiátricos. Além disso, é bem conhecido o papel fundamental do estresse como precipitante de episódios de transtornos psiquiátricos em indivíduos predispostos.1 Essas anormalidades parecem estar relacionadas às mudanças na capacidade dos glicocorticóides circulantes em exercer seu feedback negativo na secreção dos hormônios do eixo HPA por meio da ligação aos receptores de mineralocorticóides (RM) e glicocorticóides (RG) nos tecidos do eixo HPA.2,3-6 De fato, na depressão melancólica é descrito que há desregulação do feedback negativo no eixo HPA levando à hipercortisolemia.2,6 Assim como a depressão melancólica, um espectro de outros transtornos pode estar associado à ativação aumentada e prolongada do eixo HPA, incluindo anorexia nervosa, com ou sem desnutrição, transtorno obsessivo-compulsivo, pânico, alcoolismo crônico, abstinência de álcool e narcóticos, exercício excessivo, diabetes mellitus mal controlado, abuso sexual na infância e hipertiroidismo.7 Outro grupo de doenças é caracterizado pela hipoativação do sistema de estresse, em vez de ativação permanente, na qual a secreção cronicamente reduzida de hormônio liberador de corticotropina (HLC) pode resultar em hiporreatividade patológica e feedback negativo intensificado do eixo HPA. Pacientes com transtorno de estresse pós-traumático, depressão sazonal, depressão atípica e síndrome da fadiga crônica entram nessa categoria (ver Tabela 1). Da mesma forma, pacientes com fibromialgia apresentam excreção urinária de cortisol livre diminuída e freqüentemente queixam-se de fadiga. Pacientes com hipotiroidismo também possuem clara evidência de hiposecreção de HLC.8-11 Finalmente, a hipótese de que os antidepressivos exerçam seus efeitos clínicos por meio da modulação direta dos hormônios glicocorticóides e de seus receptores é um dos mais impactantes e inovadores modelos dos mecanismos de ação dessa classe de fármacos.12-13

Revisaremos as evidências que embasam que: 1) a hiperatividade do eixo HPA exerce um importante papel na patogênese da depressão maior; 2) essa hiperatividade deve-se, em especial, à deficiência de feedback negativo no eixo HPA pelos hormônios glicocorticóides circulantes; 3) essa deficiência na inibição por feedback relaciona-se com uma função diminuída dos receptores de glicocorticóides (RG), que medeiam os efeitos dos hormônios glicocorticóides, incluindo o feedback negativo no eixo HPA; e 4) os antidepressivos atuam revertendo essas alterações na função dos RGs e dessa forma normalizam a hiperatividade do eixo HPA em pacientes com depressão maior.
A regulação do eixo HPA
A atividade do eixo HPA é governada pela secreção de HLC e vasopressina (AVP) pelo hipotálamo, os quais, por sua vez, ativam a secreção do hormônio adrenocorticotrópico (ACTH) pela pituitária, que finalmente estimula a secreção de glicocorticóides pelo córtex adrenal.2 Os glicocorticóides, então, interagem com seus receptores em múltiplos tecidos-alvo, incluindo o eixo HPA, onde são responsáveis pela inibição negativa por feedback da secreção do ACTH pela pituitária e do HLC a partir do hipotálamo. Embora os glicocorticóides regulem a função de quase todos os tecidos do corpo, o efeito fisiológico mais conhecido desses hormônios é a regulação do metabolismo energético. Os efeitos antiinflamatórios e imunossupressores dos glicocorticóides são evidentes em doses farmacológicas, ao passo que, fisiologicamente, esses hormônios possuem um importante papel regulatório no sistema imunológico.5
Vários fatores regulam a atividade do eixo HPA. Há evidência de inervação direta catecolaminérgica, serotonérgica e dopaminérgica nos neurônios produtores de HLC no hipotálamo e esses e outros neurotransmissores parecem influenciar a liberação de HLC. Por exemplo, a serotonina exerce uma influência estimuladora no HLC, por meio dos subtipos de receptores 5-HT1A, 5-HT1B, 5-HT1C e 5-HT2. A norepinefrina possui um efeito mais variável, sendo estimulatória em doses baixas (via receptores alpha1) e inibitória em doses altas (via receptores beta).14
Anormalidades do eixo HPA na depressão
A hiperatividade do eixo HPA na depressão maior é um dos achados mais consistentes em psiquiatria. Um percentual significativo de pacientes com depressão maior apresenta concentrações aumentadas de cortisol (o glicocorticóide endógeno nos seres humanos) no plasma, na urina e no fluido cerebrospinal (LCR); resposta exagerada de cortisol após estimulação com hormônio adrenocortitrópico (ACTH); e aumento tanto da pituitária como das glândulas adrenais.2,3,6,14-15 Hipertrofia da adrenal tem sido encontrada em pacientes deprimidos e esse achado provavelmente explica porque a resposta do cortisol ao HLC é similar em indivíduos deprimidos e em controles, já que a glândula adrenal aumentada é capaz de compensar a resposta achatada de ACTH ao HLC, geralmente observada em pacientes deprimidos.2 Também foi observado volume pituitário aumentado nesses pacientes, o que pode ser considerado um marcador da ativação excessiva do eixo HPA.16 Em um recente estudo, o primeiro episódio de psicose foi associado ao volume aumentado da pituitária, sugerindo que isso se deva à hiperativação do eixo HPA. Menor volume pituitário em indivíduos com psicose estabelecida pode também ser conseqüência de repetidos episódios de hiperatividade do eixo HPA.17
Em geral, as alterações no eixo HPA aparecem em pacientes com depressão crônica e episódios depressivos graves. Além disso, essas alterações parecem ser estado-dependentes, tendendo a se resolver com a resolução da síndrome depressiva.12
Achados provindos de múltiplas linhas de pesquisa têm fornecido evidências de que, durante a depressão, a disfunção de estruturas límbicas, incluindo o hipotálamo e o hipocampo, resulta na hipersecreção de HLC e AVP, o que, por sua vez, induz a ativação pituitário-adrenal. Além disso, as concentrações de HLC no líquido cefalo raquidiano (LCR) estão aumentadas em pacientes deprimidos não-medicados e foi encontrado um menor número de receptores de HLC no córtex frontal de vítimas de suicídio.18 Vários estudos forneceram evidências de que o HLC pode desempenhar um papel nos sinais e sintomas comportamentais da depressão (libido diminuída, apetite diminuído, alterações psicomotoras e distúrbios de sono).

Ainda que os mecanismos pelos quais o HLC extra-hipotalâmico está elevado na depressão não tenham sido entendidos, os níveis elevados de HLC no hipotálamo são considerados como relacionados à alteração de inibição por feedback pelos glicocorticóides endógenos. Dados confirmando a noção de que a inibição por feedback mediada por glicocorticóides encontra-se deficiente na depressão maior provêm de vários estudos3 que demonstram não haver supressão da secreção de cortisol. Esses estudos foram complementados por muitos testes da função neuroendócrina que examinaram a supressão de ACTH e de corticosteróides pelo glicocorticóide sintético dexametasona (DEX) teste de supressão da dexametasona (TSD). O TSD revelou que uma alta proporção de pacientes com diferentes transtornos afetivos possuem níveis de cortisol elevados,19 escapando dessa forma do efeito supressor do DEX. Depois que o HLC foi descoberto e caracterizado por Vale et al,20 estudos iniciais empregando HLC ovino ou humano em indivíduos deprimidos demonstraram que a resposta do ACTH após injeção desse neuropeptídeo encontrava-se diminuída, sugerindo que os receptores de HLC estavam desensibilizados devido à hiposensibilização homóloga pelo HLC hiper-secretado.21-22 O teste da função neuroendócrina mais sensível para detectar a desregulação do eixo HPA combina o TSD e o teste de estimulação do HLC (teste DEX/HLC).23-25 De fato, Heuser et al25 concluíram, a partir de seus estudos, que a sensibilidade desse teste é maior que 80%, dependendo da idade e sexo. Enquanto a resposta do ACTH após estímulo pelo HLC está achatada em indivíduos deprimidos, o tratamento prévio com DEX produz o efeito oposto e, paradoxalmente, aumenta a secreção de ACTH após HLC. De forma semelhante, a secreção de cortisol induzido pelo HLC é muito maior em pacientes que foram tratados previamente com DEX do que naqueles não tratados. A interpretação dos achados acima é a seguinte: o DEX, devido a sua baixa ligação à globulina carregadora de corticosteróide e seu menor acesso ao cérebro,26 atua primariamente na pituitária para suprimir o ACTH. A diminuição subseqüente de cortisol e a incapacidade do DEX em compensar níveis mais baixos de cortisol no tecido nervoso criam uma situação que é detectada pelos elementos reguladores centrais do sistema HPA como uma adrenalectomia parcial e transitória. Em resposta a essa situação, a secreção de neuropeptídeos centrais capazes de ativar a secreção do ACTH em especial o HLC e a vasopressina é aumentada. Quando a vasopressina é infundida em baixa freqüência em controles tratados previamente com DEX, infusão concomitante de HLC induz uma resposta do ACTH e do cortisol que é similar ao perfil de secreção hormonal dos indivíduos depressivos que recebem o teste combinado DEX/HLC, mas sem tratamento simultâneo com vasopressina.27
Há, no entanto, algumas limitações a esse teste. Em particular, a farmacodinâmica e a farmacocinética do DEX diferem notavelmente da do cortisol. O DEX se acopla somente ao RG (não ao RM) in vivo, não se acopla à globulina carregadora de cortisol (CBG) e possui uma meia-vida muito maior comparada ao cortisol.4,28 Em resposta a essas preocupações, desenvolvemos recentemente um teste supressor do eixo HPA utilizando prednisolona, que é similar aos glicocorticóides endógenos.29 A prednisolona é um glicocorticóide sintético semelhante ao cortisol em sua farmacodinâmica (liga-se tanto ao RM e RG, quanto ao CBG) e farmacocinética (sua meia-vida é similar à do cortisol).28 Propomos que a prednisolona em dosagem de 5 mg (que acarreta a supressão parcial do eixo HPA), junto com a avaliação do cortisol salivar, pode ser utilizada para investigar tanto o feedback negativo mediado pelo glicocorticóide deficiente quanto o intensificado, em amostras maiores de pacientes com transtornos psiquiátricos.
É interessante que estudos recentes encontraram que o cortisol, por si próprio, tem um acesso limitado ao cérebro humano.30 Portanto, não está claro até o momento se o DEX é ou não capaz de compensar no cérebro os níveis séricos baixos de cortisol, já que não está claro, em primeiro lugar, quanto do cortisol circulante é capaz de entrar no cérebro em circunstâncias normais.
