Abstracts
OBJECTIVE: Mounting evidence suggests that the limbic system is pathologically involved in cases of psychiatric comorbidities in temporal lobe epilepsy (TLE) patients. Our objective was to develop a conceptual framework describing how neuropathological and connectivity changes might contribute to the development of psychosis and to the potential neurobiological mechanisms that cause schizophrenia-like psychosis in TLE patients. METHODS: In this review, clinical and neuropathological findings, especially brain circuitry of the limbic system, were examined together to enhance our understanding of the association between TLE and psychosis. Finally, the importance of animal models in epilepsy and psychiatric disorders was discussed. CONCLUSIONS: TLE and psychiatric symptoms coexist more frequently than chance would predict. Damage and deregulation among critical anatomical regions, such as the hippocampus, amygdala, thalamus, and the temporal, frontal and cingulate cortices, might predispose TLE brains to psychosis. Studies of the effects of kindling and injection of neuroactive substances on behavior and electrophysiological patterns may offer a model of how limbic seizures in humans increase the vulnerability of TLE patients to psychiatric symptoms.
Temporal Lobe Epilepsy; Psychosis; Psychiatric Comorbidities; Neuropathology; Limbic Circuits
OBJETIVO: Existem cada vez mais evidências de que o sistema límbico está envolvido na patologia das comorbidades psiquiátricas em pacientes com epilepsia do lobo temporal (ELT). Nosso objetivo foi elaborar um desenho conceitual descrevendo como aspectos neuropatológicos e de conectividade podem contribuir para o desenvolvimento de psicose em pacientes com ELT. MÉTODOS: Nesta revisão, achados clínicos e neuropatológicos, e especialmente os aspectos da circuitaria límbica, foram examinados em conjunto para auxiliar nossa compreensão sobre a associação entre ELT e psicose. Achados em modelos animais de epilepsia e esquizofrenia também foram levados em consideração. CONCLUSÕES: ELT e comorbidades psiquiátricas coexistem com maior frequência que o predito pela associação ao acaso. Dano e desregulação entre estruturas anatômicas críticas, como hipocampo, amígdala, tálamo, e córtices temporal, frontal e cingulado podem predispor o cérebro com ELT à psicose. Estudos sobre efeitos comportamentais e eletrofisiológicos do abrasamento elétrico e injeções de substâncias neuroativas em modelos animais podem oferecer pistas sobre como crises límbicas em humanos aumentam a vulnerabilidade de pacientes com ELT a sintomas psiquiátricos.
Epilepsia do lobo temporal; Psicose; Esquizofrenia; Comorbidades psiquiátricas; Neuropatologia; Circuitos límbicos
SPECIAL ARTICLE
Psychiatric comorbidities in temporal lobe epilepsy: ossible relationships between psychotic disorders and involvement of limbic circuits
Comorbidades psiquiátricas na epilepsia do lobo temporal: Possíveis relações entre desordens psicóticas e comprometimento de circuitos límbicos
Ludmyla KandrataviciusI,III; Cleiton Lopes-AguiarI; Lézio Soares Bueno-JúniorI; Rodrigo Neves Romcy-PereiraII; Jaime Eduardo Cecilio HallakI,III João Pereira LeiteI
IFaculdade de Medicina de Ribeirão Preto, Department of Neurosciences and Behavior, Universidade de São Paulo, Brazil
IIBrain Institute, Universidade Federal do Rio Grande do Norte, Natal, Brazil
IIINational Institute of Science and Technology in Translational Medicine (Instituto Nacional de Ciência e Tecnologia - INCT-TM; CNPq)
Corresponding author Corresponding author: Ludmyla Kandratavicius PhD Faculdade de Medicina de Ribeirão Preto, Department of Neurosciences and Behavior, Universidade de São Paulo Av Bandeirantes 3900 CEP 14049-900, Ribeirao Preto, SP, Brazil Phone: (+55 16) 3602 2796; Fax: (+55 16) 3633 0866 E-mail: ludykandra@gmail.com
ABSTRACT
OBJECTIVE:Mounting evidence suggests that the limbic system is pathologically involved in cases of psychiatric comorbidities in temporal lobe epilepsy (TLE) patients. Our objective was to develop a conceptual framework describing how neuropathological and connectivity changes might contribute to the development of psychosis and to the potential neurobiological mechanisms that cause schizophrenia-like psychosis in TLE patients.
METHODS: In this review, clinical and neuropathological findings, especially brain circuitry of the limbic system, were examined together to enhance our understanding of the association between TLE and psychosis. Finally, the importance of animal models in epilepsy and psychiatric disorders was discussed.
CONCLUSIONS: TLE and psychiatric symptoms coexist more frequently than chance would predict. Damage and deregulation among critical anatomical regions, such as the hippocampus, amygdala, thalamus, and the temporal, frontal and cingulate cortices, might predispose TLE brains to psychosis. Studies of the effects of kindling and injection of neuroactive substances on behavior and electrophysiological patterns may offer a model of how limbic seizures in humans increase the vulnerability of TLE patients to psychiatric symptoms.
Descriptors: Temporal Lobe Epilepsy; Psychosis; Psychiatric Comorbidities; Neuropathology; Limbic Circuits.
RESUMO
OBJETIVO: Existem cada vez mais evidências de que o sistema límbico está envolvido na patologia das comorbidades psiquiátricas em pacientes com epilepsia do lobo temporal (ELT). Nosso objetivo foi elaborar um desenho conceitual descrevendo como aspectos neuropatológicos e de conectividade podem contribuir para o desenvolvimento de psicose em pacientes com ELT.
MÉTODOS: Nesta revisão, achados clínicos e neuropatológicos, e especialmente os aspectos da circuitaria límbica, foram examinados em conjunto para auxiliar nossa compreensão sobre a associação entre ELT e psicose. Achados em modelos animais de epilepsia e esquizofrenia também foram levados em consideração.
CONCLUSÕES: ELT e comorbidades psiquiátricas coexistem com maior frequência que o predito pela associação ao acaso. Dano e desregulação entre estruturas anatômicas críticas, como hipocampo, amígdala, tálamo, e córtices temporal, frontal e cingulado podem predispor o cérebro com ELT à psicose. Estudos sobre efeitos comportamentais e eletrofisiológicos do abrasamento elétrico e injeções de substâncias neuroativas em modelos animais podem oferecer pistas sobre como crises límbicas em humanos aumentam a vulnerabilidade de pacientes com ELT a sintomas psiquiátricos.
Descritores: Epilepsia do lobo temporal; Psicose; Esquizofrenia; Comorbidades psiquiátricas; Neuropatologia; Circuitos límbicos.
Background
Temporal lobe epilepsy (TLE) is a focal neurological condition, in which seizures are able to spread and compromise a whole set of limbic structures and neighboring cortices.1 In contrast, various forms of psychosis are not clearly related to any consistent pathological localization. Instead, they have been attributed to neurochemical and ultrastructural dysfunctions in a much wider set of limbic circuits, including the ones directly affected by TLE.2 However, it is noteworthy that the high prevalence of psychosis in TLE patients3-7 indicates a possible shared mechanism between the two etiologies, despite their distinct degree of substrate commitment and localization. While the specific mechanisms generating psychosis in TLE are still poorly understood, neuroanatomical knowledge of limbic network connectivity might help to define potential brain targets for investigation and lead to a better understanding of this pathophysiological issue. The present article conducts a survey of TLE and psychosis, analyzing historical, clinical and neuropathological observations within the framework of limbic connectivity. Additionally, we reviewed experimental studies with electrical kindling or pharmacological treatment of the hippocampal formation, the amygdaloid complex, and the prefrontal cortex circuits. We focused on the dopaminergic and glutamatergic mechanisms that are potentially involved in both TLE and psychosis.
Early observations and clinical aspects
In 1825, two of Esquirol's assistants reported that the Ammon's horn was strikingly abnormal in the brains of some epileptic and a few psychotic patients.8 Fifty-three years later, Paul Broca would describe the great limbic lobe as "the seat of those lower faculties which predominate in the beast."8 This ample region regained attention more than thirty years later, especially with the study of Papez stating that, "the hypothalamus, the anterior thalamic nuclei, the gyrus cinguli, the hippocampus and their interconnections constitute a harmonious mechanism which may elaborate the functions of central emotion, as well as participate in emotional expression".9 The amygdala was not included in Papez's original theory of emotion, but after Klüver and Bucy's findings,10 MacLean made the amygdala one of the epicenters of a more extensive system, later called the limbic system.11,12 Beyond the limbic lobe structures cited above, the existence of anatomical connections among the frontal lobes, the hypothalamus and temporal lobes, as well as the effects of experimental lesions, suggested that the frontal lobes influence the basic instinctive and emotional drives.13 In addition to emotional disturbances, another important feature of several types of epilepsy is the presence of periods of short, intermittent lack of the awareness of the environment and of the self, indicating that the brainstem and most of the thalamus are also affected.14
A distinct syndrome of behavior abnormalities occurs in many patients with temporal lobe epilepsy. This constellation of interictal personality changes includes hyposexuality, hyperreligiosity/deeply held convictions, viscosity (a striking preoccupation with detail, especially concerned with moral and ethical issues), and, occasionally, hypergraphia or the urge to express in forms other than writing.15 Frequent symptoms of TLE patients also include psychic or experiential phenomena: intellectual aurae or dreamy states, complex visual or auditory hallucinations or illusions, memory "flashbacks", déjà vu, jamais vu, and emotions, most commonly fear.16 It is important to stress that these patients, who characteristically exhibit a preserved or even deepened affect, do not fall into any established nosologic category and often do not appear schizophrenic.15 Indeed, as described by Gloor et al.,17 "the patients are never in doubt that these phenomena occur incongruously, that is, out of context, as if they were superimposed upon the ongoing stream of consciousness (...). This insight clearly distinguishes these phenomena from psychotic hallucinations and illusions".17
In 1957, Desmond Pond made the first explicit clinical recognition of schizophrenic-like psychoses of epilepsy, as reviewed by Beard and Slater.18 Typically, the psychotic state closely resembles schizophrenia, with paranoid ideas which might become systematized, ideas of influence, and auditory hallucinations often of a menacing quality. The points of difference with classic schizophrenia are the common religious coloring of the paranoid ideas, the tendency of the affect to remain warm and appropriate, and the lack of typical deterioration to the hebephrenic state 18. Although auditory hallucinations are common, visual hallucinations are relatively rare. Other forms of delusions, including grandiose, referential, religious and Schneiderian symptoms, have also been reported, especially when a history of traumatic brain injury is present.19
The nature of the relationship between psychosis and epilepsy is controversial, and existent hypotheses are not mutually exclusive. One hypothesis suggests that there is a basic antagonism between epilepsy and psychosis. Psychosis in TLE is regularly associated with fewer or no psychomotor seizures,3,20 and normalization of the EEG and elimination of seizures through anticonvulsant medication - though they relieve epileptic symptomatology - often exacerbate an underlying psychiatric disorder and lead to the emergence of a psychotic state.21 Indeed, this supposed mutual antagonism was the basis for the development of electroconvulsive therapy, wherein seizure induction is used to treat psychosis, as reviewed by Pollock.22 Gamma-amino butyric acid (GABA) and dopamine also exert antagonistic effects in epilepsy and psychosis. For instance, dopamine antagonists are commonly used as antipsychotic drugs, and the drugs can trigger seizures.23 On the other hand, dopamine agonists are able to exacerbate or trigger psychotic symptoms, while having anticonvulsant properties.24,25 In contrast to this antagonist relationship, epilepsy also predisposes a patient to the development of schizophrenia-like psychosis. This last view is in line with the extensive works of Slater, Beard and Glithero,26 and of Kristensen and Sindrup,20 which recognize epileptic psychoses as truly organic phenomena, caused by structural damage to the limbic parts of the temporal lobe responsible for both the epilepsy and the psychosis.
Psychiatric disorders in epilepsy can be classified into ictal (the psychiatric symptoms are a clinical manifestation of the seizure), periictal (symptoms precede and/or follow the seizure occurrence) and interictal (symptoms occur independently of the seizure occurrence) disorders. In the present review, our focus will be primarily interictal psychosis, which includes schizophrenia-like psychosis of epilepsy, as defined by the International League Against Epilepsy (ILAE, Commission on Neuropsychiatric Aspects).27 Because interictal symptoms are not related to any "seizure collateral effect", psychiatric manifestations are much like the pure form of the psychopathology. For instance, chronic interictal psychosis of epilepsy is also referred to as schizophrenia-like psychosis of epilepsy, due to its resemblance to schizophrenia's phenomenological manifestations.28 Interictal psychosis is usually prodigal in florid symptoms, whereas postictal psychosis exhibits few common schizophreniform psychotic traits such as perceptual delusions or voices.29 Of note, recurrent postictal psychosis in human TLE is considered to be a risk factor for the development of interictal psychosis,30 which makes animal models of postictal psychosis suitable for the study of the possible gradual commitment of the limbic circuits. The increased risk of psychiatric symptoms in epilepsy may be related to several risk factors, such as genetic background and illness chronicity, which are liable to facilitate psychopathological manifestations. In the next two sections, the contributions of structural neurologic factors and their possible imbalance among connected neuroanatomical regions will be examined.
