Abstract
Objective:
In the present study, we aimed to examine the effects of repeated D-amphetamine (AMPH) exposure, a well-accepted animal model of acute mania in bipolar disorder (BD), and histone deacetylase (HDAC) inhibitors on locomotor behavior and HDAC activity in the prefrontal cortex (PFC) and peripheral blood mononuclear cells (PBMCs) of rats. Moreover, we aimed to assess brain-derived neurotrophic factor (BDNF) protein and mRNA levels in these samples.
Methods:
We treated adult male Wistar rats with 2 mg/kg AMPH or saline intraperitoneally for 14 days. Between the 8th and 14th days, rats also received 47.5 mg/kg lithium (Li), 200 mg/kg sodium valproate (VPT), 2 mg/kg sodium butyrate (SB), or saline. We evaluated locomotor activity in the open-field task and assessed HDAC activity in the PFC and PBMCs, and BDNF levels in the PFC and plasma.
Results:
AMPH significantly increased locomotor activity, which was reversed by all drugs. This hyperactivity was associated with increased HDAC activity in the PFC, which was partially reversed by Li, VPT, and SB. No differences were found in BDNF levels.
Conclusion:
Repeated AMPH administration increases HDAC activity in the PFC without altering BDNF levels. The partial reversal of HDAC increase by Li, VPT, and SB may account for their ability to reverse AMPH-induced hyperactivity.
Bipolar disorder; mood stabilizer; sodium butyrate; histone deacetylase; BDNF
Introduction
A growing body of evidence suggests an association between epigenetic mechanisms and the pathophysiology of bipolar disorder (BD), given the importance of long-lasting changes in neuroplasticity and gene expression to the mechanism of action of mood stabilizers.11. Wu X, Chen PS, Dallas S, Wilson B, Block ML, Wang CC, et al. Histone deacetylase inhibitors up-regulate astrocyte GDNF and BDNF gene transcription and protect dopaminergic neurons. Int J Neuropsychopharmacol. 2008;11:1123-34. The importance of gene-environment interaction in the development of different psychiatric disorders has been discussed elsewhere, in that external stimuli can cause modifications in gene expression without actually altering the genetic code.22. Caspi A, Moffitt TE. Gene-environment interactions in psychiatry: joining forces with neuroscience. Nat Rev Neurosci. 2006;7:583-90. These long-lasting changes based on epigenetic mechanisms would be desirable in a treatment approach as well. Among such mechanisms, a change in the methylation and acetylation state of genes and histones is the most well known.33. Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci. 2007;8:355-67. Hyperacetylation of histones generally promotes chromatin decondensation and increased gene activity, whereas hypoacetylation leads to condensation and reduced activity.44. Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074-80. Given that such modifications can alter and regulate the expression of specific genes, this mechanism could be potentially associated with acute mood symptoms in BD and with the mechanisms of action of psychotropic drugs, altering protein levels in parallel with behavioral changes. Sodium valproate (VPT) inhibits the enzyme histone deacetylase (HDAC), which catalyzes the removal of acetyl groups from histones.55. Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem. 2001;276:36734-41. Lithium (Li) seems to share some mechanism with HDAC inhibitors (HDACi), but whether it can inhibit this enzyme directly remains unclear.66. Bordonaro M, Lazarova DL, Sartorelli AC. The activation of beta-catenin by Wnt signaling mediates the effects of histone deacetylase inhibitors. Exp Cell Res. 2007;313:1652-66. So far, little is known about the association between HDAC activity and the behavioral effects of mood stabilizers in vivo.