Acredita-se que a hiperatividade do eixo HPA na depressão maior é secundária à hipersecreção de HLC. O HLC possui efeitos comportamentais similares em animais e em pacientes deprimidos, incluindo alterações na atividade, apetite e sono.14 Além disso, pacientes deprimidos exibem concentrações mais elevadas de HLC no LCR, assim como de HLC mRNA e proteína no núcleo paraventricular (PVN) do hipotálamo (amostras pós-mortem), e uma resposta achatada de ACTH ao estímulo com HLC.2,6 Os resultados de resposta achatada do hormônio adrenocorticotrópico (ACTH) ao HLC humano22 e ovino,26 os níveis elevados de HLC no LCR,31 o menor número de sítios receptores de HLC no córtex frontal de pacientes com depressão que cometeram suicídio,32 o maior número de neurônios produtores de HLC no núcleo paraventricular hipotalâmico de pacientes com depressão,33 e o achado de que concentrações de HLC no fluído medular diminuem durante tratamento de longo prazo com fluoxetina ou amitriptilina34 dão sustentação à idéia de que o HLC é o neuropeptídeo-chave responsável pelas alterações do eixo HPA na depressão.
Intensas pesquisas pré-clínicas têm investigado se os sinais e sintomas psicológicos da depressão estão também relacionados à hipersecreção de HLC e vasopressina e como os antidepressivos funcionam para tratar tanto o aspecto neuroendócrino como o comportamental dessa desregulação. Vários grupos de pesquisa formularam uma hipótese relacionando a desregulação hormonal aberrante de resposta ao estresse à etiologia da depressão e propuseram que os antidepressivos podem atuar por meio da normalização dessas alterações do eixo HPA.3,5,15 Enquanto a não-supressão por DEX no TSD e no teste DEX/HLC provavelmente representa uma deficiência da inibição por feedback na pituitária,4,35 a responsividade deficiente ao teste de hidrocortisona em pacientes deprimidos sugere que essas alterações de feedback também ocorram no cérebro.36
Estudos realizados tanto em animais como em humanos sugerem que o estresse nas fases iniciais de desenvolvimento pode induzir alterações persistentes na capacidade do eixo HPA em responder ao estresse na vida adulta e que esse mecanismo pode levar a uma maior suscetibilidade à depressão.37 É interessante que a hiperatividade persistente do eixo HPA também tem sido associada a índices mais altos de recaída.38-40 Estudos conduzidos com pacientes que estavam recebendo uma gama de antidepressivos mostraram que aqueles que não tiveram uma normalização no nível do cortisol após tratamento com DEX tenderam a ter pior desfecho em termos de re-hospitalização, suicídio e recorrência de depressão.38 Dois relatos recentes descreveram um estudo prospectivo analisando a relação entre os resultados do teste DEX/HLC e o desfecho clínico. Especificamente, Zobel et al39-40 descreveram uma coorte de pacientes que receberam o teste DEX/HLC em duas ocasiões distintas: na primeira semana após a admissão (ou após começarem o primeiro tratamento com antidepressivos) e alguns dias antes da alta. Os pacientes foram seguidos por seis meses após a alta. O estudo encontrou que aqueles pacientes que tiveram um aumento nos níveis de cortisol após o teste DEX/HLC entre a admissão e a alta tenderam a recair durante o período de seguimento, ao passo que aqueles que mostraram uma diminuição nos níveis de cortisol após o teste DEX/HLC tenderam a permanecer clinicamente estáveis no período de seguimento. Dessa forma, esses estudos sugerem que a avaliação do eixo HPA durante o tratamento antidepressivo pode ser útil para identificar aqueles com maior risco de recaída (ver Tabela 3).2,6,14-15,25
Devido a uma ampla variedade de estressores ativarem comprovadamente o eixo hipotálamo-pituitária-adrenal (HPA) e devido aos glicocorticóides serem o produto final da ativação do eixo HPA, esses hormônios têm sido mais comumente vistos como os agents provocateurs, ou até em casos extremos como a corporificação da patologia induzida pelo estresse. De fato, tem sido sugerido que a prolongada superprodução de glicocorticóides seja como resultado de um estresse em curso ou de uma predisposição genética para a hiperatividade do eixo HPA danifica as estruturas cerebrais (especialmente o hipocampo) essenciais para o controle do eixo HPA.41 Tem-se levantado a hipótese de que esse dano, por sua vez, leve a um circuito de pró-alimentação (feedforward) em que os estressores permanentes estimulem a superprodução de glicocorticóides indefinidamente (a "hipótese da cascata de glicocorticóides"). Devido à capacidade das altas concentrações de glicocorticóides alterarem o funcionamento celular podendo levar a um grande número de enfermidades, considera-se que essa superprodução de hormônios glicocorticóides contribua diretamente para muitas das seqüelas comportamentais e psicológicas associadas ao estresse crônico.41-42
No entanto, apesar da popularidade da hipótese da cascata de glicocorticóides, cada vez mais dados fornecem evidências de que, além do excesso de glicocorticóides, sua sinalização insuficiente pode desempenhar um papel significativo no desenvolvimento e expressão da patologia dos transtornos relacionados ao estresse.
Ainda que não ocorram conjuntamente, tanto o hipocortisolismo como a responsividade reduzida aos glicocorticóides (como determinado por testes de supressão com dexametasona) foram encontrados de forma confiável. Transtornos psiquiátricos relacionados ao estresse também foram associados à inflamação/ativação do sistema autoimune, à elevação do tônus do SNC e à hipersecreção de HLC, que são todos consistentes com regulação insuficiente de hiper-responsividade ao estresse mediada por glicocorticóide. Finalmente, os antidepressivos, fundamentais no tratamento de transtornos relacionados ao estresse, foram regularmente associados à intensificação da sinalização por glicocorticóides.
Definimos a sinalização insuficiente por glicocorticóides como qualquer estado em que a capacidade de sinalização por glicocorticóides seja inadequada para controlar sistemas responsivos ao estresse relevantes, seja como resultado de uma diminuição da biodisponibilidade hormonal (e.g., hipocortisolismo) ou como resultado de responsividade atenuada aos glicocorticóides (e.g., secundária à redução da sensibilidade do receptor dos glicocorticóides). Assim definida, a sinalização insuficiente pelos glicocorticóides não implica nenhum mecanismo específico ou deficiência absoluta, mas, ao contrário, tem seu foco no ponto final da atividade glicocorticóide. A pergunta fundamental é se a mensagem do glicocorticóide está chegando de uma forma adequada ao ambiente (externo e interno) em que um organismo se encontra. Dessa forma, mesmo no caso de hipersecreção de glicocorticóides, pode existir uma insuficiência glicocorticóide se a sensibilidade reduzida ao glicocorticóide em tecidos-alvo relevantes sobrepujar o excesso de hormônio circulante.43
Estudos neuroendócrinos recentes fornecem evidência de que há insuficiente sinalização glicocorticóide em transtornos neuropsiquiátricos relacionados ao estresse. Alteração da regulação por feedback das principais respostas ao estresse, especialmente inflamação/ativação imunológica, por outro lado, pode contribuir para a patologia relacionada ao estresse, incluindo alterações no comportamento, sensibilidade à insulina, metabolismo ósseo e respostas de imunidade adquirida. Desde uma perspectiva evolutiva, a sinalização glicocorticóide reduzida, tanto alcançada no nível do hormônio ou de seu receptor, pode impulsionar a prontidão imune e aumentar a reatividade. A ênfase na sinalização glicocorticóide insuficiente em patologias relacionadas ao estresse encoraja o desenvolvimento de estratégias terapêuticas para intensificar as vias de sinalização glicocorticóide.44
O receptor glicocorticóide
Os hormônios esteróides (e.g., glicocorticóides, estrógeno, testosterona e mineralocorticóides) são pequenas moléculas liposolúveis que se difundem através das membranas celulares. Ao contrário dos receptores dos hormônios protéicos, que estão localizados na membrana celular, os receptores desses ligantes estão localizados no citoplasma. Em resposta ao acoplamento ao ligante, os receptores de hormônios esteróides translocam-se para o núcleo, onde regulam a expressão de certos genes por meio da ligação a elementos de resposta hormonal específicos (ERHs) em suas regiões regulatórias. Os glicocorticóides mediam suas ações, incluindo a regulação do feedback do eixo HPA, por meio de dois subtipos distintos de receptores de corticosteróide intracelular, referidos como receptor mineralocorticóide (RM) e RG.4,45 O RM tem alta afinidade pelos corticosteróides endógenos e considera-se que desempenhem um papel na regulação das flutuações circadianas desses hormônios (especialmente na secreção de ACTH durante a queda progressiva diurna na secreção do cortisol). Ao contrário do RM, o RG possui uma alta afinidade pelo DEX e uma menor afinidade pelos corticosteróides endógenos. Portanto, acredita-se que o RG seja mais importante na regulação da resposta ao estresse quando os níveis endógenos de glicocorticóides estão altos. Spencer et al46 e de Kloet et al4 esclareceram que a ativação do RG é necessária para a regulação por feedback do HPA quando os níveis de glicocorticóides estão altos (resposta ao estresse, pico circadiano), mas que o RM também desempenha um papel importante na modulação da regulação dependente de RG.