Neuropathological aspects
Schizophrenic patients often have abnormal electrical activity in the temporal lobes and sometimes in the frontal lobes.31 Early descriptions suggested that the sites of maximum abnormalities were in the hippocampus, amygdala, septum, uncus, anterior-temporal, and orbito-frontal areas, even in patients with no history of epilepsy.32 For more than 50 years, studies have suggested that any given brain tumor affecting the limbic system can present as classical schizophrenia.5 In Malamud's studies of eighteen patients with temporal lobe tumors, ten had been diagnosed and treated as schizophrenics, three as melancholics, one as a psychotic depressive, one as maniac, two as psychoneurotics, and one as anxious.33 In Taylor's series of TLE post-mortem brains, 23% of the cases with "alien tissue" (tumors, hamartomas and focal dysplasias) were psychotic, contrasting with only 5% in the mesial temporal sclerosis group.7 Although some authors agree that psychiatric symptoms presage temporal lobe tumors and that presence of mesial temporal sclerosis is protective against schizophrenia-like psychosis in patients with epilepsy,34 recent neuropathological evidence suggests that there is a structural basis for psychiatric symptoms in TLE patients with hippocampal sclerosis.35 In our series of patients35 with no dual pathology or alien tissue, we did not observe a predominance of female patients or of left handedness in the psychosis group (as did Taylor)7 nor a predominance of patients with left or bilateral temporal foci (as did Flor-Henry).3 Other studies also indicate that the presence of hippocampal sclerosis is not protective against psychosis. In Roberts's series of 249 TLE patients, the largest epilepsy series analyzed to date for their neuropathological features and schizophrenia-like psychosis, 6.4% had a preoperative diagnosis of TLE and psychosis; 40% of the cases with TLE and psychosis had hippocampal sclerosis with left sided foci and approximately 20% had gangliogliomas.4 It is noteworthy that gangliogliomas usually have origin in and predilection for the temporal lobe,4 meaning that, more important than the type of lesion, the location of a lesion within the hippocampal-amygdalar-temporal gyri may represent a true predisposition factor for psychosis.
With a focal brain lesion, hallucinations may arise after compensatory over-activation of nearby sensory pathways. Complex auditory hallucinations are most common in TLE and schizophrenia, and the affected areas mainly comprise the primary auditory pathway (the pons, inferior colliculus, medial geniculate body and temporal lobe).36 In fact, fMRI studies in schizophrenic patients during auditory hallucinations have revealed activation of areas related not only to the auditory pathway but also to inner speech as follows: transverse temporal gyrus of the dominant hemisphere, posterior superior temporal gyrus, middle temporal gyrus, frontoparietal operculum, orbitofrontal cortex, hippocampus, amygdala, sensorimotor cortex, right inferior colliculus, right and left insula, left parahippocampal gyrus, right temporal gyrus, right thalamus, middle frontal and anterior cingulate gyri.31 The structural lesions most often seen in auditory verbal hallucinations are a reduction in grey matter volume in the superior temporal and middle temporal gyri of the dominant hemisphere, and in the insula, temporal pole, thalamus and right prefrontal cortex.36 Interestingly, auditory hallucinations have been associated with impaired effective connectivity between the left superior temporal, frontal and anterior cingulate cortex, suggesting that such patients tend to misattribute their own speech to an external source (external speech).36 In addition to hallucinations, another classic feature exhibited by patients with psychosis is a reduction in the operational index of sensorimotor gating. This index is obtained by measuring the prepulse inhibition (PPI) of the startle response, which refers to the ability of a weak prestimulus to transiently inhibit the response to a closely following strong sensory stimulus. Both schizophrenia patients and their relatives show gating deficits that are significantly heritable, consistent with the endophenotype criteria.37,38 In fact, reduced PPI has been observed in subjects with schizotypal personality and in normal subjects with high scores of self-transcendence in Personality Questionnaire scales.39 TLE patients with psychosis40 and non-psychotic patients with psychogenic "non-epileptic" seizures41 also exhibit reductions in PPI. It is important to stress that, although patients with psychogenic seizures may not exhibit epileptiform brain activity, all of them present with hysterical features.42 Often - in more than 90% of the cases43 - they also present with psychiatric comorbidities. Definite neuropathological substrates adjunct to PPI impairment in humans are not known, but experimental data suggest that kindling of the amygdala44 and precommissural lesions of the fornix45 are effective ways to disrupt PPI in rats.
Neuropathological abnormalities are frequently observed in the brains of schizophrenic46 and TLE patients.47 TLE patients with hippocampal sclerosis exhibit hippocampal neuronal loss, often accompanied by neuronal loss and gliosis in the amygdala and entorhinal cortex.48 The same set of mesial structures may appear atrophic under magnetic resonance imaging, in addition to being consistently recruited at the onset of electrographical seizure activity.49 The search for an organic basis in the post-mortem brains of schizophrenic patients has resulted in controversial findings, mostly because methodological approaches are rarely consistent in their selection of patients and controls.46 Common findings include neuronal loss, shrinkage or disarray in the cortical layers and occasional gliosis; affected areas comprise prefrontal cortical areas, the pons, the nucleus accumbens, the hypothalamus, the substantia innominata (part of the basal forebrain), the cingulate, superior, middle and inferior temporal gyri, the amygdala and the hippocampus.46,50,51 The hippocampal formation has been the subject of intensive study. Left hemisphere neuronal loss in Ammon's horn without gliosis, entorhinal cortex layer II neuronal loss and reduced density of the interneurons have been described in the post-mortem brains of schizophrenic patients, the former being more noticeable in paranoid than in catatonic patients.50,52 As reviewed by Roberts and Bruton,50 although, in the early 1900's, Kraepelin believed schizophrenia had an organic cause and the characteristics of a degenerative process, the majority of contemporary neuropathological studies have not shown signs of progressive features, such as reactive gliosis or correlations between structural abnormalities and the length of illness. Current theories consider prenatal and perinatal effects, as well as environmental factors, to be risk factors for disease manifestation.52
Excision of parts of the limbic system or the temporal lobe of schizophrenic patients usually decreases the florid symptoms (i.e., delusions, hallucinations, disorganized speech or thinking, and chaotic or confused behavior in their fully developed form), but it leaves the psychosis untouched.5 Therefore, it is not the complete destruction which gives rise to psychosis, but possibly the irritation process during the development of destruction.32 On some occasions, psychosis or deep personality changes may arise after temporal lobectomy in TLE patients who were previously psychiatrically normal.34,53 Not only the recruitment and/or partial damage of the limbic structures are necessary to generate psychiatric disturbances. As postulated by Racine,54 the kindling process may gradually develop and spread throughout brain structures distal to the stimulated focus. In patients with epilepsy, there is evidence of a time-dependent spread of epileptic excitability that is independent of tissue pathology.55 It is generally assumed that a human limbic seizure disorder results in enhancement of affective limbic functions, rather than their flattening.56 Again, the relatively long interval between TLE onset and the onset of comorbid psychiatric symptoms suggests that damage to key structures is necessary and that it builds up over time.19,57 Such gradual commitment of connected systems might be determinant to the sum of neurobiological events that eventually will lower the psychopathological threshold of a normal or prodromal psychiatric state.
Limbic lobe and major connections
Through clinical and pathological observation, it is well accepted that TLE is attributable to focal mesial structures of the temporal lobe, especially the amygdaloid complex and the hippocampal formation. The hippocampal formation consists of the hippocampus proper - the dentate gyrus (DG) and Ammon's horn/Cornus Ammonis (CA) subfields - along with the subicular and rhinal cortices.58 Despite the focal nature of TLE, it is possible that recurrent limbic seizures gradually compromise remote encephalic areas, which may be due to the axonal connectivity of the mesial temporal lobe. Indeed, according to tract-tracing studies in non-human primates and rodents, the hippocampus, amygdaloid complex, and neighboring cortices are embedded within a broad network of limbic connections, whose dysfunctions are involved in at least some of the psychiatric morbidities that are related to TLE. Therefore, it seems reasonable to consider that efferent axons arising from the mesial temporal lobe may convey paroxysmal activity to the entire limbic circuitry, ultimately promoting ictal and periictal psychiatric symptoms. In the same vein, it is possible that seizure recurrence is responsible for the chronic pathological commitment of subcortical and neocortical sites targeted by mesial temporal lobe axons, which would explain the increased susceptibility of TLE patients to interictal psychiatric symptoms.
Hippocampal formation connectivity
The hippocampal formation is the largest structure of the mesial temporal lobe. Intrinsic connections between the DG and CA subfields constitute a well-known tri-synaptic circuit, primarily represented by a unidirectional sequence from DG granule cells to CA3 pyramidal cells (mossy fibers) and then from CA3 to CA1 pyramidal cells (Schaffer axonal collaterals).59 Despite evidence for some CA3-DG back-projections,60 the DG acts as the major starting point for the circuit, receiving most of its extrahippocampal afferents from superficial cells of the entorhinal cortex via the perforant path of the angular bundle.61,62 At the other end of the sequence, CA1 pyramidal cells provide the main hippocampal output, which may or may not be relayed through subiculum cells.63,64 Interestingly, part of the CA1 and subiculum axons project back to the entorhinal cortex, specifically to its deep layers, making the entorhinal cortex a critical link in the excitatory loop of the hippocampal formation.65 Intrinsically, in the entorhinal cortex an inhibitory system between deep layers receiving CA1/subiculum axons and DG-projecting superficial layers seems to gate the flow of information from, and to, the hippocampus, thereby controlling its excitatory loop.66,67 Finally, in addition to the strong entorhinal-DG unidirectional pathway, there are lighter projections from the entorhinal cortex to all CA subfields and to the subiculum, which means that entorhinal cortex reciprocates the CA1 and subiculum inputs.65,68 The hippocampal formation and its main connections are shown in Figures 1 and 2.
In turn, subicular cells that do not project back to the hippocampal formation send axons through the fimbria-fornix bundles, forwarding hippocampal output to several limbic sites, including specific regions of the basal forebrain (e.g., vertical limb of diagonal band), septum, nucleus accumbens, amygdaloid complex (with particularly dense projections to the basolateral division), mammillary bodies of hypothalamus and adjacent nuclei, midline thalamus (especially nucleus reuniens), retrosplenial cortex, and prefrontal cortex.63,69,72 The CA1 subfield, itself, also sends direct projections to the vertical limb of diagonal band, septum, nucleus accumbens, basolateral amygdaloid complex, and prefrontal cortex,63,70,71,73,74 as well as weaker efferents to retrosplenial cortex and sensorial neocortical areas.74,75 Roughly, all these efferents are accompanied by returning afferents, except for from the nucleus accumbens, retrosplenial cortex, and prefrontal cortex, which do not send projections back to the hippocampus proper.68,76 In addition to the preferential hippocampal output by the CA1/subiculum system, CA3 also sends direct efferents to the vertical limb of the diagonal band, septum, and nucleus accumbens, with returning axons from the septum.70,77,78 Additionally, CA3-2 receives projections - presumably unidirectional - from basolateral amygdala,71 and substance P-containing afferents from the supramammilary hypothalamic nucleus.79 In contrast with CA subfields, the DG is a mere receiver of extrahippocampal afferents, projecting axons solely to CA3.68,80 Beyond the substantial input from the entorhinal cortex, few subcortical sites target the DG (the septum and some parts of the hypothalamus, mainly its supramammilary area, are among these sites).81,82 In spite of the mammillary nuclei and adjacencies that are reciprocally connected with the subiculum, none of the CA subfields project to the hypothalamus,83 although CA2 and CA3 do receive hypothalamic projections.79 Mostly, the non-entorhinal afferents directed to the DG and CA subfields use the fimbria-fornix system as their main route, along with the majority of hippocampal efferents described above.68 Last but not least, the entire hippocampal formation (Figure 1) receives noradrenergic, serotoninergic, and dopaminergic inputs, respectively from locus coeruleus, raphe nuclei and ventral tegmental area of the brainstem.84,86 Particularly in the DG and CA subfields, dopaminergic innervation is much more meager than the others, preferentially targeting the entorhinal cortex.68
Because there are unknown efferents from the DG and CA2-3 to the neocortex, it seems that CA1 unidirectional projections to retrosplenial and prefrontal cortices are the strongest communication between the hippocampus proper and neocortical areas outside the hippocampal formation.63,75 Unlike the hippocampus proper, the entorhinal cortex is reciprocally connected with a wider neocortical domain, with a preference for higher-order association areas. Therefore, the entorhinal cortex acts as the principal link between the polymodal neocortical inputs and the hippocampus.68,87 Although this neuroanatomical and functional distinction exists, subcortical connections of the entorhinal cortex are similar to those of the hippocampus proper, including bidirectional pathways with the septum and basolateral amygdala, as well as unidirectional projections to nucleus accumbens, and unidirectional projections from distributed parts of the hypothalamus and midline thalamus, once again with major participation of nucleus reuniens.72,88-91 Among the neocortical sites reciprocally connected to the entorhinal cortex are the perirhinal, postrhinal, and prefrontal cortices, including a weak communication with the retrosplenial cortex.68 As part of the hippocampal formation, the entorhinal cortex is also subjected to neuromodulatory influence by the monoaminergic ascending systems, including significant dopaminergic afferents from the ventral tegmental area.90,92 In Figure 2, the arrows outside the hippocampal formation represent the extrinsic connections depicted here.