One gene whose expression has been linked to mood-stabilizing effects is brain-derived neurotrophic factor (BDNF). Among its functions, BDNF acts to promote neuronal survival and synaptic plasticity77. Kuipers SD, Bramham CR. Brain-derived neurotrophic factor mechanisms and function in adult synaptic plasticity: new insights and implications for therapy. Curr Opin Drug Discov Devel. 2006;9:580-6. and has been extensively studied as a biomarker of illness activity in BD.88. Grande I, Fries GR, Kunz M, Kapczinski F. The role of BDNF as a mediator of neuroplasticity in bipolar disorder. Psychiatry Investig. 2010;7:243-50. Serum BDNF levels are reduced in BD patients during manic and depressive episodes, whereas no differences are found in euthymic patients compared to healthy controls.99. Cunha AB, Frey BN, Andreazza AC, Goi JD, Rosa AR, Goncalves CA, et al. Serum brain-derived neurotrophic factor is decreased in bipolar disorder during depressive and manic episodes. Neurosci Lett. 2006;398:215-9. Moreover, BDNF levels seem to increase along with remission of symptoms1010. Tramontina JF, Andreazza AC, Kauer-Sant'anna M, Stertz L, Goi J, Chiarani F, et al. Brain-derived neurotrophic factor serum levels before and after treatment for acute mania. Neurosci Lett. 2009;452:111-3. and after mood stabilizer treatment in vitro and in vivo.1111. Yasuda S, Liang MH, Marinova Z, Yahyavi A, Chuang DM. The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Mol Psychiatry. 2009;14:51-9.,1212. Frey BN, Andreazza AC, Cereser KM, Martins MR, Valvassori SS, Reus GZ, et al. Effects of mood stabilizers on hippocampus BDNF levels in an animal model of mania. Life Sci. 2006;79:281-6. Of note, BDNF transcription is at least partially modulated by epigenetic mechanisms.1313. Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, et al. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890-3. Evidence suggests that BDNF increases after treatment with HDACi in vitro and in vivo, and this increase seems to occur in a promoter-specific way.1414. Fukuchi M, Nii T, Ishimaru N, Minamino A, Hara D, Takasaki I, et al. Valproic acid induces up- or down-regulation of gene expression responsible for the neuronal excitation and inhibition in rat cortical neurons through its epigenetic actions. Neurosci Res. 2009;65:35-43.
Based on previous findings, the ability to inhibit HDAC and modulate behavior could be a way to screen for potential novel drugs with mood-stabilizing properties. Studies investigating other drugs capable of inhibiting HDAC as compared with known mood stabilizers in vitro suggest that the short-chain fatty acid sodium butyrate (SB) may be a promising candidate.1111. Yasuda S, Liang MH, Marinova Z, Yahyavi A, Chuang DM. The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Mol Psychiatry. 2009;14:51-9.,1515. Kwon B, Houpt TA. Phospho-acetylation of histone H3 in the amygdala after acute lithium chloride. Brain Res. 2010;1333:36-47. SB has been shown to exhibit antidepressant properties1616. Yamawaki Y, Fuchikami M, Morinobu S, Segawa M, Matsumoto T, Yamawaki S. Antidepressant-like effect of sodium butyrate (HDAC inhibitor) and its molecular mechanism of action in the rat hippocampus. World J Biol Psychiatry. 2012;13:458-67.,1717. Schroeder FA, Lin CL, Crusio WE, Akbarian S. Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biol Psychiatry. 2007;62:55-64. and can increase BDNF levels in vitro.1111. Yasuda S, Liang MH, Marinova Z, Yahyavi A, Chuang DM. The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Mol Psychiatry. 2009;14:51-9. Moreover, it has recently been shown that SB reverses and prevents D-amphetamine (AMPH)-induced hyperactivity in an animal model of mania,1818. Moretti M, Valvassori SS, Varela RB, Ferreira CL, Rochi N, Benedet J, et al. Behavioral and neurochemical effects of sodium butyrate in an animal model of mania. Behav Pharmacol. 2011;22:766-72. but the extent to which it is associated with HDAC activity requires further research.