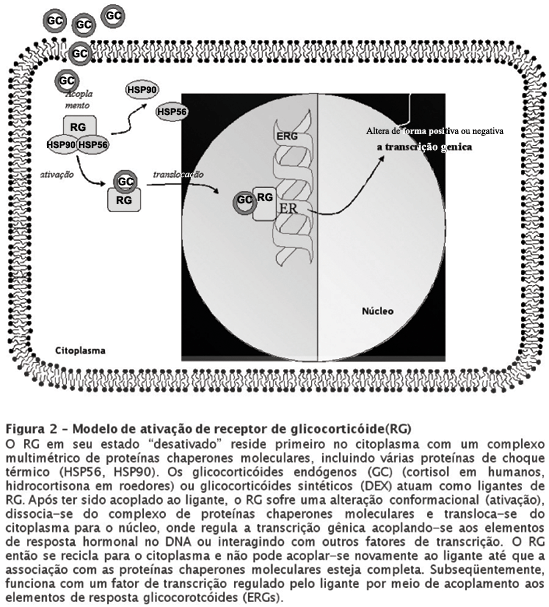
Como mencionamos antes, de acordo com o modelo de "tráfego nucleo-citoplasmático" da ação do RG (ver Figura 2), o RG em sua forma "inativa" reside essencialmente no citoplasma em associação com um complexo multimétrico de proteínas chaperones moleculares, incluindo diversas proteínas de choque térmico (HSPs).47 Após ser acoplado ao esteróide, o RG sofre uma alteração de conformação ("ativação"), dissocia-se do complexo das proteínas chaperones moleculares e transloca-se do citoplasma para o núcleo, onde pode acoplar-se aos elementos de resposta glicocorticóide (ERGs) no DNA ou interagir com outros fatores de transcrição.48 Os ERGs podem designar tanto uma regulação positiva como negativa aos genes aos quais estão acoplados. O RG ativado não pode se acoplar novamente ao ligante já que, para isso, é necessária a associação com o complexo das proteínas chaperone moleculares para manter o receptor em um estado conformacional receptivo ao hormônio.47 Os RGs possuem uma baixa afinidade mas alta capacidade para cortisol e são muito responsivos a alterações nas concentrações de cortisol. Enquanto se considera que os RMs possam estar envolvidos na atividade inibitória tônica no eixo HPA, os RGs parecem "desligar" a produção de cortisol em períodos de estresse.49
Vários grupos de pesquisa sugeriram que a hiper-reatividade do eixo HPA na depressão pode ser devido a uma anormalidade do RG no nível límbico-hipocampal.3,5,15,50 Essa anormalidade resulta numa falta de ou resistência ao glicocorticóide. De fato, vários achados em depressão são consistentes com uma anormalidade do RG. Mais notável é o fato de que os pacientes com depressão não exibam a maioria dos sintomas físicos do excesso de corticosteróide, apesar da presença freqüente de hipercortisolismo,51 sugerindo que os RGs periféricos possam ser anormais ou insensíveis na depressão. Consistentemente com o fato de que o RG é mais importante na regulação do eixo HPA quando os níveis endógenos dos glicocorticóides estão elevados,4 e que os pacientes com depressão maior exibem feedback negativo prejudicado do eixo HPA no contexto de níveis elevados de cortisol circulante,2 vários estudos têm descrito uma função reduzida do RG em pacientes deprimidos (resistência do RG) e que os antidepressivos atuam revertendo essas supostas alterações do RG.5
Receptores de glicocorticóide na depressão
Vários estudos avaliaram o RG em pacientes com depressão maior. Em geral, esses estudos mediram o número de RG diretamente ou examinaram a influência in vitro ou in vivo dos glicocorticóides nas funções conhecidas por serem reguladas pelo RG. Há informações limitadas com relação ao número e função do RG no sistema nervoso central.
Em geral, os estudos não encontraram alterações na expressão total do RG, mas encontraram menos RG na fração citosólica celular (revisada em cinco deles). Esses estudos sugerem que as alterações do RG observadas na depressão são provavelmente secundárias à compartimentalização nuclear do RG (ativação e, portanto, translocação para o núcleo e, conseqüentemente, reduzido nível de RG no citoplasma). É certamente matéria de especulação até que ponto se podem fazer inferências sobre a função dos receptores de corticosteróides no SNC a partir de estudos sobre RGs periféricos, tais como RGs de linfócitos, que podem não refletir precisamente os RGs na pituitária e no cérebro.52 Recentemente, um estudo cerebral pós-mortem encontrou uma reduzida expressão gênica de RG no lobo frontal e hipocampo e uma expressão gênica reduzida de RG e RM no lobo frontal, não somente em pacientes com depressão maior, mas também em pacientes com esquizofrenia e transtorno bipolar.53 Esses estudos fornecem evidências sugestivas de que o eixo HPA pode estar anormal em alguns pacientes com transtorno bipolar e esquizofrenia e dão sustentação à visão de que a desregulação do eixo HPA pode ter um papel em diferentes transtornos psiquiátricos.54 No entanto, outro estudo pós-mortem realizado por Lopez et al55 não encontrou diferenças no RG mRNA (mas menos RM mRNA) no hipocampo de seis vítimas de suicídio com história de depressão, comparados com um grupo de seis controles.
O estresse crônico também tem sido associado à hiperativação do eixo HPA e à função alterada do RG. Este estudo avaliou o impacto dos glicocorticóides na função de células periféricas (função imune), sabidamente inibidas pela ativação do RG, e, em particular, a conhecida capacidade do DEX de inibir a capacidade das células mononucleares sanguíneas periféricas de proliferarem em resposta a mitógenos policlonais. Por exemplo, tem sido sugerido que níveis de cortisol cronicamente elevados podem produzir um estado de resistência aos esteróides, habilitando os linfócitos a responderem com menos intensidade aos GCs. Um trabalho recentemente produzido por nosso grupo56 revelou que o estresse crônico (i.e., cuidar de pacientes dementes) em seres humanos está associado a elevações significativas nos níveis de cortisol e reduzida sensibilidade linfocítica aos GCs in vitro. Esses dados sugerem que as elevações crônicas no cortisol podem ser subjacentes à resistência dos RGs em humanos.
Estudos explorando as diferenças entre a função dos RGs em indivíduos depressivos e controles encontraram resultados bem mais consistentes, deparando-se com o fato de que os linfócitos de indivíduos não-respondedores à supressão por DEX foram mais consistentemente resistentes ao efeito inibitório da DEX administrada in vitro.3,5 Além disso, os poucos estudos que investigaram alterações na sensibilidade do RG in vitro e in vivo nos mesmos pacientes encontraram uma notável consistência de resposta. Kok et al57 demonstraram que o cortisol estimula a produção de imunoglobulinas (IgG e IgM) in vitro em controles saudáveis, ao passo que somente a produção de IgG estava aumentada em pacientes deprimidos.
Na maior parte desses estudos, os linfócitos de pacientes deprimidos não-respondedores à supressão por DEX, de acordo com o TSD, são mais resistentes ao efeito inibitório do DEX administrado in vitro comparado aos pacientes deprimidos que respondem à supressão no TSD; somado a isso, parece haver uma correlação inversa entre a concentração de cortisol plasmático e a inibição da resposta proliferativa induzida pela DEX, sugerindo um vínculo entre a hipercortisolemia e a resistência a respostas mediadas por RG in vitro. Após a recuperação clínica, a hipercortisolemia tende a se resolver e a sensibilidade dos linfócitos à DEX retorna aos níveis controle. No entanto, pesquisas em nosso laboratório relataram recentemente que a resistência adquirida ao RG pode ser demonstrada em pacientes deprimidos resistentes ao tratamento (DRT) na ausência de níveis de cortisol salivar basal elevados. Foi observado que a supressão induzida por GC da proliferação de células T e da produção de citocinas in vitro é geralmente menos marcada na depressão resistente ao tratamento (DRT), comparada com controles saudáveis.58 Em outro estudo, observouse que o impacto da administração in vivo de DEX na redistribuição linfocitária é maior no grupo controle do que em pacientes DRT.59 Em geral, as medidas da função linfocitária in vitro ou in vivo demonstram que as células de pacientes DRT parecem ser menos sensíveis aos esteróides. É tentador, no entanto, especular que a resistência aos antidepressivos nessa amostra de pacientes possa estar relacionada à resistência aos esteróides. Ainda que comumente agrupados, o hipercortisolismo e a resistência aos glicocórticóides não necessariamente ocorrem conjuntamente e podem representar estados distintos de disfunção do eixo HPA ou, pelo menos, diferentes pontos ao longo de uma evolução da patologia do eixo HPA.60-61
É interessante que os achados desses estudos tenham sido confirmados recentemente por um estudo in vivo, mostrando que pacientes deprimidos possuem uma resposta vasoconstritora reduzida à aplicação tópica de beclometasona em comparação a controles saudáveis emparelhados.62 Esse achado é sugestivo de uma sensibilidade disfuncional dos RGs periféricos na medida em que a resposta vasoconstritora à beclometasona é mediada por esses receptores e fornece uma confirmação adicional para a hipótese de uma anormalidade do RG na depressão.50 Novamente, utilizando o TSD como uma medida da ativação do eixo HPA, não foi encontrada diferença na sensibilidade dérmica à beclometasona entre pacientes com resultados normais e anormais nos resultados do TSD. Esses achados sugerem que a função RG periférica é anormal na depressão, mas que a resposta vasoconstritora à beclometasona não é necessariamente um efeito secundário da hipercortisolemia ou da hiperatividade do eixo HPA.62 Consistente com a presença de resistência do RG na depressão maior, Maguire et al63 encontraram que, apesar de terem maiores concentrações de cortisol plasmático em comparação aos controles, pacientes deprimidos melancólicos não exibem aumento nos níveis de sialiltransferases plasmáticas. As sialitransferases são uma família de enzimas que participam do metabolismo da cadeia de oligossacarídeos e são conhecidas por serem estimuladas pelos glicocorticóides por meio do RG. Não foram encontradas alterações no acoplamento de RG entre os grupos. Esses achados sugerem que a função deficiente dos RGs, e não seu número, está relacionada à sensibilidade diminuída dos níveis de sialiltransferase plasmática ao cortisol em pacientes deprimidos.
Apesar dos dados acima fornecerem sólida evidência de que há resistência aos glicocorticóides na depressão maior, existem alguns dados que sugerem que a sensibilidade aos glicocorticóides em pacientes deprimidos permanece intacta, pelo menos em alguns compartimentos do corpo. Especificamente, foi demonstrado que pacientes deprimidos exibem deposição de gordura intra-abdominal,64 que também é observada em enfermidades médicas caracterizadas por hipercortisolemia, como por exemplo a síndrome de Cushing e após o tratamento prolongado com glicocorticóides. Esses achados sugerem que os RGs intra-abdominais podem manter sua sensibilidade aos glicocorticóides, ao passo que outros tipos de tecidos/células são resistentes. Em apoio a essa possibilidade, estudos também mostraram densidade mineral óssea diminuída em pacientes deprimidos,65-66 já que glicocorticóides elevados também têm sido associados à perda óssea.