Amygdaloid complex and its efferents and afferents
Another important part of the mesial temporal lobe is represented by the amygdaloid complex, a group of functionally and structurally diverse nuclei, with intricate interconnections and widely distributed communication with cortical and subcortical limbic sites.93 In particular, the basolateral group projects strong efferents to CA3, CA1, the subiculum, entorhinal cortex and perirhinal cortex, receiving back-projections from all sites except CA3,72,94,95 as seen in Figure 3. The same basolateral group is responsible for most of the amygdaloid innervation of the prefrontal cortex (which is also a recipient of CA1/subiculum axons, as previously mentioned), nucleus accumbens (a target of the whole hippocampal formation), and the striatum.96-98 Specifically, the prefrontal cortex sends returning projections to the basolateral complex99 (Figure 3). Considering the emotional roles of the amygdala, as well as the mnemonic/attentive functions of the hippocampus and the prefrontal cortex, this basolateral-hippocampal reciprocity, along with its common limbic targets, comprises a wide network that is well positioned to control cognitive aspects of behavior.100-102
In contrast, the centromedial group of amygdaloid nuclei preferentially sends projections to neuromodulator sites, including brainstem ascending monoaminergic centers (noradrenergic locus coeruleus, dopaminergic substantia nigra and ventral tegmental area, and serotoninergic raphe nuclei), and cholinergic regions of the basal forebrain and septum.103-106 Moreover, this centromedial division of the amygdala sends efferents to several different autonomic, reproductive, and defensive-related sites, including some brainstem regions (periaqueductal grey, parabrachial nucleus, and nucleus of the solitary tract), and various portions of the hypothalamus.105,107-110 In particular, the medial nucleus of the amygdala sends projections to the nucleus reuniens of midline thalamus.104 An important conduit of the amygdala fibers that are directed to the brainstem, the basal forebrain, and the diencephalon is the stria terminalis, whose bed nucleus contributes to additional innervation of these sites.111 Finally, the corticomedial nuclei of the amygdala, which are also referred to as cortical-like nuclei due to their superficial location and layered structure, are the main source of efferents to the olfactory system.93 In general, all of these efferent connections of the centromedial division are reciprocated93 (Figure 3).
The amygdaloid complex, as a whole, receives afferents from regions that are not preferentially targeted by amygdala projections, such as higher-order areas of somatosensory, auditory, and visual cortices, as well as primary areas of the olfactory system, and gustatory and visceral portions of the insular cortex.112-115 Thalamic nuclei directly related to specific sensorial functions also contribute with afferents to the amygdala,116,117 along with the paraventricular and reuniens nuclei of the midline thalamus 118. Overall, the various afferents terminate onto virtually all the amygdaloid divisions, despite some intricate patterns of specificity not detailed here. Nevertheless, the basolateral group acts as the main input receiver, which processes the information locally, forwarding it to the centromedial nuclei as a preferential output station93 (Figure 3).
Each one of the projection systems mentioned above have specific patterns of ipsilateral and/or contralateral connectivity not detailed in the present article. The majority of connections are mediated by glutamatergic neurotransmission, regardless of termination onto glutamatergic projection cells or GABAergic interneurons, with specific exceptions, for instance, monoaminergic and cholinergic projections arising from the brainstem and basal forebrain, GABAergic projections from the septum that coexist with septal cholinergic axons, and GABAergic projections from central amygdala. There are differential patterns of topographical connectivity throughout the long axis of the hippocampal formation, which are not explained here. Because comparative neuroanatomy is not the main purpose of the present review, information on differences between mammalian species was omitted for clarity.
According to Bear,57 psychopathological states in TLE would derive from intrinsic mechanisms leading to anatomical sensory limbic hyper connectivity. This psychiatric manifestation would be the result of a long epileptogenic process throughout the limbic system, and progressive changes in limbic structures secondary to seizure activity would be the underlying physiopathology.57 Such changes would explain the fact that some patients have incomplete seizure remission after surgical resection of the mesial temporal lobe.1 The extra-temporal commitment would be due to the spread of seizures by critical subcortical synchronizers, possibly represented by thalamo-hippocampal circuits.1 Specifically, the midline nuclei of the thalamus are well positioned to act as major synchronizers,118 because some of them are interconnected with both the hippocampal formation and the amygdaloid complex (Figures 2 and 3). Importantly, the same thalamic nuclei project to the nucleus accumbens and are interconnected with the prefrontal cortex, forming a behaviorally flexible circuit whose dysfunction contributes to the pathophysiology of psychosis and schizophrenia.119 Therefore, the hippocampal and amygdala connectivity summarized here may be the common context for TLE and psychotic-like symptoms. Although these network dysfunctions are not fully understood, there is increasing evidence from animal models showing how particular pathways of the limbic circuitry may underlie the relationship between psychosis and TLE.
Evidence from animal models
The relationship between psychosis and epilepsy has been investigated experimentally through electrical kindling in specific circuits and through single electrically evoked hippocampal afterdischarge protocols,120-123 dopaminergic sensitization by pharmacological challenge,124,125 and acute or chronic treatments with non-competitive antagonists of N-methyl D-aspartate (NMDA) receptors.126,127
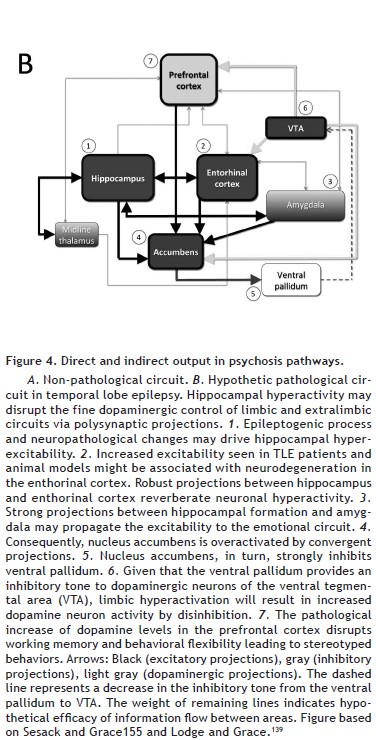
Kindling by electrical stimulation in specific circuits provides evidence for dopaminergic involvement in the putative shared mechanisms between epilepsy and psychosis. For example, kindling in the ventral tegmental area generates progressive fearfulness, hiding and loss of social behavior in cats,128 enhancing amphetamine- or methamphetamine-induced locomotor activity in rodents.129,130 Hippocampal kindling increases the density of dopaminergic receptors, as well as dopamine release in nucleus accumbens.131,132 These dopaminergic changes in mesolimbic circuits after ictal episodes seem to depend on the hippocampal activity. Hyperlocomotion, PPI deficits and aberrant increases of fast oscillations elicited by prefrontal or nucleus accumbens kindling are reverted if the hippocampus is previously inactivated.122 Unlike to hippocampal stimulation, prefrontal cortex stimulation is unable to significantly depolarize nucleus accumbens neurons.133,134 Other studies have shown that kindling in the basolateral amygdala down-regulates dopamine transporters in the striatum, while disrupting PPI.44,135 Altogether, kindling data reinforce the idea that, in experimental psychosis, the hippocampus becomes unable to regulate the source of convergent information flow to the nucleus accumbens, especially those from the prefrontal cortex, ventral tegmental area and amygdala136 (Figure 4).

Abnormalities in the cortico-frontal-mesolimbic circuits observed in experimental models may be useful to understand how dopaminergic dysfunctions could be related to the high rates of psychosis in TLE patients.137 Postictal psychosis models based on single electrically evoked hippocampal afterdischarge also induce psychotic-like behaviors, such as hyperlocomotion and stereotyped movements.121 This postictal locomotor activity is mediated by D2 dopaminergic receptors in the nucleus accumbens and requires ventral pallidum activity, suggesting that the nucleus accumbens-ventral pallidum pathway is critical for the expression of postictal abnormal behaviors.120 Impairment of sensorimotor gating induced by afterdischarge is related to increased neural activity in the medial septum and increased hippocampal gamma waves induced by local afterdischarges through GABAergic, not cholinergic, septo-hippocampal neurons.123 Experimental studies on seizure-induced pathological sensitization of the dopaminergic system have also been useful to elucidate the relationship between mesolimbic circuits, psychosis and epilepsy.124 Seizure activity transiently elevates extracellular dopamine levels at various brain sites, including the hippocampus, striatum, nucleus accumbens and prefrontal cortex132,138,139 (Figure 4). In fact, rats subjected to the pilocarpine model of TLE display significantly higher spontaneous firing rates of ventral tegmental area neurons compared to control rats.124 This population activity of dopaminergic neurons, defined as the proportion of spontaneously firing neurons, is regulated in intact rats by a ventral subicular-nucleus accumbens-ventral pallidal-ventral tegmental area pathway.119 However, the underlying mechanisms by which hippocampal hyperactivity influences the dopaminergic neurons in TLE are not well recognized. Neonatal seizures affect both dopaminergic and glutamatergic systems in the prefrontal-striatal circuitry, resulting in enhanced behavioral sensitization to methamphetamine in adolescence.125 The increased excitability observed in patients and in TLE models might be associated with a pronounced neurodegeneration in layer III of the medial entorhinal cortex.140,141 This pattern of neuronal loss eventually results in hippocampal and parahippocampal hyperexcitability, which have an important role in epileptogenesis and enhanced susceptibility to seizures and epileptiform discharges.142,143 Such hyperexcitability could be related to the cognitive deficits and psychiatric comorbidities observed in some cases of TLE because entorhinal-hippocampus projections are crucial to temporal association memory in mice.144 Their dysfunction is involved in the potentiation of drug-induced locomotor hyperactivity in pilocarpine-treated rats.124 In fact, recent data on human TLE suggest that decreased neuronal density is found in entorhinal cortex layer III of TLE patients with hippocampal sclerosis and psychosis.35 Therefore, according to kindling and hippocampal afterdischarge studies, the increased sensitization to psychostimulants observed in TLE models is likely attributable to an increase in tonic dopaminergic transmission, secondary to an augmented activity within the hippocampal and parahippocampal circuits145 (Figure 4B). Increased hippocampal and parahippocampal drive is also found in patients with schizophrenia.146
Despite the available data, putative shared mechanisms between epilepsy and psychosis remain poorly understood. The non-competitive NMDA antagonists ketamine (KET), phencyclidine (PCP), and dizocilpine (MK-801) produce robust psychotic-like behaviors, PPI deficits, memory impairments and hippocampal synaptic plasticity disruptions probably associated with increased excitability in limbic thalamo-cortical circuits.147,148 Non-competitive NMDA antagonists generate disinhibition of specific circuits by blocking the action of NMDA receptors on GABAergic neurons, resulting in decreased firing of GABAergic neurons (mainly parvalbumin-positive chandelier cells) and increased excitability of limbic circuits and the prefrontal cortex.149-153 Treatment with non-competitive NMDA antagonists can also decrease parvalbumin expression154 and produce epileptiform activity in limbic circuits.155 NMDA antagonists can also damage and kill cortical neurons by increasing cytoplasmic vacuoles in pyramidal neurons of the posterior cingulate and retrosplenial cortex of adult rats.156 In the same vein, KET significantly potentiates kainic acid-elicited gamma power in slices of the mouse prefrontal cortex.157 Moreover, the dopaminergic system is also involved in the behavioral changes observed in NMDA-antagonist-based models.158 Impairment of executive tasks in rodents treated with KET is mediated by D2 receptors.159 In addition, a single dose of PCP promotes an increase of dopamine levels in the prefrontal cortex.160 Jackson et al.152 demonstrated that a single MK-801 injection increases random firing and decreases burst firing activity in the prefrontal cortex. The reduction in burst activity is associated with a decrease in synaptic efficacy, which in turn could affect the control of behavioral outputs. Similarly, increases in the random firing rates of prefrontal neurons are associated with deficits in PPI and working memory.152 Suzuki et al.127 showed that systemic injection with PCP significantly changes neuronal firing rates in the prefrontal cortex of rats. No changes were seen after local frontal cortex microinjection. However, when PCP was microinjected into the ventral hippocampus, prefrontal neurons showed responses similar to those observed after systemic injection.161 Altogether, a set of evidence indicates the involvement of the dopaminergic system and points to a possible role of hippocampal to prefrontal cortex projections in the expression of aberrant behaviors induced by non-competitive NMDA antagonists in rats. Considering that the main source of direct hippocampal afferents to the cortex (CA1 subfield of the hippocampus) is especially affected in TLE, and that psychotic-like effects produced by the administration of NMDA antagonists require a preserved communication between the hippocampus (CA1) and the prefrontal cortex, we speculate that synaptic plasticity in this pathway may represent a point of vulnerability for the development of psychosis in TLE patients.