Considering that chromatin remodeling can lead to alterations in the expression of neuroprotective genes, the effects of AMPH and mood stabilizers on such parameters would be of great interest to the field of BD research and therapy. AMPH exposure has been consistently put forward as a model for mania due to its ability to increase dopamine levels upon administration.1919. Machado-Vieira R, Kapczinski F, Soares JC. Perspectives for the development of animal models of bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28:209-24. AMPH-treated rats become hyperactive1212. Frey BN, Andreazza AC, Cereser KM, Martins MR, Valvassori SS, Reus GZ, et al. Effects of mood stabilizers on hippocampus BDNF levels in an animal model of mania. Life Sci. 2006;79:281-6. and exhibit several memory deficits,2020. de Lima MN, Presti-Torres J, Vedana G, Alcalde LA, Stertz L, Fries GR, et al. Early life stress decreases hippocampal BDNF content and exacerbates recognition memory deficits induced by repeated D-amphetamine exposure. Behav Brain Res. 2012;224:100-6. some of which are reversed by administration of mood stabilizers.2121. Pinheiro RM, de Lima MN, Fries GR, Garcia VA, Presti-Torres J, Hallmenschlager LH, et al. Early life stress exacerbates cognitive dysfunction induced by d-amphetamine: amelioration by valproic acid. J Neural Transm. 2012;119:627-37. Moreover, the construct validity of this model has been increasing significantly over time, with several biomarkers found in human BD patients also altered in animals subjected to this model.1212. Frey BN, Andreazza AC, Cereser KM, Martins MR, Valvassori SS, Reus GZ, et al. Effects of mood stabilizers on hippocampus BDNF levels in an animal model of mania. Life Sci. 2006;79:281-6. The present study aimed to evaluate if repeated exposure to AMPH would induce alterations in HDAC activity and if these alterations would be reversed by the administration of a classic HDACi (SB or VPT) or Li. Furthermore, we evaluated whether such HDAC alterations would be associated with a modulation of BDNF levels. To that end, we assessed the effects of the treatments on the locomotor behavior of rats and measured the activity of HDAC and BDNF protein and mRNA levels in the prefrontal cortex (PFC), a brain region known to be associated with BD pathophysiology.2222. Brambilla P, Stanley JA, Nicoletti MA, Sassi RB, Mallinger AG, Frank E, et al. 1H magnetic resonance spectroscopy investigation of the dorsolateral prefrontal cortex in bipolar disorder patients. J Affect Disord. 2005;86:61-7.,2323. Yurgelun-Todd DA, Ross AJ. Functional magnetic resonance imaging studies in bipolar disorder. CNS Spectr. 2006;11:287-97. We also evaluated HDAC modulation in peripheral blood mononuclear cells (PBMCs) and plasma BDNF levels to analyze whether the effects of these drugs would be mainly central or could also be systemic.
Methods
Animals