Mecanismos moleculares de resistência dos RGs na depressão
Como discutido previamente, os dados sobre RGs não são convincentes a favor da hiporegulação (downregulation) dos RGs secundária ao hipercortisolismo na depressão maior. No entanto, é concebível que o hipercortisolismo possa sobrecarregar a capacidade de reciclagem dos RGs com conseqüente diminuição da capacidade da célula de responder à estimulação subseqüente. No entanto, uma segunda possibilidade é que a função RG esteja alterada na depressão maior por meio de mecanismos independentes de ligação.3,5 O conceito da regulação "independente de ligação covalente" da função dos RGs deriva de achados de que a função receptora de esteróide é regulada não somente pela ligação ao esteróide, mas também pelas vias de transdução de sinal, impulsionadas por compostos não relacionados a esteróides.67 Por exemplo, pesquisas demonstraram que a função dos RGs pode ser influenciada por uma miríade de compostos não-esteróides, incluindo citocinas pró-inflamatórias, tais como interleucina-1,68-69 participantes da cascata de AMPc, como a proteína quinase A (PKA).70 O restante desta revisão será focado em dois mecanismos que têm sido investigados em nosso laboratório e que são alvos potenciais do tratamento com antidepressivos: as citocinas pró-inflamatórias e os transportadores de hormônios esteróides. No entanto, outros fatores não revisados aqui poderiam também estar envolvidos na resistência adquirida pelos RGs na depressão. A fosforilação do RG e/ou de outros coativadores de receptores de esteróides pela proteína quinase dependente de AMPc possui um papel relevante na regulação da função dos RGs. Esses achados são particularmente intrigantes tendo em conta o fato de que pacientes deprimidos exibem função de proteína G reduzida em células mononucleares71 e reduzida atividade de proteína quinase dependente de AMPc em culturas de fibroblastos.72 Dessa forma, é possível que uma interrupção na via AMPc/PKA descrita na depressão maior esteja ligada à resistência dos RGs nesse transtorno e que os antidepressivos possam superar essas alterações nos receptores por meio de um efeito direto nessa via. Vale ressaltar que foi recentemente demonstrado que uma ligação não-covalente b-isoforme do RG humano (hGRb) pode também estar implicada na resistência adquirida aos esteróides. O hGRb heterodimerisa com o hGRa ligado covalentemente e se transloca para o núcleo para atuar como um inibidor negativo dominante do receptor clássico. Dessa forma, uma alta expressão de hGRb poderia participar no desenvolvimento da resistência adquirida ao esteróide, ao passo que uma expressão anormalmente alta de hGRa e baixa de hGRb poderiam levar ao estado de hipersensibilidade ao glicocorticóide.73 É possível que a proporção GRa/GRb possa estar alterada nos não-respondedores ao TSD, levando à resistência adquirida dos GRs. Não podemos excluir a participação das alterações no sistema de transdução de RG (e.g., expressão alterada de AP-1 e NF-kB, proteínas de choque térmico) na promoção da sensibilidade dos tecidos aos glicocorticóides.74 Em resumo, vários mecanismos podem mediar a "resistência adquirida ao esteróide" (ver Tabela 3) e exploraremos alguns desses mecanismos em maiores detalhes a seguir.
Antidepressivos e o receptor de glicocorticóide
A hipótese de que os antidepressivos exerçam seus efeitos clínicos por meio da modulação direta do receptor de glicocorticóide (RG) é um dos mais notáveis e inovadores modelos do mecanismo de ação dessa classe de drogas.5,12,75-77 Especificamente, estudos em pacientes deprimidos, modelos animais e celulares demonstraram que os antidepressivos aumentam a expressão de RG, intensificam sua função, e promovem sua translocação nuclear; isso, por sua vez, está associado ao feedback negativo intensificado pelos glicocorticóides endógenos e, portanto, ao repouso reduzido e atividade estimulada do eixo HPA5 (ver Tabela 4). Esses efeitos, por sua vez, podem contribuir para a ação terapêutica dessa classe de drogas (ver Figura 3). No entanto, a relação entre a estrutura química, os mecanismos farmacológicos e os efeitos conhecidos no RG ainda têm que ser melhor esclarecidos.
Trabalhos desenvolvidos em nosso laboratório e em outras partes no decorrer dos últimos anos têm tentado compreender os mecanismos pelos quais os antidepressivos regulam o RG, examinando essa interação in vitro. Descrevemos em células L929s (fibroblastos de ratos) que a incubação com o antidepressivo tricíclico desipramina induz a translocação do RG do citoplasma para o núcleo na ausência de esteróides.75,78 Além disso, encontramos que a incubação concomitante de desipramina e DEX leva a uma intensificação da transcrição gênica mediada por RG, ao passo que a incubação prévia com desipramina seguida por DEX leva a uma redução da transcrição gênica mediada por RG.75,78 Este último achado foi recentemente reproduzido por Budziszewska et al,77 que também encontraram que a incubação prévia de células L929 de fibroblastos de ratos com vários antidepressivos (incluindo desipramina) reduz a transcrição gênica mediada por RG induzida por um tratamento subseqüente com corticosterona ou DEX.
Alguns de nossos trabalhos mais recentes sugerem um possível papel dos transportadores de esteróides da membrana, tais como o complexo de resistência a múltiplas drogas glicoproteína-p (MDR PGP), na regulação da função de RG durante o tratamento com antidepressivos e, possivelmente, na depressão maior. Alguns ligantes de RG, como cortisol e DEX (mas não a hidrocortisona), são ativamente excretados das células pelo MDR PGP e outros transportadores da membrana que pertencem à família dos transportadores de esteróides dependentes de ATP.4,75,79 Tem sido extensivamente descrito que o MDR PGP regula as concentrações intracelulares de esteróides, secreta os metabólitos que naturalmente ocorrem e as substâncias tóxicas diretamente nos tratos urinário e gastrointestinal, e confere resistência ao tratamento de tumores, pois facilita a excreção dos agentes antimitóticos.80-81 Além disso, o MDR PGP localizado na membrana apical das células endoteliais da barreira hemato-encefálica limita o acesso do DEX e do cortisol (mas não da hidrocortisona) ao cérebro humano, bem como às células periféricas, tais como linfócitos.82-83 A expressão in vitro de MDR PGP pode induzir a resistência do RG em uma linhagem de células de timoma,83 reproduzindo dessa forma uma condição similar à descrita em linfócitos de pacientes com depressão maior. Além disso, alguns antidepressivos são capazes inibir o MDR PGP em células tumorais84-86 ao serem transportadas por esse complexo.87 Baseados nessa evidência, formulamos a hipótese de que um mecanismo pelo qual os antidepressivos regulam a função dos RGs in vitro (e, teoricamente, in vivo) é através da regulação da função do MDR PGP e, portanto, do acesso intracelular aos glicocorticóides.
Nós exploramos recentemente essa hipótese examinando os efeitos de uma gama de antidepressivos na função RG (transcrição gênica mediada por RG) na presença de esteróides que são afetados de forma diferenciada pelo transportador de esteróide da membrana L929. Além disso, avaliamos a capacidade do inibidor do transportador de esteróide da membrana, verapamil, em reverter os efeitos dos antidepressivos na função do RG. Ainda que não esteja claro se o transportador de esteróide da membrana das células L929 é idêntico ao MDR PGP,88-89 nós e outros autores temos demonstrado que eles compartilham do mesmo perfil de substratos enzimáticos.75,89-90 De fato, nossos achados sugerem fortemente que uma inibição induzida por antidepressivo (ou hiposensibilização) do transportador de esteróide da membrana L929 é relevante para a intensificação in vitro da função do RG.75 Na realidade, encontramos que três antidepressivos distintos (desipramina, clomipramina e paroxetina) aumentam a função do RG na presença de DEX e cortisol (que são excretados das células pelo MDR PGP), mas não de hidrocortisona (que não é excretada por esse transportador). Além disso, a clomipramina (o antidepressivo que provoca a mais forte potencialização da transcrição gênica mediada por RG na presença de DEX ou cortisol) não possui nenhum efeito na presença de DEX, após o bloqueio do transportador de esteróide com verapamil.
É importante notar que nossos dados in vitro são consistentes com estudos em animais. Por exemplo, a clomipramina em dose de 10 mg/Kg/dia, durante dois dias, superou totalmente a resistência a drogas anticancerígenas por tumores subcutâneos em camundongos.85 Ressalte-se que a dose utilizada neste estudo está dentro dos limites (10-20 mg/Kg/dia) utilizados na maioria dos estudos em animais que demonstram hiper-sensibilização de RG por antidepressivos tricíclicos.5 Um artigo recente de Uhr et al87 descreveu que a amitriptilina, mas não a fluoxetina, é transportada pelo MDR PGP. Por outro lado, nossos dados são também consistentes com o estudo de Przegalinski et al9,1 que mostram que o tratamento prévio de ratos com nifedipina (outro inibidor de MDR PGP) evita a hiper-sensibilização de RG do hipocampo induzida pelo tratamento crônico com desipramina, amitriptilina ou eletroconvulsoterapia. Mais ainda, nossa hipótese de que a modulação dos transportadores de esteróides da membrana é importante para os efeitos dos antidepressivos nos RGs é uma explicação potencial sobre como drogas química e farmacologicamente não relacionadas podem ter efeitos similares nos RGs. Na verdade, os antidepressivos, assim como outros inibidores de transportadores de esteróides da membrana, parecem modular o MDR PGP, interagindo diretamente com os fosfolipídios da membrana um efeito que não é mediado pelos receptores e está relacionado às propriedades fisico-químicas das drogas, ou seja, liposolubilidade e carga elétrica.79 Finalmente, nossos dados são consistentes com todas as evidências in vivo, em humanos e animais, dando sustentação à noção de que o tratamento com antidepressivos aumenta a função dos RGs.5 Portanto, parece plausível que os efeitos dos antidepressivos in vivo estejam relacionados principalmente aos efeitos nos transportadores de esteróides da membrana, levando a uma função dos RGs aumentada, e propomos que os transportadores de esteróides de membrana, especialmente os que regulam o acesso dos glicocorticóides ao cérebro in vivo, como a MDR PGP, podem ser um alvo fundamental para a ação antidepressiva.

Em síntese, os efeitos dos glicocorticóides são mediados pelos RGs. Vários estudos têm demonstrado que a função do RG encontra-se prejudicada na depressão maior, resultando em reduzido feedback negativo mediado por RG no eixo HPA e produção e secreção aumentadas de HLC em várias regiões cerebrais possivelmente envolvidas na etiologia da depressão. O conceito da sinalização deficiente pelo RG é um mecanismo chave na patogênese da depressão. Os dados indicam que os antidepressivos têm efeitos diretos no RG, conduzindo à função intensificada e à expressão aumentada do RG. O mecanismo de alteração desses receptores envolve também componentes não-esteróides, tais como citocinas e neurotransmissores. Além disso, evidências que sugerem que os transportadores de esteróides da membrana, como a glicoproteína-p MDR, podem ser alvos fundamentais do tratamento antidepressivo. As pesquisas nesta área estão conduzindo a novos insights na fisiopatologia e tratamento dos transtornos afetivos.
Referências
1. Checkley S. The neuroendocrinology of depression and chronic stress. Br Med Bull. 1996;52(3):597-617.
2. Nemeroff CB. The corticotropin-releasing factor (CRF hypothesis of depression: new findings and new directions. Mol Psychiatry. 1996;1(4):336.
3. Juruena MF, Cleare AJ, Bauer ME, Pariante CM. Molecular mechanism of GR sensitivity and relevance for affective disorders for special issue Acta Neuropsychiatrica. 2003;15(3):354-67.
4. de Kloet ER, Vreugdenhil E, Oitzl MS, Joels M. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 1998;19(3):269-301.
5. Pariante CM, Miller AH. Glucocorticoid receptors in major depression: relevance to pathophysiology and treatment. Biol Psychiatry. 2001;49(5):391-404.
6. Gold PW, Goodwin FK, Chrousos GP. Clinical and biochemical manifestation of depression. Relation to the neurobiology of stress. N Engl J Med. 1988;319(7):;413-20. Erratum in: N Engl J Med. 1988;319921):1428.