Conclusions
The investigation on TLE and psychosis has independently provided important pathophysiological hallmarks. The best-known observations include hippocampal sclerosis in TLE, related to a chronic state of recurrent seizures, and heterogeneous cortico-striato-pallido-thalamic disturbances in psychosis, possibly due to interactions between genetic and environmental influences on the maturation of these circuits. The relatively high prevalence of psychotic-like symptoms in TLE patients suggests shared mechanisms and/or substrates between these two conditions. However, few studies have addressed this issue, despite the interesting reports on electrical kindling, dopaminergic manipulation, and NMDA antagonism reviewed here. Considering mesial temporal lobe connectivity along with the midline thalamus, the prefrontal cortex and the nucleus accumbens, as well as the ascending neuromodulatory influence on these circuits, future studies involving a convergence of approaches would be useful. For instance, investigations into the relationship between behavioral symptoms (e.g., sensorimotor gating), synaptic plasticity (e.g., long-term potentiation/depression in limbic pathways), and pathology in animal models of TLE (e.g., lithium-pilocarpine or electrical kindling in the hippocampus or amygdala) are scant. Nevertheless, such animal studies would be necessary to gain a better understanding of clinical cases. In summary, present clinical, neurophysiological and neuropathological data demonstrate the existence of an intricate mesh of both antagonistic and also shared mechanisms between TLE and psychosis. As once mentioned by Slater et al. "like effects commonly have similar causes" 26, and the underlying pathogenesis of psychosis in TLE still needs clarification.
Acknowledgments
Supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), Conselho Nacional de Desenvolvimento Científico e Tecnológico (National Counsel of Technological and Scientific Development - CNPq) and Fundação de Amparo à Pesquisa do Estado do Rio Grande do Norte (FAPERN), Brazil.
References
1. Bertram EH. Temporal lobe epilepsy: where do the seizures really begin? Epilepsy Behav. 2009;14(Suppl 1):32-7.
2. Swerdlow NR. Integrative circuit models and their implications for the pathophysiologies and treatments of the schizophrenias. Curr Top Behav Neurosci. 2010;4:555-83.
3. Flor-Henry P. Psychosis and temporal lobe epilepsy. A controlled investigation. Epilepsia. 1969;10(3):363-95.
4. Roberts GW, Done DJ, Bruton C, Crow TJ. A "mock up" of schizophrenia: temporal lobe epilepsy and schizophrenia-like psychosis. Biol Psychiatry. 1990;28(2):127-43.
5. Serafetinides EA, Falconer MA. The effects of temporal lobectomy in epileptic patients with psychosis. J Ment Sci. 1962;108:584-93.
6. Stevens JR. Epilepsy, psychosis and schizophrenia. Schizophr Res. 1988;1(1):79-89.
7. Taylor DC. Factors influencing the occurrence of schizophrenia-like psychosis in patients with temporal lobe epilepsy. Psychol Med. 1975;5(3):249-54.
8. Editorial: Epilepsy, schizophrenia, and limbic system. Lancet. 1974;2(7886):935-6.
9. Papez JW. A proposed mechanism of emotion. J Neuropsychiatry Clin Neurosci. 1937;7(1):103-12.
10. Klüver H, Bucy P. "Psychic blindness" and other symtoms following bilateral temporal lobectomy in rhesus monkeys. American Journal of Physiology. 1937;119:352-3.
11. MacLean P. Psychosomatic disease and the visceral brain; recent developments bearing on the Papez theory of emotion. Psychosom Med. 1949;11(6):338-53.
12. Heimer L, Van Hoesen GW, Trimble M, Zahn DS. The limbic system: a concept in perpetual search for a definition. In: Heimer L, Van Hoesen GW, Trimble M, Zahn DS, eds. Anatomy of Neuropsychiatry. U.S.A.: Academic Press Elsevier; 2008:1-13.
13. Brain L. Psychosomatic Medicine and the Brain-Mind Relationship. Lancet. 1964;2(7355):325-8.
14. Cairns H. Disturbances of consciousness with lesions of the brain-stem and diencephalon. Brain. 1952;75(2):109-46.
15. Waxman SG, Geschwind N. The interictal behavior syndrome of temporal lobe epilepsy. Arch Gen Psychiatry. 1975;32(12):1580-6.
16. Penfield W, Perot P. The Brain's Record of Auditory and Visual Experience. A Final Summary and Discussion. Brain. 1963;86:595-696.
17. Gloor P, Olivier A, Quesney LF, Andermann F, Horowitz S. The role of the limbic system in experiential phenomena of temporal lobe epilepsy. Ann Neurol. 1982;12(2):129-44.
18. Beard AW, Slater E. The schizophrenic-like psychoses of epilepsy. Proc R Soc Med. 1962;55:311-6.
19. Fujii D, Ahmed I. Psychotic disorder following traumatic brain injury: a conceptual framework. Cogn Neuropsychiatry. 2002;7(1):41-62.
20. Kristensen O, Sindrup EH. Psychomotor epilepsy and psychosis. I. Physical aspects. Acta Neurol Scand. 1978;57(5):361-9.
21. Landolt H. Some correlations between the electroencephalogram and normal and pathologic mental processes. Epilepsy Behav. 1963;14(3):448-51. 22. Pollock DC. Models for understanding the antagonism between seizures and psychosis. Prog Neuropsychopharmacol Biol Psychiatry. 1987;11(4):483-504.
23. Trimble M. The relationship between epilepsy and schizophrenia: a biochemical hypothesis. Biol Psychiatry. 1977;12(2):299-304.
24. Ogren SO, Pakh B. Effects of dopamine D1 and D2 receptor agonists and antagonists on seizures induced by chemoconvulsants in mice. Pharmacol Toxicol. 1993;72(4-5):213-20.
25. Turski L, Cavalheiro EA, Bortolotto ZA, Ikonomidou-Turski C, Kleinrok Z, Turski WA. Dopamine-sensitive anticonvulsant site in the rat striatum. J Neurosci. 1988;8(11):4027-37.
26. Slater E, Beard AW, Glithero E. The schizophrenia-like psychoses of epilepsy. Br J Psychiatry. 1963;109:95-150.
27. Krishnamoorthy ES, Trimble MR, Blumer D. The classification of neuropsychiatric disorders in epilepsy: a proposal by the ILAE Commission on Psychobiology of Epilepsy. Epilepsy Behav. 2007;10(3):349-53.
28. Toone BK. The psychoses of epilepsy. J Neurol Neurosurg Psychiatry. 2000;69(1):1-3.
29. Kanemoto K, Kawasaki J, Kawai I. Postictal psychosis: a comparison with acute interictal and chronic psychoses. Epilepsia. 1996;37(6):551-6.
30. Tarulli A, Devinsky O, Alper K. Progression of postictal to interictal psychosis. Epilepsia. 2001;42(11):1468-71.
31. Braun CM, Dumont M, Duval J, Hamel-Hebert I, Godbout L. Brain modules of hallucination: an analysis of multiple patients with brain lesions. J Psychiatry Neurosci. 2003;28(6):432-49.
32. Torrey EF, Peterson MR. Schizophrenia and the limbic system. Lancet. 1974;2(7886):942-6.
33. Malamud N. Psychiatric disorder with intracranial tumors of limbic system. Arch Neurol. 1967;17(2):113-23.
34. Taylor DC. Mental state and temporal lobe epilepsy. A correlative account of 100 patients treated surgically. Epilepsia. 1972;13(6):727-65.
35. Kandratavicius L, Hallak JE, Young LT, Assirati JA, Carlotti CG, Jr., Leite JP. Differential aberrant sprouting in temporal lobe epilepsy with psychiatric co-morbidities. Psychiatry Res. 2012;195(3):144-50.
36. Allen P, Laroi F, McGuire PK, Aleman A. The hallucinating brain: a review of structural and functional neuroimaging studies of hallucinations. Neurosci Biobehav Rev. 2008;32(1):175-91.
37. Braff DL. Gating in schizophrenia: from genes to cognition (to real world function?). Biol Psychiatry. 2011;69(5):395-6.
38. Quednow BB, Ettinger U, Mossner R et al. The schizophrenia risk allele C of the TCF4 rs9960767 polymorphism disrupts sensorimotor gating in schizophrenia spectrum and healthy volunteers. J Neurosci. 2011;31(18):6684-91.
39. Takahashi H, Iwase M, Yasuda Y et al. Relationship of prepulse inhibition to temperament and character in healthy Japanese subjects. Neurosci Res. 2012;72(2):187-93.
40. Braff DL, Geyer MA, Swerdlow NR. Human studies of prepulse inhibition of startle: normal subjects, patient groups, and pharmacological studies. Psychopharmacology (Berl). 2001;156(2-3):234-58.
41. Pouretemad HR, Thompson PJ, Fenwick PB. Impaired sensorimotor gating in patients with non-epileptic seizures. Epilepsy Res. 1998;31(1):1-12.
42. Syed TU, LaFrance WC, Jr., Kahriman ES et al. Can semiology predict psychogenic nonepileptic seizures? A prospective study. Ann Neurol. 2011;69(6):997-1004.
43. Mokleby K, Blomhoff S, Malt UF, Dahlstrom A, Tauboll E, Gjerstad L. Psychiatric comorbidity and hostility in patients with psychogenic nonepileptic seizures compared with somatoform disorders and healthy controls. Epilepsia. 2002;43(2):193-8.
44. Howland JG, Hannesson DK, Barnes SJ, Phillips AG. Kindling of basolateral amygdala but not ventral hippocampus or perirhinal cortex disrupts sensorimotor gating in rats. Behav Brain Res. 2007;177(1):30-6.
45. Miller EJ, Saint Marie LR, Breier MR, Swerdlow NR. Pathways from the ventral hippocampus and caudal amygdala to forebrain regions that regulate sensorimotor gating in the rat. Neuroscience. 2010;165(2):601-11.
46. Weinberger DR, Wagner RL, Wyatt RJ. Neuropathological studies of schizophrenia: a selective review. Schizophr Bull. 1983;9(2):193-212.
47. Thom M. Hippocampal sclerosis: progress since Sommer. Brain Pathol. 2009;19(4):565-72.
48. Cavanagh JB, Meyer A. Aetiological aspects of Ammon's horn sclerosis associated with temporal lobe epilepsy. Br Med J. 1956;2(5006):1403-7.
49. Bonilha L, Rorden C, Appenzeller S, Coan AC, Cendes F, Li LM. Gray matter atrophy associated with duration of temporal lobe epilepsy. Neuroimage. 2006;32(3):1070-9.
50. Roberts GW, Bruton CJ. Notes from the graveyard: neuropathology and schizophrenia. Neuropathol Appl Neurobiol. 1990;16(1):3-16.
51. Sokolov BP, Tcherepanov AA, Haroutunian V, Davis KL. Levels of mRNAs encoding synaptic vesicle and synaptic plasma membrane proteins in the temporal cortex of elderly schizophrenic patients. Biol Psychiatry. 2000;48(3):184-96.
52. Falkai P, Parlapani E, Gruber O, Schmitt A. The Neuropathology of Schizophrenia: Central Role for the Hippocampus? In: Gattaz WF, Busatto G, eds. Advances in Schizophrenia Research 2009. New York: Springer Science; 2010:149-65.
53. Shaw P, Mellers J, Henderson M, Polkey C, David AS, Toone BK. Schizophrenia-like psychosis arising de novo following a temporal lobectomy: timing and risk factors. J Neurol Neurosurg Psychiatry. 2004;75(7):1003-8.
54. Morimoto K, Fahnestock M, Racine RJ. Kindling and status epilepticus models of epilepsy: rewiring the brain. Prog Neurobiol. 2004;73(1):1-60.
55. Flor-Henry P. Lateralized temporal-limbic dysfunction and psychopathology. Ann N Y Acad Sci. 1976;280:777-97.
56. Adamec RE. Does kindling model anything clinically relevant? Biol Psychiatry. 1990;27(3):249-79.
57. Bear DM. Temporal lobe epilepsy--a syndrome of sensory-limbic hyperconnection. Cortex. 1979;15(3):357-84.
58. Duvernoy HM. The Human Hippocampus. Berlin: Springer-Verlag; 1988.
59. Andersen P, Soleng AF, Raastad M. The hippocampal lamella hypothesis revisited. Brain Res. 2000;886(1-2):165-71.
60. Scharfman HE. The CA3 "backprojection" to the dentate gyrus. Prog Brain Res. 2007;163:627-37.
61. Heinemann U, Schmitz D, Eder C, Gloveli T. Properties of entorhinal cortex projection cells to the hippocampal formation. Ann N Y Acad Sci. 2000;911:112-26.
62. Witter MP, Wouterlood FG, Naber PA, Van Haeften T. Anatomical organization of the parahippocampal-hippocampal network. Ann N Y Acad Sci. 2000;911:1-24.
63. Aggleton JP. Multiple anatomical systems embedded within the primate medial temporal lobe: Implications for hippocampal function. Neurosci Biobehav Rev. 2012;36(7):1579-96.
64. Gigg J. Constraints on hippocampal processing imposed by the connectivity between CA1, subiculum and subicular targets. Behav Brain Res. 2006;174(2):265-71.
65. Naber PA, Lopes da Silva FH, Witter MP. Reciprocal connections between the entorhinal cortex and hippocampal fields CA1 and the subiculum are in register with the projections from CA1 to the subiculum. Hippocampus. 2001;11(2):99-104.
66. Canto CB, Wouterlood FG, Witter MP. What does the anatomical organization of the entorhinal cortex tell us? Neural Plast. 2008;2008:381243.
67. de Curtis M, Pare D. The rhinal cortices: a wall of inhibition between the neocortex and the hippocampus. Prog Neurobiol. 2004;74(2):101-10.
68. Amaral DG, Lavenex P. Hippocampal Neuroanatomy. In: Andersen P, Morris RG, Amaral DG, Bliss TV, O'Keefe J, eds. The Hippocampus Book. New York: Oxford University Press; 2007:37-114.
69. Finch DM, Nowlin NL, Babb TL. Demonstration of axonal projections of neurons in the rat hippocampus and subiculum by intracellular injection of HRP. Brain Res. 1983;271(2):201-16.
70. Friedman DP, Aggleton JP, Saunders RC. Comparison of hippocampal, amygdala, and perirhinal projections to the nucleus accumbens: combined anterograde and retrograde tracing study in the Macaque brain. J Comp Neurol. 2002;450(4):345-65.
71. Pitkanen A, Pikkarainen M, Nurminen N, Ylinen A. Reciprocal connections between the amygdala and the hippocampal formation, perirhinal cortex, and postrhinal cortex in rat. A review. Ann N Y Acad Sci. 2000;911:369-91.
72. Saunders RC, Rosene DL, Van Hoesen GW. Comparison of the efferents of the amygdala and the hippocampal formation in the rhesus monkey: II. Reciprocal and non-reciprocal connections. J Comp Neurol. 1988;271(2):185-207.
73. Jay TM, Witter MP. Distribution of hippocampal CA1 and subicular efferents in the prefrontal cortex of the rat studied by means of anterograde transport of Phaseolus vulgaris-leucoagglutinin. J Comp Neurol. 1991;313(4):574-86.
74. van Groen T, Wyss JM. Extrinsic projections from area CA1 of the rat hippocampus: olfactory, cortical, subcortical, and bilateral hippocampal formation projections. J Comp Neurol. 1990;302(3):515-28.
75. Cenquizca LA, Swanson LW. Spatial organization of direct hippocampal field CA1 axonal projections to the rest of the cerebral cortex. Brain Res Rev. 2007;56(1):1-26.
76. Vertes RP, Hoover WB, Szigeti-Buck K, Leranth C. Nucleus reuniens of the midline thalamus: link between the medial prefrontal cortex and the hippocampus. Brain Res Bull. 2007;71(6):601-9.
77. Gaykema RP, van der Kuil J, Hersh LB, Luiten PG. Patterns of direct projections from the hippocampus to the medial septum-diagonal band complex: anterograde tracing with Phaseolus vulgaris leucoagglutinin combined with immunohistochemistry of choline acetyltransferase. Neuroscience. 1991;43(2-3):349-60.
78. Swanson LW, Cowan WM. An autoradiographic study of the organization of the efferent connections of the hippocampal formation in the rat. J Comp Neurol. 1977;172(1):49-84.
79. Leranth C, Nitsch R. Morphological evidence that hypothalamic substance P-containing afferents are capable of filtering the signal flow in the monkey hippocampal formation. J Neurosci. 1994;14(7):4079-94.
80. Swanson LW, Wyss JM, Cowan WM. An autoradiographic study of the organization of intrahippocampal association pathways in the rat. J Comp Neurol. 1978;181(4):681-715.
81. Vertes RP. PHA-L analysis of projections from the supramammillary nucleus in the rat. J Comp Neurol. 1992;326(4):595-622.
82. Wyss JM, Swanson LW, Cowan WM. Evidence for an input to the molecular layer and the stratum granulosum of the dentate gyrus from the supramammillary region of the hypothalamus. Anat Embryol (Berl). 1979;156(2):165-76.
83. Swanson LW, Cowan WM. Hippocampo-hypothalamic connections: origin in subicular cortex, not ammon's horn. Science. 1975;189(4199):303-4.
84. Bobillier P, Seguin S, Degueurce A, Lewis BD, Pujol JF. The efferent connections of the nucleus raphe centralis superior in the rat as revealed by radioautography. Brain Res. 1979;166(1):1-8.
85. Loughlin SE, Foote SL, Grzanna R. Efferent projections of nucleus locus coeruleus: morphologic subpopulations have different efferent targets. Neuroscience. 1986;18(2):307-19.
86. Oades RD, Halliday GM. Ventral tegmental (A10) system: neurobiology. 1. Anatomy and connectivity. Brain Res. 1987;434(2):117-65.
87. Insausti R, Herrero MT, Witter MP. Entorhinal cortex of the rat: cytoarchitectonic subdivisions and the origin and distribution of cortical efferents. Hippocampus. 1997;7(2):146-83.
88. Insausti R, Amaral DG, Cowan WM. The entorhinal cortex of the monkey: III. Subcortical afferents. J Comp Neurol. 1987;264(3):396-408.
89. Ohtake T, Yamada H. Efferent connections of the nucleus reuniens and the rhomboid nucleus in the rat: an anterograde PHA-L tracing study. Neurosci Res. 1989;6(6):556-68.
90. Room P, Groenewegen HJ. Connections of the parahippocampal cortex in the cat. II. Subcortical afferents. J Comp Neurol. 1986;251(4):451-73.
91. Witter MP, Groenewegen HJ. Connections of the parahippocampal cortex in the cat. IV. Subcortical efferents. J Comp Neurol. 1986;252(1):51-77.
92. Beckstead RM, Domesick VB, Nauta WJ. Efferent connections of the substantia nigra and ventral tegmental area in the rat. Brain Res. 1979;175(2):191-17.
93. Sah P, Faber ES, Lopez De Armentia M, Power J. The amygdaloid complex: anatomy and physiology. Physiol Rev. 2003;83(3):803-34.
94. Canteras NS, Swanson LW. Projections of the ventral subiculum to the amygdala, septum, and hypothalamus: a PHAL anterograde tract-tracing study in the rat. J Comp Neurol. 1992;324(2):180-94.
95. McDonald AJ, Mascagni F. Projections of the lateral entorhinal cortex to the amygdala: a Phaseolus vulgaris leucoagglutinin study in the rat. Neuroscience. 1997;77(2):445-59.
96. Mitrano DA, Pare JF, Smith Y. Ultrastructural relationships between cortical, thalamic, and amygdala glutamatergic inputs and group I metabotropic glutamate receptors in the rat accumbens. J Comp Neurol. 2010;518(8):1315-29.
97. Porrino LJ, Crane AM, Goldman-Rakic PS. Direct and indirect pathways from the amygdala to the frontal lobe in rhesus monkeys. J Comp Neurol. 1981;198(1):121-36.
98. McDonald AJ. Organization of amygdaloid projections to the prefrontal cortex and associated striatum in the rat. Neuroscience. 1991;44(1):1-14.
99. Gabbott PL, Warner TA, Jays PR, Salway P, Busby SJ. Prefrontal cortex in the rat: projections to subcortical autonomic, motor, and limbic centers. J Comp Neurol. 2005;492(2):145-77.
100. Ghashghaei HT, Barbas H. Pathways for emotion: interactions of prefrontal and anterior temporal pathways in the amygdala of the rhesus monkey. Neuroscience. 2002;115(4):1261-79.
101. Ishikawa A, Nakamura S. Convergence and interaction of hippocampal and amygdalar projections within the prefrontal cortex in the rat. J Neurosci. 2003;23(31):9987-95.
102. Vertes RP. Interactions among the medial prefrontal cortex, hippocampus and midline thalamus in emotional and cognitive processing in the rat. Neuroscience. 2006;142(1):1-20.
103. Caffe AR, van Leeuwen FW, Luiten PG. Vasopressin cells in the medial amygdala of the rat project to the lateral septum and ventral hippocampus. J Comp Neurol. 1987;261(2):237-52.
104. Price JL, Amaral DG. An autoradiographic study of the projections of the central nucleus of the monkey amygdala. J Neurosci. 1981;1(11):1242-59.
105. Usunoff KG, Schmitt O, Itzev DE et al. Efferent projections of the anterior and posterodorsal regions of the medial nucleus of the amygdala in the mouse. Cells Tissues Organs. 2009;190(5):256-85.
106. Wallace DM, Magnuson DJ, Gray TS. Organization of amygdaloid projections to brainstem dopaminergic, noradrenergic, and adrenergic cell groups in the rat. Brain Res Bull. 1992;28(3):447-54.
107. Jia HG, Zhang GY, Wan Q. A GABAergic projection from the central nucleus of the amygdala to the parabrachial nucleus: an ultrastructural study of anterograde tracing in combination with post-embedding immunocytochemistry in the rat. Neurosci Lett. 2005;382(1-2):153-7.
108. Petrovich GD, Canteras NS, Swanson LW. Combinatorial amygdalar inputs to hippocampal domains and hypothalamic behavior systems. Brain Res Brain Res Rev. 2001;38(1-2):247-89.
109. Rizvi TA, Ennis M, Behbehani MM, Shipley MT. Connections between the central nucleus of the amygdala and the midbrain periaqueductal gray: topography and reciprocity. J Comp Neurol. 1991;303(1):121-31.
110. Saha S, Batten TF, Henderson Z. A GABAergic projection from the central nucleus of the amygdala to the nucleus of the solitary tract: a combined anterograde tracing and electron microscopic immunohistochemical study. Neuroscience. 2000;99(4):613-26.
111. Dong HW, Swanson LW. Projections from bed nuclei of the stria terminalis, dorsomedial nucleus: implications for cerebral hemisphere integration of neuroendocrine, autonomic, and drinking responses. J Comp Neurol. 2006;494(1):75-107.
112. LeDoux JE, Farb CR, Romanski LM. Overlapping projections to the amygdala and striatum from auditory processing areas of the thalamus and cortex. Neurosci Lett. 1991;134(1):139-44.
113. Scalia F, Winans SS. The differential projections of the olfactory bulb and accessory olfactory bulb in mammals. J Comp Neurol. 1975;161(1):31-55.
114. Yasui Y, Breder CD, Saper CB, Cechetto DF. Autonomic responses and efferent pathways from the insular cortex in the rat. J Comp Neurol. 1991;303(3):355-74.
115. McDonald AJ. Cortical pathways to the mammalian amygdala. Prog Neurobiol. 1998;55(3):257-332.
116. Doron NN, Ledoux JE. Organization of projections to the lateral amygdala from auditory and visual areas of the thalamus in the rat. J Comp Neurol. 1999;412(3):383-409.
117. Turner BH, Herkenham M. Thalamoamygdaloid projections in the rat: a test of the amygdala's role in sensory processing. J Comp Neurol. 1991;313(2):295-325.
118. Su HS, Bentivoglio M. Thalamic midline cell populations projecting to the nucleus accumbens, amygdala, and hippocampus in the rat. J Comp Neurol. 1990;297(4):582-93.
119. Floresco SB, Zhang Y, Enomoto T. Neural circuits subserving behavioral flexibility and their relevance to schizophrenia. Behav Brain Res. 2009;204(2):396-409.
120. Ma J, Brudzynski SM, Leung LW. Involvement of the nucleus accumbens-ventral pallidal pathway in postictal behavior induced by a hippocampal afterdischarge in rats. Brain Res. 1996;739(1-2):26-35.
121. Ma J, Leung LS. Schizophrenia-like behavioral changes after partial hippocampal kindling. Brain Res. 2004;997(1):111-8.
122. Ma J, Leung LS. Kindled seizure in the prefrontal cortex activated behavioral hyperactivity and increase in accumbens gamma oscillations through the hippocampus. Behav Brain Res. 2010;206(1):68-77.
123. Ma J, Leung LW. Medial septum mediates the increase in post-ictal behaviors and hippocampal gamma waves after an electrically induced seizure. Brain Res. 1999;833(1):51-7.
124. Cifelli P, Grace AA. Pilocarpine-induced temporal lobe epilepsy in the rat is associated with increased dopamine neuron activity. Int J Neuropsychopharmacol. 2011:1-8.
125. Lin TC, Huang LT, Huang YN, Chen GS, Wang JY. Neonatal status epilepticus alters prefrontal-striatal circuitry and enhances methamphetamine-induced behavioral sensitization in adolescence. Epilepsy Behav. 2009;14(2):316-23.
126. Olney JW, Newcomer JW, Farber NB. NMDA receptor hypofunction model of schizophrenia. J Psychiatr Res. 1999;33(6):523-33.
127. Suzuki Y, Jodo E, Takeuchi S, Niwa S, Kayama Y. Acute administration of phencyclidine induces tonic activation of medial prefrontal cortex neurons in freely moving rats. Neuroscience. 2002;114(3):769-79.
128. Stevens JR, Livermore A, Jr. Kindling of the mesolimbic dopamine system: animal model of psychosis. Neurology. 1978;28(1):36-46.
129. Ehlers CL, Koob GF. Locomotor behavior following kindling in three different brain sites. Brain Res. 1985;326(1):71-9.
130. Watanabe T, Morimoto K, Nakamura M et al. Kindling of the ventral tegmental area induces supersensitivity in the central dopamine system. Brain Res. 2004;1003(1-2):194-8.
131. Csernansky JG, Kerr S, Pruthi R, Prosser ES. Mesolimbic dopamine receptor increases two weeks following hippocampal kindling. Brain Res. 1988;449(1-2):357-60.