7. Tsigos C, Chrousos GP. Hypothalamicpituitaryadrenal axis, neuroendocrine factors and stress. J Psychosom Res. 200253(4):865-71.
8. Kellner M, Yehuda R. Do panic disorder and posttraumatic stress disorder share a common psychoneuroendocrinology? Psychoneuroendocrinology. 1999;24(5):485-504.
9. Cleare AJ, Blair D, Chambers S, Wessely S. Urinary free cortisol in chronic fatigue syndrome. Am J Psychiatry. 2001;158(4):641-3.
10. Cleare AJ, Miell J, Heap E, Sookdeo S, Young L, Malhi GS, et al. Hypothalamo -pituitary-adrenal axis function in chronic fatigue syndrome, and the effects of low-dose hydrocortisone therapy. J Clin Endocrinol Metab. 2001;86(8):3545-54.
11. Gold PW, Chrousos GP. Organization of the stress system and its dysregulation in melancholic and atypical depression: high vs low CRH/NE states. Mol Psychiatry. 2002;7(3):254-75.
12. Holsboer F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology. 2000;23(5):477-501.
13. McQuade R, Young AH. Future therapeutic targets in mood disorders: the glucocorticoid receptor. Br J Psychiatry. 2000;177:390-5.
14. Owens MJ, Nemeroff CB. The ole of HLC in the pathophysiology of affective disorders. New York: John Wiley & Sons; 1993. (Laboratory and Clinical Studies, 172).
15. Holsboer F, Barden N. Antidepressants and hypothalamic-pituitaryadrenocortical regulation. Endocr Rev. 1996;17:187-205.
16. Axelson DA, Doraiswamy PM, Boyko OB, Rodrigo EP, McDonald WM, Ritchie JC, et al. In vivo assessment of pituitary volume with magnetic resonance imaging and systematic stereology: relationship to dexamethasone suppression test results in patients. Psychiatry Res. 1992;44(1):63-70.
17. Pariante CM, Vassilopoulou K, Velakoulis D, Phillips L, Soulsby B, Wood SJ, et al. Pituitary volume in psychosis. Br J Psychiatry. 2004;185:5-10.
18. Heuser IJ, Bissette G, Dettling M, Schweiger U, Gotthardt U, Schmider J, et al. Cerebrospinal fluid concentrations of corticotropin-releasing-hormone vasopressin, and somatostatin in depressed patients and healthy controls: response to amitriptyline treatment. Depress Anxiety. 1998;8(2):71-9.
19. Carroll J. Clinical applications of the dexamethasone suppression test for endogenous depression. Pharmacopsychiatria. 1982;15(1):19-24.
20. Vale W, Spiess I, Rivier C, Rivier J. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and betaendorphin. Science. 1981;213(4514):1394-7.
21. Gold PW, Loriaux DL, Roy A, Kling MA, Calabrese JR, Kellner CH, et al. Responses to corticotrophin-releasing hormone in the hyporcortisolism of depression and Cushing's disease. N Engl J Med.1986;314(21):1329-35.
22. Holsboer F, Gerken A, von Bardeleben U, Grimm W, Beyer H, Muller OA, Stalla GK. Human corticotropinreleasing hormone (HLC) in patients with depression, alcoholism and panic disorder. Biol Psychiatry. 1986;21(7):601-11.
23. von Bardeleben U, Holsboer F. Cortisol response to a combined dexamethasone- hHLC challenge in patients with depression. J Neuroendocrinol. 1989;1:485-8.
24. von Bardeleben U, Holsboer F. Effect of age upon the cortisol response to human corticotropin-releasing hormone in depressed patients pretreated with dexamethasone. Biol Psychiatry. 1991;29(10):1042-50.
25. Heuser I, Yassouridis A, Holsboer F. The combined dexamethasone/CRH test: A refined laboratory test for psychiatric disorders. J Psychiatr Res. 1994;28(4):341-56.
26. Meijer OC, de Lange ECM, Breimer DD, de Boer AC, Workel JO, de Kloet ER. Penetration of dexamethasone into brain glucocorticoid targets is enhanced in mdrlAP-glycoprotein knockout mice. Endocrinology. 1998;139(4):1789-93.
27. von Bardeleben U, Holsboer F, Stalla GK, Muller OA. Combined administration of human corticotropin-releasing factor and lysine vasopressin induces cortisol escape from dexamethasone suppression in healthy subjects. Life Sci. 1985;37(17):1613-9.
28. Orth DN, Kovacs WJ. The adrenal cortex. In: Wilson JD, Foster DW, Kronenberg HM, Larsen PR, editors. Williams textbook of endocrinology, 9th ed. Philadelphia: W.B. Saunders 1998. p. 517-664.
29. Pariante CM, Papadopoulos AS, Poon L, Checkley SA, English J, Kerwin RW, et al. A novel prednisolone suppression test for the hypothalamic-pituitary-adrenal axis. Biol Psychiatry. 2002;51(11):922-30.
30. Karssen AM, Meijer OC, van der Sandt IC, Lucassen PJ, de Lange EC, de Boer AG, et al. Multidrug resistance P-glycoprotein hampers the access of cortisol but not of corticosterone to mouse and human brain. Endocrinology. 2001;142(6):2686-94.
31. Nemeroff CB, Widerlov E, Bissette C, Walleus H, Karlsson I, EkLund K, et al. Elevated concentrations of CSF corticotropin-releasing factor-like immunoreactivity in depressed patients. Science. 1984;226(4680):1342-4.
32. Nemeroff CB, Owens MJ, Bissette G, Andorn AC, Stanley M. Reduced corticotropin- releasing factor (HLC) binding sites in the frontal cortex of suicide victims. Arch Gen Psychiatry. 1988;45(6):377-9.
33 Raadsheer FC, Hoogendijk WIG, Stam FC, Tilders FHJ, Swaab DF. Increased numbers of corticotropinreleasing hormone expressing neurons in the hypothalamic paraventricular nucleus of depressed patients. Neuroendocrinology. 1994;60:436-44.
34. de Bellis MD, Gold PW, Geracioti TD Jr, Listwak SI, Kling MA. Association of fluoxetine treatment with reductions in CSF concentrations of corticotropinreleasing hormone and arginine vasopressin in patients with major depression. Am J Psychiatry. 1993;150(4):656-7.
35. Miller AH, Spencer RL, Pulera M, Kang S, McEwen BS, Stein M. Adrenal steroid receptor activation in rat brain and pituitary following dexamethasone: Implications for the dexamethasone suppression test. Biol Psychiatry. 1992;32(10):850-69.
36. Young EA, Haskett RF, Murphy.Weinberg V, Watson SI, Akil H. Loss of glucocorticoid fast feedback in depression. Arch Gen Psychiatry. 1991;48(8):693-9.
37. Glover V, O'Connor TG. Effects of antenatal stress and anxiety: Implications for development and psychiatry Br J Psychiatry. 2002;180:389-91.
38. Ribeiro SCM, Tandon R, Grunhaus L, Greden JF. The DST as a predictor of outcome in depression: a meta-analysis. Am J Psychiatry. 1993;150(11):1618-29.
39. Zobel AW, Yassouridis A, Frieboes RM, Holsboer F. Prediction of medium-term outcome by cortisol response to the combined dexamethasone-CRH test in patients with remitted depression. Am J Psychiatry. 1999;156(6):949-51.
40. Zobel AW, Nickel T, Sonntag A, Uhr M, Holsboer F, Ising M. Cortisol response in the combined dexamethasone/CRH test as predictor of relapse in patients with remitted depression a prospective study. J Psychiatr Res. 2001;35(2):83-94.
41. Sapolsky RM. Glucocorticoid toxicity in the hippocampus: reversal by supplementation with brain fuels. J Neurosci. 1986;6(8):2240-4.
42. McEwen BS, Seeman T. Protective and damaging effects of mediators of stress. Elaborating and testing the concepts of allostasis and allostatic load. Ann N Y Acad Sci. 1999;896:30-47.
43. Sapolsky RM, Romero M, Munck AU. How do glucocorticoids influence stress responses? integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev. 2000;21(1):55-89.
44. Raison CL, Miller AH. When not enough is too much: the role of insufficient glucocorticoid signaling in the pathophysiology of stress-related disorders. Am J Psychiatry. 2003;160(9):1554-65.
45. McEwen BS. The neurobiology of stress: from serendipity to clinical relevance. Brain Res. 2000;886(1-2):172-89.
46. Spencer RL, Kim PJ, Kalman BA, Cole MA. Evidence for mineralocorticoid receptor facilitation of glucocorticoid receptor-dependent regulation of hypothalamic-pituitary-adrenal axis activity. Endocrinology. 1998;139(6):2718-26.
47. Pratt WB. The role of heat shock proteins in regulating the function, folding, and trafficking of the glucocorticoid receptor. J Biol Chem. 1993;268(29):21455-8.
48. Guiochon-Mantel A, Delabre K, Lescop P, Milgrom E. The Ernst Schering Poster Award. Intracellular traffic of steroid hormone receptors. J Steroid Biochem Mol Biol. 1996;56(1-6):3-9.
49. Reul JM, de Kloet ER. Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology. 1985;117(6):2505-11.
50. Modell S, Yassouridis A, Huber I, Holsboer F. Corticosteroid receptor function is decreased in depressed patients. Neuroendocrinology. 1997;65(3):216-22.
51. Murphy BEP. Treatment of major depression with steroid suppressive drugs. J Steroid Biochem Mol Biol. 1991;39(92):239-44.
52. Gametchu B. Glucocorticoid receptor-like antigen in lymphoma cell membranes: correlation to cell lysis. Science. 1987;236(4800):456-61.
53. Webster MJ, O'Grady J, Orthmann C, Weickert C. Decreased glucocorticoid receptor mRNA levels in individuals with depression, bipolar disorder and schizophrenia [abstract] Schizophr Res. 2000;41(1):111.[Presented 10th Biennial Winter Workshop on Schizophrenia. Davos, Switzerland, February 5-11, 2000].
54. Cotter D, Pariante CM. Stress on the progress of the developmental hypothesis of schizophrenia? Br J Psychiatry. 2002;181:363-5.
55. Lopez JF, Chalmers DT, Little KI, Watson SJ. E. Bennett Research Award. Regulation of serotonin 1A, glucocorticoid and mineralocorticoid receptor in rat and human hippocampus: Implications for the neurobiology of depression. Biol Psychiatry. 1998;43(8):547-73.
56. Bauer M, Vedhara K, Perks P, Wilcock G, Lightman S, Shanks N. Chronic stress in caregivers of dementia patients is associated with reduced lymphocyte sensitivity to glucocorticoids. J Neuroimmunol. 2000;103(1):84-92.