132. Strecker RE, Moneta ME. Electrical stimulation of the kindled hippocampus briefly increases extracellular dopamine in the nucleus accumbens. Neurosci Lett. 1994;176(2):173-7.
133. Goto Y, O'Donnell P. Network synchrony in the nucleus accumbens in vivo. J Neurosci. 2001;21(12):4498-504.
134. O'Donnell P, Grace AA. Synaptic interactions among excitatory afferents to nucleus accumbens neurons: hippocampal gating of prefrontal cortical input. J Neurosci. 1995;15(5 Pt 1):3622-39.
135. Gordon I, Mintz M, Rosenne E, Rehavi M. Long-term effects of amygdaloid kindling on striatal dopaminergic terminals. Brain Res Bull. 1995;36(3):235-9.
136. Grace AA. Gating of information flow within the limbic system and the pathophysiology of schizophrenia. Brain Res Brain Res Rev. 2000;31(2-3):330-41.
137. Leung LS, Ma J, McLachlan RS. Behaviors induced or disrupted by complex partial seizures. Neurosci Biobehav Rev. 2000;24(7):763-75.
138. Dazzi L, Serra M, Porceddu ML, Sanna A, Chessa MF, Biggio G. Enhancement of basal and pentylenetetrazol (PTZ)-stimulated dopamine release in the brain of freely moving rats by PTZ-induced kindling. Synapse. 1997;26(4):351-8.
139. Meurs A, Clinckers R, Ebinger G, Michotte Y, Smolders I. Seizure activity and changes in hippocampal extracellular glutamate, GABA, dopamine and serotonin. Epilepsy Res. 2008;78(1):50-9.
140. Du F, Eid T, Lothman EW, Kohler C, Schwarcz R. Preferential neuronal loss in layer III of the medial entorhinal cortex in rat models of temporal lobe epilepsy. J Neurosci. 1995;15(10):6301-13.
141. Du F, Whetsell WO, Jr., Abou-Khalil B, Blumenkopf B, Lothman EW, Schwarcz R. Preferential neuronal loss in layer III of the entorhinal cortex in patients with temporal lobe epilepsy. Epilepsy Res. 1993;16(3):223-33.
142. Spencer SS, Spencer DD. Entorhinal-hippocampal interactions in medial temporal lobe epilepsy. Epilepsia. 1994;35(4):721-7.
143. Wozny C, Gabriel S, Jandova K, Schulze K, Heinemann U, Behr J. Entorhinal cortex entrains epileptiform activity in CA1 in pilocarpine-treated rats. Neurobiol Dis. 2005;19(3):451-60.
144. Suh J, Rivest AJ, Nakashiba T, Tominaga T, Tonegawa S. Entorhinal cortex layer III input to the hippocampus is crucial for temporal association memory. Science. 2011;334(6061):1415-20.
145. Mitchell SN, Yee BK, Feldon J, Gray JA, Rawlins JN. Activation of the retrohippocampal region in the rat causes dopamine release in the nucleus accumbens: disruption by fornix section. Eur J Pharmacol. 2000;407(1-2):131-38.
146. Lodge DJ, Grace AA. Hippocampal dysregulation of dopamine system function and the pathophysiology of schizophrenia. Trends Pharmacol Sci. 2011;32(9):507-13.
147. Manahan-Vaughan D, von Haebler D, Winter C, Juckel G, Heinemann U. A single application of MK801 causes symptoms of acute psychosis, deficits in spatial memory, and impairment of synaptic plasticity in rats. Hippocampus. 2008;18(2):125-34.
148. Sharp FR, Tomitaka M, Bernaudin M, Tomitaka S. Psychosis: pathological activation of limbic thalamocortical circuits by psychomimetics and schizophrenia? Trends Neurosci. 2001;24(6):330-4.
149. Carlen M, Meletis K, Siegle JH et al. A critical role for NMDA receptors in parvalbumin interneurons for gamma rhythm induction and behavior. Mol Psychiatry. 2012;17(5):537-48.
150. Gunduz-Bruce H. The acute effects of NMDA antagonism: from the rodent to the human brain. Brain Res Rev. 2009;60(2):279-86.
151. Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci. 2007;27(43):11496-500.
152. Jackson ME, Homayoun H, Moghaddam B. NMDA receptor hypofunction produces concomitant firing rate potentiation and burst activity reduction in the prefrontal cortex. Proc Natl Acad Sci U S A. 2004;101(22):8467-72.
153. Kiss T, Hoffmann WE, Hajos M. Delta oscillation and short-term plasticity in the rat medial prefrontal cortex: modelling NMDA hypofunction of schizophrenia. Int J Neuropsychopharmacol. 2011;14(1):29-42.
154. Abdul-Monim Z, Neill JC, Reynolds GP. Sub-chronic psychotomimetic phencyclidine induces deficits in reversal learning and alterations in parvalbumin-immunoreactive expression in the rat. J Psychopharmacol. 2007;21(2):198-205.
155. Feinberg I, Campbell IG, Marrs JC. Intraperitoneal dizocilpine induces cortical spike-wave seizure discharges in rats. Neurosci Lett. 1995;196(3):157-60.
156. Olney JW, Labruyere J, Price MT. Pathological changes induced in cerebrocortical neurons by phencyclidine and related drugs. Science. 1989;244(4910):1360-2.
157. McNally JM, McCarley RW, McKenna JT, Yanagawa Y, Brown RE. Complex receptor mediation of acute ketamine application on in vitro gamma oscillations in mouse prefrontal cortex: modeling gamma band oscillation abnormalities in schizophrenia. Neuroscience. 2011;199:51-63.
158. Javitt DC. Glutamate and schizophrenia: phencyclidine, N-methyl-D-aspartate receptors, and dopamine-glutamate interactions. Int Rev Neurobiol. 2007;78:69-108.
159. Verma A, Moghaddam B. NMDA receptor antagonists impair prefrontal cortex function as assessed via spatial delayed alternation performance in rats: modulation by dopamine. J Neurosci. 1996;16(1):373-9.
160. Hondo H, Yonezawa Y, Nakahara T, et al. Effect of phencyclidine on dopamine release in the rat prefrontal cortex; an in vivo microdialysis study. Brain Res. 1994;633(1-2):337-42.
161. Jodo E, Suzuki Y, Katayama T, et al. Activation of medial prefrontal cortex by phencyclidine is mediated via a hippocampo-prefrontal pathway. Cereb Cortex. 2005;15(5):663-9.
162. Sesack SR, Grace AA. Cortico-Basal Ganglia Reward Network: Microcircuitry. Neuropsychopharmacology. 2010;35:27-47.
Submitted on March 9, 2012; accepted on April 23, 2012

ERRATUM
In the article Psychiatric comorbidities in temporal lobe epilepsy: Possible relationships between psychotic disorders and involvement of limbic circuits, in Figure 4, where it reads "Figure based on Sesack and Grace155 and Lodge and Grace.139", it should read "Figure based on Sesack and Grace162 and Lodge and Grace146".
162. Sesack SR, Grace AA. Cortico-Basal Ganglia Reward Network: Microcircuitry. Neuropsychopharmacology. 2010;35:27-47.
- 1. Bertram EH. Temporal lobe epilepsy: where do the seizures really begin? Epilepsy Behav. 2009;14(Suppl 1):32-7.
- 2. Swerdlow NR. Integrative circuit models and their implications for the pathophysiologies and treatments of the schizophrenias. Curr Top Behav Neurosci. 2010;4:555-83.
- 3. Flor-Henry P. Psychosis and temporal lobe epilepsy. A controlled investigation. Epilepsia. 1969;10(3):363-95.
- 4. Roberts GW, Done DJ, Bruton C, Crow TJ. A "mock up" of schizophrenia: temporal lobe epilepsy and schizophrenia-like psychosis. Biol Psychiatry. 1990;28(2):127-43.
- 5. Serafetinides EA, Falconer MA. The effects of temporal lobectomy in epileptic patients with psychosis. J Ment Sci. 1962;108:584-93.
- 6. Stevens JR. Epilepsy, psychosis and schizophrenia. Schizophr Res. 1988;1(1):79-89.
- 7. Taylor DC. Factors influencing the occurrence of schizophrenia-like psychosis in patients with temporal lobe epilepsy. Psychol Med. 1975;5(3):249-54.
- 8. Editorial: Epilepsy, schizophrenia, and limbic system. Lancet. 1974;2(7886):935-6.
- 9. Papez JW. A proposed mechanism of emotion. J Neuropsychiatry Clin Neurosci. 1937;7(1):103-12.
- 10. Klüver H, Bucy P. "Psychic blindness" and other symtoms following bilateral temporal lobectomy in rhesus monkeys. American Journal of Physiology. 1937;119:352-3.
- 11. MacLean P. Psychosomatic disease and the visceral brain; recent developments bearing on the Papez theory of emotion. Psychosom Med. 1949;11(6):338-53.
- 12. Heimer L, Van Hoesen GW, Trimble M, Zahn DS. The limbic system: a concept in perpetual search for a definition. In: Heimer L, Van Hoesen GW, Trimble M, Zahn DS, eds. Anatomy of Neuropsychiatry. U.S.A.: Academic Press Elsevier; 2008:1-13.
- 13. Brain L. Psychosomatic Medicine and the Brain-Mind Relationship. Lancet. 1964;2(7355):325-8.
- 14. Cairns H. Disturbances of consciousness with lesions of the brain-stem and diencephalon. Brain. 1952;75(2):109-46.
- 15. Waxman SG, Geschwind N. The interictal behavior syndrome of temporal lobe epilepsy. Arch Gen Psychiatry. 1975;32(12):1580-6.
- 16. Penfield W, Perot P. The Brain's Record of Auditory and Visual Experience. A Final Summary and Discussion. Brain. 1963;86:595-696.
- 17. Gloor P, Olivier A, Quesney LF, Andermann F, Horowitz S. The role of the limbic system in experiential phenomena of temporal lobe epilepsy. Ann Neurol. 1982;12(2):129-44.
- 18. Beard AW, Slater E. The schizophrenic-like psychoses of epilepsy. Proc R Soc Med. 1962;55:311-6.
- 19. Fujii D, Ahmed I. Psychotic disorder following traumatic brain injury: a conceptual framework. Cogn Neuropsychiatry. 2002;7(1):41-62.
- 20. Kristensen O, Sindrup EH. Psychomotor epilepsy and psychosis. I. Physical aspects. Acta Neurol Scand. 1978;57(5):361-9.
- 21. Landolt H. Some correlations between the electroencephalogram and normal and pathologic mental processes. Epilepsy Behav. 1963;14(3):448-51.
- 22. Pollock DC. Models for understanding the antagonism between seizures and psychosis. Prog Neuropsychopharmacol Biol Psychiatry. 1987;11(4):483-504.
- 23. Trimble M. The relationship between epilepsy and schizophrenia: a biochemical hypothesis. Biol Psychiatry. 1977;12(2):299-304.
- 24. Ogren SO, Pakh B. Effects of dopamine D1 and D2 receptor agonists and antagonists on seizures induced by chemoconvulsants in mice. Pharmacol Toxicol. 1993;72(4-5):213-20.
- 25. Turski L, Cavalheiro EA, Bortolotto ZA, Ikonomidou-Turski C, Kleinrok Z, Turski WA. Dopamine-sensitive anticonvulsant site in the rat striatum. J Neurosci. 1988;8(11):4027-37.
- 26. Slater E, Beard AW, Glithero E. The schizophrenia-like psychoses of epilepsy. Br J Psychiatry. 1963;109:95-150.
- 27. Krishnamoorthy ES, Trimble MR, Blumer D. The classification of neuropsychiatric disorders in epilepsy: a proposal by the ILAE Commission on Psychobiology of Epilepsy. Epilepsy Behav. 2007;10(3):349-53.
- 28. Toone BK. The psychoses of epilepsy. J Neurol Neurosurg Psychiatry. 2000;69(1):1-3.
- 29. Kanemoto K, Kawasaki J, Kawai I. Postictal psychosis: a comparison with acute interictal and chronic psychoses. Epilepsia. 1996;37(6):551-6.
- 30. Tarulli A, Devinsky O, Alper K. Progression of postictal to interictal psychosis. Epilepsia. 2001;42(11):1468-71.
- 31. Braun CM, Dumont M, Duval J, Hamel-Hebert I, Godbout L. Brain modules of hallucination: an analysis of multiple patients with brain lesions. J Psychiatry Neurosci. 2003;28(6):432-49.
- 32. Torrey EF, Peterson MR. Schizophrenia and the limbic system. Lancet. 1974;2(7886):942-6.
- 33. Malamud N. Psychiatric disorder with intracranial tumors of limbic system. Arch Neurol. 1967;17(2):113-23.
- 34. Taylor DC. Mental state and temporal lobe epilepsy. A correlative account of 100 patients treated surgically. Epilepsia. 1972;13(6):727-65.
- 35. Kandratavicius L, Hallak JE, Young LT, Assirati JA, Carlotti CG, Jr., Leite JP. Differential aberrant sprouting in temporal lobe epilepsy with psychiatric co-morbidities. Psychiatry Res. 2012;195(3):144-50.