57. Kok F, Heijnen C, Bruijn J, Westenberg H, Van Ree J. Immunoglobulin production in vitro in major depression: a pilot study on the modulating action of endogenous cortisol. Biol Psychiatry. 1995;38(4):217-26.
58. Bauer ME, Papadopoulus A, Poon L, Perks P, Lightman SL, Checkley S, et al. Altered glucocorticoid immunoregulation in treatment resistant depression. Psychoneuroendocrinology. 2003 ;28(1):49-65.
59. Bauer M, Papadopoulos A, Poon L, Perks P, Lightman S, Checkley S, Shanks N. Dexamethasone-induced effects on lymphocyte distribution and expression of adhesion molecules in treatment resistant major depression. Psychiatry Res. 2002;113(1-2):1-15.
60. Asnis GM, Haibreich U, Ryan ND, Rabinowicz H, Puig-Antich J, Nelson B, et al. The relationship of the dexamethasone suppression test (1 mg and 2 mg) to basal plasma cortisol levels in endogenous depression. Psychoneuroendocrinology. 1987;12(4):295-301.
61. Miller AH, Sastry G, Speranza Jr AJ, Lawlor BA, Mohs RC, Ryan TM, et al. Lack of association between cortisol hypersecretion and nonsuppression on the DST in patients with Alzheimer's disease. Am J Psychiatry. 1994;151(2):267-70.
62. Cotter P, Mulligan O, Landau S, Papadopoulos A, Lightman S, Checkley S. Vasoconstrictor response to topical beclomethasone in major depression. Psychoneuroendocrinology. 2002;27(4):475-87.
63. Maguire TM, Thakore J, Dinan TG, Hopwood S, Breen KC. Plasma sialyltransferase levels in psychiatric disorders as a possible indicator of HPA axis function. Biol Psychiatry. 1997;41(11):1131-6.
64. Thakore JH, Richards PJ, Reznek RH, Martin A, Dinan TG. Increased intra-abdominal fat deposition in patients with major depression illness as measured by computed tomography. Biol Psychiatry. 1997;41(11):1140-2.
65. Schweiger U, Deuschle M, Korner A, Lammers CH, Schmider J, Gotthard U, et al. Low lumbar bone mineral density in patients with major depression. Am J Psychiatry. 1994;151(11):1691-3.
66. Michelson D, Stratakis C, Hill L, Reynolds J, Galliven E, Chrousos G, Gold P. Bone mineral density in women with depression. N Engl J Med.1996;335(16):1176-81.
67. O'Malley BW, Schrader WT, Mani S, Smith C, Weigel NL, Conneely OM, et al. An alternative ligand independent pathway for activation of steroid receptors. Recent Prog Horm Res. 1995;50:333-47.
68. Miller AH, Pariante CM, Pearce BD. Effects of cytokines on glucocorticoid receptor expression and function: Glucocorticoid resistance and relevance to depression. Adv Exp Med Biol. 1999;461:107-16.
69 Pariante CM, Pearce BD, Pisell TL, Sanchez CI, Po C, Su C, Miller AH. The proinflammatory cytokine, interleukin-1 alpha, reduces glucocorticoid receptor translocation and function. Endocrinology. 1999;140(9):4359-66.
70. Rangarajan PN, Umesono K, Evans RM. Modulation of glucocorticoid receptor function by protein kinase A Mol Endocrinol. 1992;6(9):1451-7.
71. Avissar S, Nechamkin Y, Roitman G, Schreiber G. Reduced G protein functions and immunoreactive levels in mononuclear leukocyts of patients with depression. Am J Psychiatry. 1997;154(2):211-7.
72. Shelton RC, Mainer DH, Sulser F. cAMP-dependent protein kinase activity in major depression. Am J Psychiatry. 1996;153(8):1037-42.
73. Castro M, Elliot S, Kino T, Bamberger C, Karl M, Webster E, Chrousos G. The non-ligand binding beta-isoform of the human glucocorticoid receptor (hGC-beta): tissue levels, mechanism of action, and potential physiologic role. Mol Med. 1996;2(5):597-607.
74. Bronnegard M, Stierna P, Marcus C. Glucocorticoid resistant syndromes - molecular basis and clinical presentations. J Neuroendocrinol. 1996;8(6):405-15.
75. Pariante CM, Makoff A, Lovestone S, Feroli S, Heyden A, Miller AH, Kerwin RW. Antidepressants enhance glucocorticoid receptor function in vitro by modulating the membrane steroid transporters. Br J Pharmacol. 2001;13496):1335-43.
76. Barden N. Regulation of corticosteroid receptor gene expression in depression and antidepressant action. J Psychiatry Neurosci. 1999;24(1):25-39.
77. Budziszewska B, Jaworska-Feil L, Kajta M, Lason W. Antidepressant drugs inhibit glucocorticoid receptor mediated gene transcription-a possible mechanism. Br J Pharmacol. 2000;130(6):1385-93.
78. Pariante CM, Pearce BD, Pisell TL, Owens MJ, Miller AH. Steroid-independent translocation of the glucocorticoid receptor by the antidepressant desipramine. Mol Pharmacol. 1997;52(4):571-81.
79. Pariante CM, Pearce BD, Pisell TL, Su C, Miller AH. The steroid receptor antagonists, RU486 and RU40555, activate glucocorticoid receptor translocation and are not excreted by the steroid hormone transporter in L929 cells. J Endocrinol. 2001;169(2):309-20.
80. Krishna R, Mayer LD. Multidrug resistance (MDR) in cancer. Mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. Eur J Pharm Sci. 2000;11(4):265-83.
81. Ueda K, Okamura N, Hirai M, Tanigawara Y, Saeki T, Kioka N, / et al. Human P-glycoprotein transports cortisol, aldosterone, and dexamethasone, but not progesterone. J Biol Chem. 1992;267(34):24248-52.
82. de Kloet ER, Meijer OC, Vreugdenhil E, Joels M.The Yin and Yang of nuclear receptors: symposium on nuclear receptors in brain, Oegstgeest, The Netherlands, 13-14 April 2000. Trends Endocrinol Metab. 2000;11(6):245-8.
83. Bourgeois S, Gruol DJ, Newby RF, Rajah FM. Expression of an mdr gene is associated with a new form of resistance to dexamethasone-induced apoptosis. Mol Endocrinol. 1993;7(5):840-51.
84. Varga A, Nugel H, Baehr R, Marx U, Hever A, Nacsa J, Ocsovsky I, Molnar J. Reversal of multidrug resistance by amitriptyline in vitro. Anticancer Res. 1996;16(1):209-12.
85. Merry S, Hamilton TG, Flanigan P, Freshney RI, Kaye SB. Circumvention of pleiotropic drug resistance in subcutaneous tumours in vivo with verapamil and clomipramine. Eur J Cancer. 1991; 27(1):31-4.
86. Szabó D, Szabó Jr G, Ocsovszki I, Aszalos A, Molnár J. Anti-psychotic drugs reverse multidrug resistance of tumor cell lines and human AML cell ex-vivo. Cancer Lett. 1999;1399(1):115-9.
87. Uhr M, Steckler T, Yassouridis A, Holsboer F. Penetration of amitriptyline, but not fluoxetine, into brain is enhanced in mice with blood-brain barrier deficiency due to MDR1a p-glycoprotein gene disruption. Neuropsychopharmacology. 2000;22(4):380-7.
88. Kralli A, Yamamoto KR. An FK506-sensitive transporter selectively decreases intracellular levels and potency of steroid hormones. J Biol Chem. 1996;271(29):17152-6.
89. Marsaud V, Mercier-Bodard C, Fortin D, Le Bihan S, Renoir JM. Dexamethasone and triamcinolone acetonide accumulation in mouse fibroblasts is differently modulated by the immunosuppressants cyclosporin A, FK506, rapamycin and their analogues, as well as by other p-glycoprotein ligands. J Steroid Biochem Molec Biol. 1998;66(1-2):11-25.
90. Medh RD, Lay RH, Schmidt TJ. Agonist-speciffic modulation of glucocorticoid receptor-mediated transcription by immunosuppressants. Mol Cell Endocrinol. 1998;138(1-2):11-23.
91. Przegalinski E, Budziszewska B, Siwanowicz J, Jaworska L. The effect of repeated combined treatment with nifedipine and antidepressant drugs or electroconvulsive shock on the hippocampal corticosteroid receptors in rats. Neuropharmacology. 1993;32(12):1397-400.
92. Pariante CM, Thomas SA, Lovestone S, Makoff A, Kerwin RW. Do antidepressants regulate how cortisol affects the brain? Psychoneuroendocrinology. 2004;29(4):423-47.
93. Pariante CM, Kim RB, Makoff A, Kerwin RW. Antidepressant fluoxetine enhances glucocorticoid receptor function in vitro by modulating membrane steroid transporters. Br J Pharmacol. 2003;139(6):1111-8.
94. Pariante CM, Hye A, Williamson R, Makoff A, Lovestone S, Kerwin RW. The antidepressant clomipramine regulates cortisol intracellular concentrations and glucocorticoid receptor expression in fibroblasts and rat primary neurones. Neuropsychopharmacology. 2003;28(9):1553-61.
95. Lowy M, Gormley G, Reder A, Meltzer H. Immune function, glucocorticoid receptor regu lation, and depression. In: Miller, A. (Ed.), Depressive Disorders & Immunity. American Psychiatric Press, Washington, DC, 1989, pp. 107133.
96. Moalli P & Rosen S. Glucocorticoid receptors and resistance to glucocorticoids in hematologia malignancies. Leuk Lymph 1994; 15: 363-374.
97. Cypcar D & Busse W. Steroid-resistant asthma. J Allergy Clin Immunol 1993; 92: 362-372.
98. Hennebold J & Daynes R. Microenvironmental control of glucocorticoid functions in immune regulation. In: Rook G & Lightman S. Steroid hormones and the T-cell cytokine profile. London: Springer-Verlag. 1997, pp. 101-134.
99. Rosner W. Plasma steroid-binding proteins. Endocr Metab Clin North Amer 1991; 20: 697-720.
100. Adcock I, Lane S, Brown C, Peters M, Lee T, Barnes P. Differences in binding of glucocorticoid receptor to DNA in steroid-resistant asthma. J Immunol 1995; 154; 3500-3505.
Financiamento: Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - CAPES (Nº 1517023) e National Alliance for Research on Schizophrenia and Depression - NARSAD.
Recebido em 06.06.2004
Aceito em 30.06.2004
O arquivo disponível sofreu correções conforme ERRATA publicada no Volume 26 Número 4 da revista.
- 1. Checkley S. The neuroendocrinology of depression and chronic stress. Br Med Bull. 1996;52(3):597-617.
- 2. Nemeroff CB. The corticotropin-releasing factor (CRF hypothesis of depression: new findings and new directions. Mol Psychiatry. 1996;1(4):336.
- 3. Juruena MF, Cleare AJ, Bauer ME, Pariante CM. Molecular mechanism of GR sensitivity and relevance for affective disorders for special issue Acta Neuropsychiatrica. 2003;15(3):354-67.
- 4. de Kloet ER, Vreugdenhil E, Oitzl MS, Joels M. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 1998;19(3):269-301.
- 5. Pariante CM, Miller AH. Glucocorticoid receptors in major depression: relevance to pathophysiology and treatment. Biol Psychiatry. 2001;49(5):391-404.
- 6. Gold PW, Goodwin FK, Chrousos GP. Clinical and biochemical manifestation of depression. Relation to the neurobiology of stress. N Engl J Med. 1988;319(7):;413-20.
- 7. Tsigos C, Chrousos GP. Hypothalamicpituitaryadrenal axis, neuroendocrine factors and stress. J Psychosom Res. 200253(4):865-71.
- 8. Kellner M, Yehuda R. Do panic disorder and posttraumatic stress disorder share a common psychoneuroendocrinology? Psychoneuroendocrinology. 1999;24(5):485-504.
- 9. Cleare AJ, Blair D, Chambers S, Wessely S. Urinary free cortisol in chronic fatigue syndrome. Am J Psychiatry. 2001;158(4):641-3.
- 10. Cleare AJ, Miell J, Heap E, Sookdeo S, Young L, Malhi GS, et al. Hypothalamo -pituitary-adrenal axis function in chronic fatigue syndrome, and the effects of low-dose hydrocortisone therapy. J Clin Endocrinol Metab. 2001;86(8):3545-54.
- 11. Gold PW, Chrousos GP. Organization of the stress system and its dysregulation in melancholic and atypical depression: high vs low CRH/NE states. Mol Psychiatry. 2002;7(3):254-75.
- 12. Holsboer F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology. 2000;23(5):477-501.
- 13. McQuade R, Young AH. Future therapeutic targets in mood disorders: the glucocorticoid receptor. Br J Psychiatry. 2000;177:390-5.
- 14. Owens MJ, Nemeroff CB. The ole of HLC in the pathophysiology of affective disorders. New York: John Wiley & Sons; 1993. (Laboratory and Clinical Studies, 172).
- 15. Holsboer F, Barden N. Antidepressants and hypothalamic-pituitaryadrenocortical regulation. Endocr Rev. 1996;17:187-205.
- 16. Axelson DA, Doraiswamy PM, Boyko OB, Rodrigo EP, McDonald WM, Ritchie JC, et al. In vivo assessment of pituitary volume with magnetic resonance imaging and systematic stereology: relationship to dexamethasone suppression test results in patients. Psychiatry Res. 1992;44(1):63-70.
- 17. Pariante CM, Vassilopoulou K, Velakoulis D, Phillips L, Soulsby B, Wood SJ, et al. Pituitary volume in psychosis. Br J Psychiatry. 2004;185:5-10.
- 18. Heuser IJ, Bissette G, Dettling M, Schweiger U, Gotthardt U, Schmider J, et al. Cerebrospinal fluid concentrations of corticotropin-releasing-hormone vasopressin, and somatostatin in depressed patients and healthy controls: response to amitriptyline treatment. Depress Anxiety. 1998;8(2):71-9.
- 19. Carroll J. Clinical applications of the dexamethasone suppression test for endogenous depression. Pharmacopsychiatria. 1982;15(1):19-24.
- 20. Vale W, Spiess I, Rivier C, Rivier J. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and betaendorphin. Science. 1981;213(4514):1394-7.
- 21. Gold PW, Loriaux DL, Roy A, Kling MA, Calabrese JR, Kellner CH, et al. Responses to corticotrophin-releasing hormone in the hyporcortisolism of depression and Cushing's disease. N Engl J Med.1986;314(21):1329-35.
- 22. Holsboer F, Gerken A, von Bardeleben U, Grimm W, Beyer H, Muller OA, Stalla GK. Human corticotropinreleasing hormone (HLC) in patients with depression, alcoholism and panic disorder. Biol Psychiatry. 1986;21(7):601-11.
- 23. von Bardeleben U, Holsboer F. Cortisol response to a combined dexamethasone- hHLC challenge in patients with depression. J Neuroendocrinol. 1989;1:485-8.
- 24. von Bardeleben U, Holsboer F. Effect of age upon the cortisol response to human corticotropin-releasing hormone in depressed patients pretreated with dexamethasone. Biol Psychiatry. 1991;29(10):1042-50.
- 25. Heuser I, Yassouridis A, Holsboer F. The combined dexamethasone/CRH test: A refined laboratory test for psychiatric disorders. J Psychiatr Res. 1994;28(4):341-56.
- 26. Meijer OC, de Lange ECM, Breimer DD, de Boer AC, Workel JO, de Kloet ER. Penetration of dexamethasone into brain glucocorticoid targets is enhanced in mdrlAP-glycoprotein knockout mice. Endocrinology. 1998;139(4):1789-93.
- 27. von Bardeleben U, Holsboer F, Stalla GK, Muller OA. Combined administration of human corticotropin-releasing factor and lysine vasopressin induces cortisol escape from dexamethasone suppression in healthy subjects. Life Sci. 1985;37(17):1613-9.
- 28. Orth DN, Kovacs WJ. The adrenal cortex. In: Wilson JD, Foster DW, Kronenberg HM, Larsen PR, editors. Williams textbook of endocrinology, 9th ed. Philadelphia: W.B. Saunders 1998. p. 517-664.
- 29. Pariante CM, Papadopoulos AS, Poon L, Checkley SA, English J, Kerwin RW, et al. A novel prednisolone suppression test for the hypothalamic-pituitary-adrenal axis. Biol Psychiatry. 2002;51(11):922-30.
- 30. Karssen AM, Meijer OC, van der Sandt IC, Lucassen PJ, de Lange EC, de Boer AG, et al. Multidrug resistance P-glycoprotein hampers the access of cortisol but not of corticosterone to mouse and human brain. Endocrinology. 2001;142(6):2686-94.
- 31. Nemeroff CB, Widerlov E, Bissette C, Walleus H, Karlsson I, EkLund K, et al. Elevated concentrations of CSF corticotropin-releasing factor-like immunoreactivity in depressed patients. Science. 1984;226(4680):1342-4.
- 32. Nemeroff CB, Owens MJ, Bissette G, Andorn AC, Stanley M. Reduced corticotropin- releasing factor (HLC) binding sites in the frontal cortex of suicide victims. Arch Gen Psychiatry. 1988;45(6):377-9.
- 33 Raadsheer FC, Hoogendijk WIG, Stam FC, Tilders FHJ, Swaab DF. Increased numbers of corticotropinreleasing hormone expressing neurons in the hypothalamic paraventricular nucleus of depressed patients. Neuroendocrinology. 1994;60:436-44.
- 34. de Bellis MD, Gold PW, Geracioti TD Jr, Listwak SI, Kling MA. Association of fluoxetine treatment with reductions in CSF concentrations of corticotropinreleasing hormone and arginine vasopressin in patients with major depression. Am J Psychiatry. 1993;150(4):656-7.
- 35. Miller AH, Spencer RL, Pulera M, Kang S, McEwen BS, Stein M. Adrenal steroid receptor activation in rat brain and pituitary following dexamethasone: Implications for the dexamethasone suppression test. Biol Psychiatry. 1992;32(10):850-69.
- 36. Young EA, Haskett RF, Murphy.Weinberg V, Watson SI, Akil H. Loss of glucocorticoid fast feedback in depression. Arch Gen Psychiatry. 1991;48(8):693-9.
- 37. Glover V, O'Connor TG. Effects of antenatal stress and anxiety: Implications for development and psychiatry Br J Psychiatry. 2002;180:389-91.
- 38. Ribeiro SCM, Tandon R, Grunhaus L, Greden JF. The DST as a predictor of outcome in depression: a meta-analysis. Am J Psychiatry. 1993;150(11):1618-29.
- 39. Zobel AW, Yassouridis A, Frieboes RM, Holsboer F. Prediction of medium-term outcome by cortisol response to the combined dexamethasone-CRH test in patients with remitted depression. Am J Psychiatry. 1999;156(6):949-51.
- 40. Zobel AW, Nickel T, Sonntag A, Uhr M, Holsboer F, Ising M. Cortisol response in the combined dexamethasone/CRH test as predictor of relapse in patients with remitted depression a prospective study. J Psychiatr Res. 2001;35(2):83-94.
- 41. Sapolsky RM. Glucocorticoid toxicity in the hippocampus: reversal by supplementation with brain fuels. J Neurosci. 1986;6(8):2240-4.
- 42. McEwen BS, Seeman T. Protective and damaging effects of mediators of stress. Elaborating and testing the concepts of allostasis and allostatic load. Ann N Y Acad Sci. 1999;896:30-47.
- 43. Sapolsky RM, Romero M, Munck AU. How do glucocorticoids influence stress responses? integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev. 2000;21(1):55-89.
- 44. Raison CL, Miller AH. When not enough is too much: the role of insufficient glucocorticoid signaling in the pathophysiology of stress-related disorders. Am J Psychiatry. 2003;160(9):1554-65.
- 45. McEwen BS. The neurobiology of stress: from serendipity to clinical relevance. Brain Res. 2000;886(1-2):172-89.
- 46. Spencer RL, Kim PJ, Kalman BA, Cole MA. Evidence for mineralocorticoid receptor facilitation of glucocorticoid receptor-dependent regulation of hypothalamic-pituitary-adrenal axis activity. Endocrinology. 1998;139(6):2718-26.
- 47. Pratt WB. The role of heat shock proteins in regulating the function, folding, and trafficking of the glucocorticoid receptor. J Biol Chem. 1993;268(29):21455-8.
- 48. Guiochon-Mantel A, Delabre K, Lescop P, Milgrom E. The Ernst Schering Poster Award. Intracellular traffic of steroid hormone receptors. J Steroid Biochem Mol Biol. 1996;56(1-6):3-9.
- 49. Reul JM, de Kloet ER. Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology. 1985;117(6):2505-11.
- 50. Modell S, Yassouridis A, Huber I, Holsboer F. Corticosteroid receptor function is decreased in depressed patients. Neuroendocrinology. 1997;65(3):216-22.
- 51. Murphy BEP. Treatment of major depression with steroid suppressive drugs. J Steroid Biochem Mol Biol. 1991;39(92):239-44.
- 52. Gametchu B. Glucocorticoid receptor-like antigen in lymphoma cell membranes: correlation to cell lysis. Science. 1987;236(4800):456-61.
- 54. Cotter D, Pariante CM. Stress on the progress of the developmental hypothesis of schizophrenia? Br J Psychiatry. 2002;181:363-5.