- 36. Allen P, Laroi F, McGuire PK, Aleman A. The hallucinating brain: a review of structural and functional neuroimaging studies of hallucinations. Neurosci Biobehav Rev. 2008;32(1):175-91.
- 37. Braff DL. Gating in schizophrenia: from genes to cognition (to real world function?). Biol Psychiatry. 2011;69(5):395-6.
- 38. Quednow BB, Ettinger U, Mossner R et al. The schizophrenia risk allele C of the TCF4 rs9960767 polymorphism disrupts sensorimotor gating in schizophrenia spectrum and healthy volunteers. J Neurosci. 2011;31(18):6684-91.
- 39. Takahashi H, Iwase M, Yasuda Y et al. Relationship of prepulse inhibition to temperament and character in healthy Japanese subjects. Neurosci Res. 2012;72(2):187-93.
- 40. Braff DL, Geyer MA, Swerdlow NR. Human studies of prepulse inhibition of startle: normal subjects, patient groups, and pharmacological studies. Psychopharmacology (Berl). 2001;156(2-3):234-58.
- 41. Pouretemad HR, Thompson PJ, Fenwick PB. Impaired sensorimotor gating in patients with non-epileptic seizures. Epilepsy Res. 1998;31(1):1-12.
- 42. Syed TU, LaFrance WC, Jr., Kahriman ES et al. Can semiology predict psychogenic nonepileptic seizures? A prospective study. Ann Neurol. 2011;69(6):997-1004.
- 43. Mokleby K, Blomhoff S, Malt UF, Dahlstrom A, Tauboll E, Gjerstad L. Psychiatric comorbidity and hostility in patients with psychogenic nonepileptic seizures compared with somatoform disorders and healthy controls. Epilepsia. 2002;43(2):193-8.
- 44. Howland JG, Hannesson DK, Barnes SJ, Phillips AG. Kindling of basolateral amygdala but not ventral hippocampus or perirhinal cortex disrupts sensorimotor gating in rats. Behav Brain Res. 2007;177(1):30-6.
- 45. Miller EJ, Saint Marie LR, Breier MR, Swerdlow NR. Pathways from the ventral hippocampus and caudal amygdala to forebrain regions that regulate sensorimotor gating in the rat. Neuroscience. 2010;165(2):601-11.
- 46. Weinberger DR, Wagner RL, Wyatt RJ. Neuropathological studies of schizophrenia: a selective review. Schizophr Bull. 1983;9(2):193-212.
- 47. Thom M. Hippocampal sclerosis: progress since Sommer. Brain Pathol. 2009;19(4):565-72.
- 48. Cavanagh JB, Meyer A. Aetiological aspects of Ammon's horn sclerosis associated with temporal lobe epilepsy. Br Med J. 1956;2(5006):1403-7.
- 49. Bonilha L, Rorden C, Appenzeller S, Coan AC, Cendes F, Li LM. Gray matter atrophy associated with duration of temporal lobe epilepsy. Neuroimage. 2006;32(3):1070-9.
- 50. Roberts GW, Bruton CJ. Notes from the graveyard: neuropathology and schizophrenia. Neuropathol Appl Neurobiol. 1990;16(1):3-16.
- 51. Sokolov BP, Tcherepanov AA, Haroutunian V, Davis KL. Levels of mRNAs encoding synaptic vesicle and synaptic plasma membrane proteins in the temporal cortex of elderly schizophrenic patients. Biol Psychiatry. 2000;48(3):184-96.
- 52. Falkai P, Parlapani E, Gruber O, Schmitt A. The Neuropathology of Schizophrenia: Central Role for the Hippocampus? In: Gattaz WF, Busatto G, eds. Advances in Schizophrenia Research 2009. New York: Springer Science; 2010:149-65.
- 53. Shaw P, Mellers J, Henderson M, Polkey C, David AS, Toone BK. Schizophrenia-like psychosis arising de novo following a temporal lobectomy: timing and risk factors. J Neurol Neurosurg Psychiatry. 2004;75(7):1003-8.
- 54. Morimoto K, Fahnestock M, Racine RJ. Kindling and status epilepticus models of epilepsy: rewiring the brain. Prog Neurobiol. 2004;73(1):1-60.
- 55. Flor-Henry P. Lateralized temporal-limbic dysfunction and psychopathology. Ann N Y Acad Sci. 1976;280:777-97.
- 56. Adamec RE. Does kindling model anything clinically relevant? Biol Psychiatry. 1990;27(3):249-79.
- 57. Bear DM. Temporal lobe epilepsy--a syndrome of sensory-limbic hyperconnection. Cortex. 1979;15(3):357-84.
- 58. Duvernoy HM. The Human Hippocampus. Berlin: Springer-Verlag; 1988.
- 59. Andersen P, Soleng AF, Raastad M. The hippocampal lamella hypothesis revisited. Brain Res. 2000;886(1-2):165-71.
- 60. Scharfman HE. The CA3 "backprojection" to the dentate gyrus. Prog Brain Res. 2007;163:627-37.
- 61. Heinemann U, Schmitz D, Eder C, Gloveli T. Properties of entorhinal cortex projection cells to the hippocampal formation. Ann N Y Acad Sci. 2000;911:112-26.
- 62. Witter MP, Wouterlood FG, Naber PA, Van Haeften T. Anatomical organization of the parahippocampal-hippocampal network. Ann N Y Acad Sci. 2000;911:1-24.
- 63. Aggleton JP. Multiple anatomical systems embedded within the primate medial temporal lobe: Implications for hippocampal function. Neurosci Biobehav Rev. 2012;36(7):1579-96.
- 64. Gigg J. Constraints on hippocampal processing imposed by the connectivity between CA1, subiculum and subicular targets. Behav Brain Res. 2006;174(2):265-71.
- 65. Naber PA, Lopes da Silva FH, Witter MP. Reciprocal connections between the entorhinal cortex and hippocampal fields CA1 and the subiculum are in register with the projections from CA1 to the subiculum. Hippocampus. 2001;11(2):99-104.
- 66. Canto CB, Wouterlood FG, Witter MP. What does the anatomical organization of the entorhinal cortex tell us? Neural Plast. 2008;2008:381243.
- 67. de Curtis M, Pare D. The rhinal cortices: a wall of inhibition between the neocortex and the hippocampus. Prog Neurobiol. 2004;74(2):101-10.
- 68. Amaral DG, Lavenex P. Hippocampal Neuroanatomy. In: Andersen P, Morris RG, Amaral DG, Bliss TV, O'Keefe J, eds. The Hippocampus Book. New York: Oxford University Press; 2007:37-114.
- 69. Finch DM, Nowlin NL, Babb TL. Demonstration of axonal projections of neurons in the rat hippocampus and subiculum by intracellular injection of HRP. Brain Res. 1983;271(2):201-16.
- 70. Friedman DP, Aggleton JP, Saunders RC. Comparison of hippocampal, amygdala, and perirhinal projections to the nucleus accumbens: combined anterograde and retrograde tracing study in the Macaque brain. J Comp Neurol. 2002;450(4):345-65.
- 71. Pitkanen A, Pikkarainen M, Nurminen N, Ylinen A. Reciprocal connections between the amygdala and the hippocampal formation, perirhinal cortex, and postrhinal cortex in rat. A review. Ann N Y Acad Sci. 2000;911:369-91.
- 72. Saunders RC, Rosene DL, Van Hoesen GW. Comparison of the efferents of the amygdala and the hippocampal formation in the rhesus monkey: II. Reciprocal and non-reciprocal connections. J Comp Neurol. 1988;271(2):185-207.
- 73. Jay TM, Witter MP. Distribution of hippocampal CA1 and subicular efferents in the prefrontal cortex of the rat studied by means of anterograde transport of Phaseolus vulgaris-leucoagglutinin. J Comp Neurol. 1991;313(4):574-86.
- 74. van Groen T, Wyss JM. Extrinsic projections from area CA1 of the rat hippocampus: olfactory, cortical, subcortical, and bilateral hippocampal formation projections. J Comp Neurol. 1990;302(3):515-28.
- 75. Cenquizca LA, Swanson LW. Spatial organization of direct hippocampal field CA1 axonal projections to the rest of the cerebral cortex. Brain Res Rev. 2007;56(1):1-26.
- 76. Vertes RP, Hoover WB, Szigeti-Buck K, Leranth C. Nucleus reuniens of the midline thalamus: link between the medial prefrontal cortex and the hippocampus. Brain Res Bull. 2007;71(6):601-9.
- 77. Gaykema RP, van der Kuil J, Hersh LB, Luiten PG. Patterns of direct projections from the hippocampus to the medial septum-diagonal band complex: anterograde tracing with Phaseolus vulgaris leucoagglutinin combined with immunohistochemistry of choline acetyltransferase. Neuroscience. 1991;43(2-3):349-60.
- 78. Swanson LW, Cowan WM. An autoradiographic study of the organization of the efferent connections of the hippocampal formation in the rat. J Comp Neurol. 1977;172(1):49-84.
- 79. Leranth C, Nitsch R. Morphological evidence that hypothalamic substance P-containing afferents are capable of filtering the signal flow in the monkey hippocampal formation. J Neurosci. 1994;14(7):4079-94.
- 80. Swanson LW, Wyss JM, Cowan WM. An autoradiographic study of the organization of intrahippocampal association pathways in the rat. J Comp Neurol. 1978;181(4):681-715.
- 81. Vertes RP. PHA-L analysis of projections from the supramammillary nucleus in the rat. J Comp Neurol. 1992;326(4):595-622.
- 82. Wyss JM, Swanson LW, Cowan WM. Evidence for an input to the molecular layer and the stratum granulosum of the dentate gyrus from the supramammillary region of the hypothalamus. Anat Embryol (Berl). 1979;156(2):165-76.
- 83. Swanson LW, Cowan WM. Hippocampo-hypothalamic connections: origin in subicular cortex, not ammon's horn. Science. 1975;189(4199):303-4.
- 84. Bobillier P, Seguin S, Degueurce A, Lewis BD, Pujol JF. The efferent connections of the nucleus raphe centralis superior in the rat as revealed by radioautography. Brain Res. 1979;166(1):1-8.
- 85. Loughlin SE, Foote SL, Grzanna R. Efferent projections of nucleus locus coeruleus: morphologic subpopulations have different efferent targets. Neuroscience. 1986;18(2):307-19.
- 86. Oades RD, Halliday GM. Ventral tegmental (A10) system: neurobiology. 1. Anatomy and connectivity. Brain Res. 1987;434(2):117-65.
- 87. Insausti R, Herrero MT, Witter MP. Entorhinal cortex of the rat: cytoarchitectonic subdivisions and the origin and distribution of cortical efferents. Hippocampus. 1997;7(2):146-83.
- 88. Insausti R, Amaral DG, Cowan WM. The entorhinal cortex of the monkey: III. Subcortical afferents. J Comp Neurol. 1987;264(3):396-408.
- 89. Ohtake T, Yamada H. Efferent connections of the nucleus reuniens and the rhomboid nucleus in the rat: an anterograde PHA-L tracing study. Neurosci Res. 1989;6(6):556-68.
- 90. Room P, Groenewegen HJ. Connections of the parahippocampal cortex in the cat. II. Subcortical afferents. J Comp Neurol. 1986;251(4):451-73.
- 91. Witter MP, Groenewegen HJ. Connections of the parahippocampal cortex in the cat. IV. Subcortical efferents. J Comp Neurol. 1986;252(1):51-77.
- 92. Beckstead RM, Domesick VB, Nauta WJ. Efferent connections of the substantia nigra and ventral tegmental area in the rat. Brain Res. 1979;175(2):191-17.
- 93. Sah P, Faber ES, Lopez De Armentia M, Power J. The amygdaloid complex: anatomy and physiology. Physiol Rev. 2003;83(3):803-34.
- 94. Canteras NS, Swanson LW. Projections of the ventral subiculum to the amygdala, septum, and hypothalamus: a PHAL anterograde tract-tracing study in the rat. J Comp Neurol. 1992;324(2):180-94.
- 95. McDonald AJ, Mascagni F. Projections of the lateral entorhinal cortex to the amygdala: a Phaseolus vulgaris leucoagglutinin study in the rat. Neuroscience. 1997;77(2):445-59.
- 96. Mitrano DA, Pare JF, Smith Y. Ultrastructural relationships between cortical, thalamic, and amygdala glutamatergic inputs and group I metabotropic glutamate receptors in the rat accumbens. J Comp Neurol. 2010;518(8):1315-29.
- 97. Porrino LJ, Crane AM, Goldman-Rakic PS. Direct and indirect pathways from the amygdala to the frontal lobe in rhesus monkeys. J Comp Neurol. 1981;198(1):121-36.
- 98. McDonald AJ. Organization of amygdaloid projections to the prefrontal cortex and associated striatum in the rat. Neuroscience. 1991;44(1):1-14.
- 99. Gabbott PL, Warner TA, Jays PR, Salway P, Busby SJ. Prefrontal cortex in the rat: projections to subcortical autonomic, motor, and limbic centers. J Comp Neurol. 2005;492(2):145-77.
- 100. Ghashghaei HT, Barbas H. Pathways for emotion: interactions of prefrontal and anterior temporal pathways in the amygdala of the rhesus monkey. Neuroscience. 2002;115(4):1261-79.
- 101. Ishikawa A, Nakamura S. Convergence and interaction of hippocampal and amygdalar projections within the prefrontal cortex in the rat. J Neurosci. 2003;23(31):9987-95.
- 102. Vertes RP. Interactions among the medial prefrontal cortex, hippocampus and midline thalamus in emotional and cognitive processing in the rat. Neuroscience. 2006;142(1):1-20.
- 103. Caffe AR, van Leeuwen FW, Luiten PG. Vasopressin cells in the medial amygdala of the rat project to the lateral septum and ventral hippocampus. J Comp Neurol. 1987;261(2):237-52.
- 104. Price JL, Amaral DG. An autoradiographic study of the projections of the central nucleus of the monkey amygdala. J Neurosci. 1981;1(11):1242-59.
- 105. Usunoff KG, Schmitt O, Itzev DE et al. Efferent projections of the anterior and posterodorsal regions of the medial nucleus of the amygdala in the mouse. Cells Tissues Organs. 2009;190(5):256-85.
- 106. Wallace DM, Magnuson DJ, Gray TS. Organization of amygdaloid projections to brainstem dopaminergic, noradrenergic, and adrenergic cell groups in the rat. Brain Res Bull. 1992;28(3):447-54.
- 107. Jia HG, Zhang GY, Wan Q. A GABAergic projection from the central nucleus of the amygdala to the parabrachial nucleus: an ultrastructural study of anterograde tracing in combination with post-embedding immunocytochemistry in the rat. Neurosci Lett. 2005;382(1-2):153-7.
- 108. Petrovich GD, Canteras NS, Swanson LW. Combinatorial amygdalar inputs to hippocampal domains and hypothalamic behavior systems. Brain Res Brain Res Rev. 2001;38(1-2):247-89.
- 109. Rizvi TA, Ennis M, Behbehani MM, Shipley MT. Connections between the central nucleus of the amygdala and the midbrain periaqueductal gray: topography and reciprocity. J Comp Neurol. 1991;303(1):121-31.
- 110. Saha S, Batten TF, Henderson Z. A GABAergic projection from the central nucleus of the amygdala to the nucleus of the solitary tract: a combined anterograde tracing and electron microscopic immunohistochemical study. Neuroscience. 2000;99(4):613-26.
- 111. Dong HW, Swanson LW. Projections from bed nuclei of the stria terminalis, dorsomedial nucleus: implications for cerebral hemisphere integration of neuroendocrine, autonomic, and drinking responses. J Comp Neurol. 2006;494(1):75-107.
- 112. LeDoux JE, Farb CR, Romanski LM. Overlapping projections to the amygdala and striatum from auditory processing areas of the thalamus and cortex. Neurosci Lett. 1991;134(1):139-44.
- 113. Scalia F, Winans SS. The differential projections of the olfactory bulb and accessory olfactory bulb in mammals. J Comp Neurol. 1975;161(1):31-55.
- 114. Yasui Y, Breder CD, Saper CB, Cechetto DF. Autonomic responses and efferent pathways from the insular cortex in the rat. J Comp Neurol. 1991;303(3):355-74.
- 115. McDonald AJ. Cortical pathways to the mammalian amygdala. Prog Neurobiol. 1998;55(3):257-332.
- 116. Doron NN, Ledoux JE. Organization of projections to the lateral amygdala from auditory and visual areas of the thalamus in the rat. J Comp Neurol. 1999;412(3):383-409.
- 117. Turner BH, Herkenham M. Thalamoamygdaloid projections in the rat: a test of the amygdala's role in sensory processing. J Comp Neurol. 1991;313(2):295-325.
- 118. Su HS, Bentivoglio M. Thalamic midline cell populations projecting to the nucleus accumbens, amygdala, and hippocampus in the rat. J Comp Neurol. 1990;297(4):582-93.
- 119. Floresco SB, Zhang Y, Enomoto T. Neural circuits subserving behavioral flexibility and their relevance to schizophrenia. Behav Brain Res. 2009;204(2):396-409.
- 120. Ma J, Brudzynski SM, Leung LW. Involvement of the nucleus accumbens-ventral pallidal pathway in postictal behavior induced by a hippocampal afterdischarge in rats. Brain Res. 1996;739(1-2):26-35.
- 121. Ma J, Leung LS. Schizophrenia-like behavioral changes after partial hippocampal kindling. Brain Res. 2004;997(1):111-8.
- 122. Ma J, Leung LS. Kindled seizure in the prefrontal cortex activated behavioral hyperactivity and increase in accumbens gamma oscillations through the hippocampus. Behav Brain Res. 2010;206(1):68-77.
- 123. Ma J, Leung LW. Medial septum mediates the increase in post-ictal behaviors and hippocampal gamma waves after an electrically induced seizure. Brain Res. 1999;833(1):51-7.
- 124. Cifelli P, Grace AA. Pilocarpine-induced temporal lobe epilepsy in the rat is associated with increased dopamine neuron activity. Int J Neuropsychopharmacol. 2011:1-8.
- 125. Lin TC, Huang LT, Huang YN, Chen GS, Wang JY. Neonatal status epilepticus alters prefrontal-striatal circuitry and enhances methamphetamine-induced behavioral sensitization in adolescence. Epilepsy Behav. 2009;14(2):316-23.
- 126. Olney JW, Newcomer JW, Farber NB. NMDA receptor hypofunction model of schizophrenia. J Psychiatr Res. 1999;33(6):523-33.
- 127. Suzuki Y, Jodo E, Takeuchi S, Niwa S, Kayama Y. Acute administration of phencyclidine induces tonic activation of medial prefrontal cortex neurons in freely moving rats. Neuroscience. 2002;114(3):769-79.
- 128. Stevens JR, Livermore A, Jr. Kindling of the mesolimbic dopamine system: animal model of psychosis. Neurology. 1978;28(1):36-46.
- 129. Ehlers CL, Koob GF. Locomotor behavior following kindling in three different brain sites. Brain Res. 1985;326(1):71-9.
- 130. Watanabe T, Morimoto K, Nakamura M et al. Kindling of the ventral tegmental area induces supersensitivity in the central dopamine system. Brain Res. 2004;1003(1-2):194-8.
- 131. Csernansky JG, Kerr S, Pruthi R, Prosser ES. Mesolimbic dopamine receptor increases two weeks following hippocampal kindling. Brain Res. 1988;449(1-2):357-60.
- 132. Strecker RE, Moneta ME. Electrical stimulation of the kindled hippocampus briefly increases extracellular dopamine in the nucleus accumbens. Neurosci Lett. 1994;176(2):173-7.
- 133. Goto Y, O'Donnell P. Network synchrony in the nucleus accumbens in vivo. J Neurosci. 2001;21(12):4498-504.
- 134. O'Donnell P, Grace AA. Synaptic interactions among excitatory afferents to nucleus accumbens neurons: hippocampal gating of prefrontal cortical input. J Neurosci. 1995;15(5 Pt 1):3622-39.
- 135. Gordon I, Mintz M, Rosenne E, Rehavi M. Long-term effects of amygdaloid kindling on striatal dopaminergic terminals. Brain Res Bull. 1995;36(3):235-9.
- 136. Grace AA. Gating of information flow within the limbic system and the pathophysiology of schizophrenia. Brain Res Brain Res Rev. 2000;31(2-3):330-41.
- 137. Leung LS, Ma J, McLachlan RS. Behaviors induced or disrupted by complex partial seizures. Neurosci Biobehav Rev. 2000;24(7):763-75.
- 138. Dazzi L, Serra M, Porceddu ML, Sanna A, Chessa MF, Biggio G. Enhancement of basal and pentylenetetrazol (PTZ)-stimulated dopamine release in the brain of freely moving rats by PTZ-induced kindling. Synapse. 1997;26(4):351-8.
- 139. Meurs A, Clinckers R, Ebinger G, Michotte Y, Smolders I. Seizure activity and changes in hippocampal extracellular glutamate, GABA, dopamine and serotonin. Epilepsy Res. 2008;78(1):50-9.
- 140. Du F, Eid T, Lothman EW, Kohler C, Schwarcz R. Preferential neuronal loss in layer III of the medial entorhinal cortex in rat models of temporal lobe epilepsy. J Neurosci. 1995;15(10):6301-13.
- 141. Du F, Whetsell WO, Jr., Abou-Khalil B, Blumenkopf B, Lothman EW, Schwarcz R. Preferential neuronal loss in layer III of the entorhinal cortex in patients with temporal lobe epilepsy. Epilepsy Res. 1993;16(3):223-33.
- 142. Spencer SS, Spencer DD. Entorhinal-hippocampal interactions in medial temporal lobe epilepsy. Epilepsia. 1994;35(4):721-7.
- 143. Wozny C, Gabriel S, Jandova K, Schulze K, Heinemann U, Behr J. Entorhinal cortex entrains epileptiform activity in CA1 in pilocarpine-treated rats. Neurobiol Dis. 2005;19(3):451-60.
- 144. Suh J, Rivest AJ, Nakashiba T, Tominaga T, Tonegawa S. Entorhinal cortex layer III input to the hippocampus is crucial for temporal association memory. Science. 2011;334(6061):1415-20.
- 145. Mitchell SN, Yee BK, Feldon J, Gray JA, Rawlins JN. Activation of the retrohippocampal region in the rat causes dopamine release in the nucleus accumbens: disruption by fornix section. Eur J Pharmacol. 2000;407(1-2):131-38.
- 146. Lodge DJ, Grace AA. Hippocampal dysregulation of dopamine system function and the pathophysiology of schizophrenia. Trends Pharmacol Sci. 2011;32(9):507-13.
- 147. Manahan-Vaughan D, von Haebler D, Winter C, Juckel G, Heinemann U. A single application of MK801 causes symptoms of acute psychosis, deficits in spatial memory, and impairment of synaptic plasticity in rats. Hippocampus. 2008;18(2):125-34.
- 148. Sharp FR, Tomitaka M, Bernaudin M, Tomitaka S. Psychosis: pathological activation of limbic thalamocortical circuits by psychomimetics and schizophrenia? Trends Neurosci. 2001;24(6):330-4.
- 149. Carlen M, Meletis K, Siegle JH et al. A critical role for NMDA receptors in parvalbumin interneurons for gamma rhythm induction and behavior. Mol Psychiatry. 2012;17(5):537-48.
- 150. Gunduz-Bruce H. The acute effects of NMDA antagonism: from the rodent to the human brain. Brain Res Rev. 2009;60(2):279-86.
- 151. Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci. 2007;27(43):11496-500.
- 152. Jackson ME, Homayoun H, Moghaddam B. NMDA receptor hypofunction produces concomitant firing rate potentiation and burst activity reduction in the prefrontal cortex. Proc Natl Acad Sci U S A. 2004;101(22):8467-72.
- 153. Kiss T, Hoffmann WE, Hajos M. Delta oscillation and short-term plasticity in the rat medial prefrontal cortex: modelling NMDA hypofunction of schizophrenia. Int J Neuropsychopharmacol. 2011;14(1):29-42.
- 154. Abdul-Monim Z, Neill JC, Reynolds GP. Sub-chronic psychotomimetic phencyclidine induces deficits in reversal learning and alterations in parvalbumin-immunoreactive expression in the rat. J Psychopharmacol. 2007;21(2):198-205.
- 155. Feinberg I, Campbell IG, Marrs JC. Intraperitoneal dizocilpine induces cortical spike-wave seizure discharges in rats. Neurosci Lett. 1995;196(3):157-60.
- 156. Olney JW, Labruyere J, Price MT. Pathological changes induced in cerebrocortical neurons by phencyclidine and related drugs. Science. 1989;244(4910):1360-2.
- 157. McNally JM, McCarley RW, McKenna JT, Yanagawa Y, Brown RE. Complex receptor mediation of acute ketamine application on in vitro gamma oscillations in mouse prefrontal cortex: modeling gamma band oscillation abnormalities in schizophrenia. Neuroscience. 2011;199:51-63.
- 158. Javitt DC. Glutamate and schizophrenia: phencyclidine, N-methyl-D-aspartate receptors, and dopamine-glutamate interactions. Int Rev Neurobiol. 2007;78:69-108.
- 159. Verma A, Moghaddam B. NMDA receptor antagonists impair prefrontal cortex function as assessed via spatial delayed alternation performance in rats: modulation by dopamine. J Neurosci. 1996;16(1):373-9.
- 160. Hondo H, Yonezawa Y, Nakahara T, et al. Effect of phencyclidine on dopamine release in the rat prefrontal cortex; an in vivo microdialysis study. Brain Res. 1994;633(1-2):337-42.
- 161. Jodo E, Suzuki Y, Katayama T, et al. Activation of medial prefrontal cortex by phencyclidine is mediated via a hippocampo-prefrontal pathway. Cereb Cortex. 2005;15(5):663-9.
- 162. Sesack SR, Grace AA. Cortico-Basal Ganglia Reward Network: Microcircuitry. Neuropsychopharmacology. 2010;35:27-47.
Corresponding author:
Publication Dates
-
Publication in this collection
04 Apr 2013 -
Date of issue
Dec 2012
History
-
Received
09 Mar 2012 -
Accepted
23 Apr 2012