- 55. Lopez JF, Chalmers DT, Little KI, Watson SJ. E. Bennett Research Award. Regulation of serotonin 1A, glucocorticoid and mineralocorticoid receptor in rat and human hippocampus: Implications for the neurobiology of depression. Biol Psychiatry. 1998;43(8):547-73.
- 56. Bauer M, Vedhara K, Perks P, Wilcock G, Lightman S, Shanks N. Chronic stress in caregivers of dementia patients is associated with reduced lymphocyte sensitivity to glucocorticoids. J Neuroimmunol. 2000;103(1):84-92.
- 57. Kok F, Heijnen C, Bruijn J, Westenberg H, Van Ree J. Immunoglobulin production in vitro in major depression: a pilot study on the modulating action of endogenous cortisol. Biol Psychiatry. 1995;38(4):217-26.
- 58. Bauer ME, Papadopoulus A, Poon L, Perks P, Lightman SL, Checkley S, et al. Altered glucocorticoid immunoregulation in treatment resistant depression. Psychoneuroendocrinology. 2003 ;28(1):49-65.
- 59. Bauer M, Papadopoulos A, Poon L, Perks P, Lightman S, Checkley S, Shanks N. Dexamethasone-induced effects on lymphocyte distribution and expression of adhesion molecules in treatment resistant major depression. Psychiatry Res. 2002;113(1-2):1-15.
- 60. Asnis GM, Haibreich U, Ryan ND, Rabinowicz H, Puig-Antich J, Nelson B, et al. The relationship of the dexamethasone suppression test (1 mg and 2 mg) to basal plasma cortisol levels in endogenous depression. Psychoneuroendocrinology. 1987;12(4):295-301.
- 61. Miller AH, Sastry G, Speranza Jr AJ, Lawlor BA, Mohs RC, Ryan TM, et al. Lack of association between cortisol hypersecretion and nonsuppression on the DST in patients with Alzheimer's disease. Am J Psychiatry. 1994;151(2):267-70.
- 62. Cotter P, Mulligan O, Landau S, Papadopoulos A, Lightman S, Checkley S. Vasoconstrictor response to topical beclomethasone in major depression. Psychoneuroendocrinology. 2002;27(4):475-87.
- 63. Maguire TM, Thakore J, Dinan TG, Hopwood S, Breen KC. Plasma sialyltransferase levels in psychiatric disorders as a possible indicator of HPA axis function. Biol Psychiatry. 1997;41(11):1131-6.
- 64. Thakore JH, Richards PJ, Reznek RH, Martin A, Dinan TG. Increased intra-abdominal fat deposition in patients with major depression illness as measured by computed tomography. Biol Psychiatry. 1997;41(11):1140-2.
- 65. Schweiger U, Deuschle M, Korner A, Lammers CH, Schmider J, Gotthard U, et al. Low lumbar bone mineral density in patients with major depression. Am J Psychiatry. 1994;151(11):1691-3.
- 66. Michelson D, Stratakis C, Hill L, Reynolds J, Galliven E, Chrousos G, Gold P. Bone mineral density in women with depression. N Engl J Med.1996;335(16):1176-81.
- 67. O'Malley BW, Schrader WT, Mani S, Smith C, Weigel NL, Conneely OM, et al. An alternative ligand independent pathway for activation of steroid receptors. Recent Prog Horm Res. 1995;50:333-47.
- 68. Miller AH, Pariante CM, Pearce BD. Effects of cytokines on glucocorticoid receptor expression and function: Glucocorticoid resistance and relevance to depression. Adv Exp Med Biol. 1999;461:107-16.
- 69 Pariante CM, Pearce BD, Pisell TL, Sanchez CI, Po C, Su C, Miller AH. The proinflammatory cytokine, interleukin-1 alpha, reduces glucocorticoid receptor translocation and function. Endocrinology. 1999;140(9):4359-66.
- 70. Rangarajan PN, Umesono K, Evans RM. Modulation of glucocorticoid receptor function by protein kinase A Mol Endocrinol. 1992;6(9):1451-7.
- 71. Avissar S, Nechamkin Y, Roitman G, Schreiber G. Reduced G protein functions and immunoreactive levels in mononuclear leukocyts of patients with depression. Am J Psychiatry. 1997;154(2):211-7.
- 72. Shelton RC, Mainer DH, Sulser F. cAMP-dependent protein kinase activity in major depression. Am J Psychiatry. 1996;153(8):1037-42.
- 73. Castro M, Elliot S, Kino T, Bamberger C, Karl M, Webster E, Chrousos G. The non-ligand binding beta-isoform of the human glucocorticoid receptor (hGC-beta): tissue levels, mechanism of action, and potential physiologic role. Mol Med. 1996;2(5):597-607.
- 74. Bronnegard M, Stierna P, Marcus C. Glucocorticoid resistant syndromes - molecular basis and clinical presentations. J Neuroendocrinol. 1996;8(6):405-15.
- 75. Pariante CM, Makoff A, Lovestone S, Feroli S, Heyden A, Miller AH, Kerwin RW. Antidepressants enhance glucocorticoid receptor function in vitro by modulating the membrane steroid transporters. Br J Pharmacol. 2001;13496):1335-43.
- 76. Barden N. Regulation of corticosteroid receptor gene expression in depression and antidepressant action. J Psychiatry Neurosci. 1999;24(1):25-39.
- 78. Pariante CM, Pearce BD, Pisell TL, Owens MJ, Miller AH. Steroid-independent translocation of the glucocorticoid receptor by the antidepressant desipramine. Mol Pharmacol. 1997;52(4):571-81.
- 79. Pariante CM, Pearce BD, Pisell TL, Su C, Miller AH. The steroid receptor antagonists, RU486 and RU40555, activate glucocorticoid receptor translocation and are not excreted by the steroid hormone transporter in L929 cells. J Endocrinol. 2001;169(2):309-20.
- 80. Krishna R, Mayer LD. Multidrug resistance (MDR) in cancer. Mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. Eur J Pharm Sci. 2000;11(4):265-83.
- 81. Ueda K, Okamura N, Hirai M, Tanigawara Y, Saeki T, Kioka N, / et al. Human P-glycoprotein transports cortisol, aldosterone, and dexamethasone, but not progesterone. J Biol Chem. 1992;267(34):24248-52.
- 82. de Kloet ER, Meijer OC, Vreugdenhil E, Joels M.The Yin and Yang of nuclear receptors: symposium on nuclear receptors in brain, Oegstgeest, The Netherlands, 13-14 April 2000. Trends Endocrinol Metab. 2000;11(6):245-8.
- 83. Bourgeois S, Gruol DJ, Newby RF, Rajah FM. Expression of an mdr gene is associated with a new form of resistance to dexamethasone-induced apoptosis. Mol Endocrinol. 1993;7(5):840-51.
- 84. Varga A, Nugel H, Baehr R, Marx U, Hever A, Nacsa J, Ocsovsky I, Molnar J. Reversal of multidrug resistance by amitriptyline in vitro. Anticancer Res. 1996;16(1):209-12.
- 85. Merry S, Hamilton TG, Flanigan P, Freshney RI, Kaye SB. Circumvention of pleiotropic drug resistance in subcutaneous tumours in vivo with verapamil and clomipramine. Eur J Cancer. 1991; 27(1):31-4.
- 86. Szabó D, Szabó Jr G, Ocsovszki I, Aszalos A, Molnár J. Anti-psychotic drugs reverse multidrug resistance of tumor cell lines and human AML cell ex-vivo. Cancer Lett. 1999;1399(1):115-9.
- 88. Kralli A, Yamamoto KR. An FK506-sensitive transporter selectively decreases intracellular levels and potency of steroid hormones. J Biol Chem. 1996;271(29):17152-6.
- 89. Marsaud V, Mercier-Bodard C, Fortin D, Le Bihan S, Renoir JM. Dexamethasone and triamcinolone acetonide accumulation in mouse fibroblasts is differently modulated by the immunosuppressants cyclosporin A, FK506, rapamycin and their analogues, as well as by other p-glycoprotein ligands. J Steroid Biochem Molec Biol. 1998;66(1-2):11-25.
- 90. Medh RD, Lay RH, Schmidt TJ. Agonist-speciffic modulation of glucocorticoid receptor-mediated transcription by immunosuppressants. Mol Cell Endocrinol. 1998;138(1-2):11-23.
- 91. Przegalinski E, Budziszewska B, Siwanowicz J, Jaworska L. The effect of repeated combined treatment with nifedipine and antidepressant drugs or electroconvulsive shock on the hippocampal corticosteroid receptors in rats. Neuropharmacology. 1993;32(12):1397-400.
- 92. Pariante CM, Thomas SA, Lovestone S, Makoff A, Kerwin RW. Do antidepressants regulate how cortisol affects the brain? Psychoneuroendocrinology. 2004;29(4):423-47.
- 93. Pariante CM, Kim RB, Makoff A, Kerwin RW. Antidepressant fluoxetine enhances glucocorticoid receptor function in vitro by modulating membrane steroid transporters. Br J Pharmacol. 2003;139(6):1111-8.
- 94. Pariante CM, Hye A, Williamson R, Makoff A, Lovestone S, Kerwin RW. The antidepressant clomipramine regulates cortisol intracellular concentrations and glucocorticoid receptor expression in fibroblasts and rat primary neurones. Neuropsychopharmacology. 2003;28(9):1553-61.
- 95. Lowy M, Gormley G, Reder A, Meltzer H. Immune function, glucocorticoid receptor regu lation, and depression. In: Miller, A. (Ed.), Depressive Disorders & Immunity. American Psychiatric Press, Washington, DC, 1989, pp. 107133.
- 96. Moalli P & Rosen S. Glucocorticoid receptors and resistance to glucocorticoids in hematologia malignancies. Leuk Lymph 1994; 15: 363-374.
- 97. Cypcar D & Busse W. Steroid-resistant asthma. J Allergy Clin Immunol 1993; 92: 362-372.
- 98. Hennebold J & Daynes R. Microenvironmental control of glucocorticoid functions in immune regulation. In: Rook G & Lightman S. Steroid hormones and the T-cell cytokine profile. London: Springer-Verlag. 1997, pp. 101-134.
- 99. Rosner W. Plasma steroid-binding proteins. Endocr Metab Clin North Amer 1991; 20: 697-720.
- 100. Adcock I, Lane S, Brown C, Peters M, Lee T, Barnes P. Differences in binding of glucocorticoid receptor to DNA in steroid-resistant asthma. J Immunol 1995; 154; 3500-3505.
Endereço para correspondência
Datas de Publicação
-
Publicação nesta coleção
23 Fev 2005 -
Data do Fascículo
Set 2004
Histórico
-
Aceito
30 Jun 2004 -
Recebido
06 Jun 2004