All experiments were conducted with male Wistar rats (age: 3-4 months; weight: 220-310 g) obtained from our breeding colony. Rats were housed five to a cage and kept on a 12-h light/dark cycle (lights on between 7 a.m. and 7 p.m.), with water and food available ad libitum. All study procedures were in accordance with the NIH Guide for the Care and Use of Laboratory Animals and the Brazilian Society of Neuroscience and Behavior (SBNeC) recommendations for animal care, and were approved by the Hospital de Clínicas de Porto Alegre Ethics Committee.
Treatment
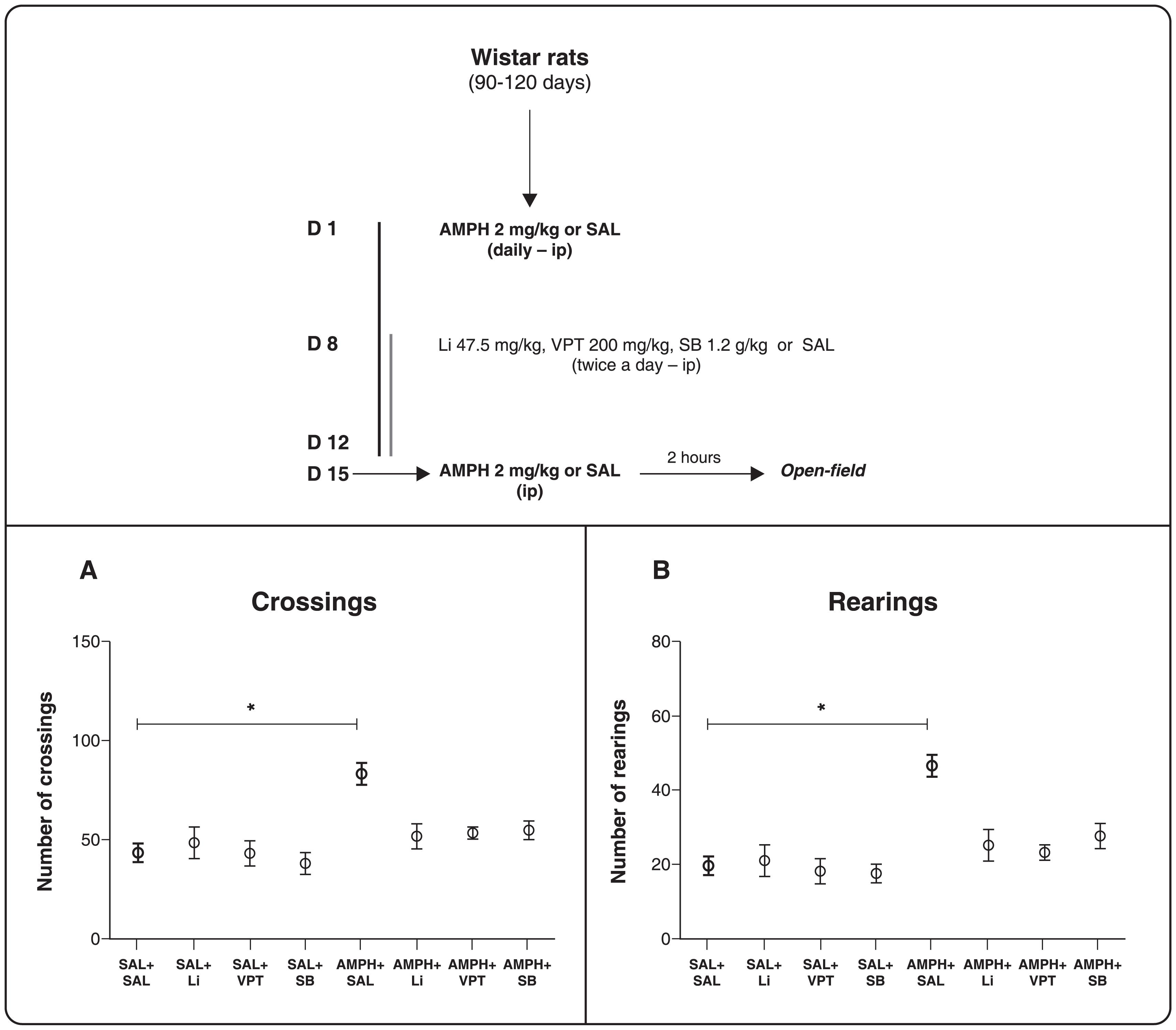
The animals received daily intraperitoneal (i.p.) injections of 2 mg/kg AMPH (Sigma, St. Louis, MO, USA) or saline solution for 14 days. Between the 8th and the 14th day, AMPH or saline-treated rats were divided into four experimental groups: treatment with 47.5 mg/kg Li i.p. twice a day (n=12), 200 mg/kg VPT i.p. twice a day (n=12), 1.2 g/kg SB i.p. twice a day (n=12), or saline (n=21). All drugs were administered in the rats' home cages. On the 15th day, rats received either AMPH or saline, and their locomotor activity was measured 2 h later. The doses of the mood stabilizers tested are within the range of doses regularly used in other behavioral and functional studies with animal models of mood disorders. Of note, animals treated with Li had plasma Li levels in the range of 0.6-1.2 mEq/L, as recommended for the treatment of BD patients.1212. Frey BN, Andreazza AC, Cereser KM, Martins MR, Valvassori SS, Reus GZ, et al. Effects of mood stabilizers on hippocampus BDNF levels in an animal model of mania. Life Sci. 2006;79:281-6.
Locomotor activity
Locomotor activity was assessed with the open-field task. The test was performed in a 40 x 60 cm open field surrounded by 50 cm-high walls made of brown plywood with a frontal glass wall. The open-field floor was divided into 12 equal rectangles by black lines. The animals were gently placed in the left rear quadrant and allowed to explore the arena freely for 5 min. Two observers who were blinded to the treatment status of the rats counted the number of times they crossed the black lines and performed rearing behavior. We considered the number of times that rats crossed the black lines to be a measurement of horizontal locomotor activity and the number of times they reared to be a measurement of vertical locomotor activity. The animals were euthanized by decapitation immediately after the open-field task.
Nuclear extraction and cytosolic fraction separation
The PFC (the anterior part of the frontal lobes of the rat brain) was fully dissected according to the stereotactic atlas of Paxinos and Watson.2424. Paxinos G, Watson C. The rat brain in stereotaxic coordinates. 6th ed. San Diego: Academic Press; 1986. The obtained samples were flash-frozen and stored at -80°C until nuclear proteins were extracted. Whole blood was collected by intracardiac puncture and PBMC separation was performed with Histopaque 1077 reagent (Sigma, St. Louis, MO, USA) according to manufacturer instructions. Tissue samples and PBMCs were subjected to a nuclear extraction protocol with a commercial Nuclear Extraction kit (Chemicon, USA). Briefly, samples were homogenized in cytoplasmic lysis buffer containing dithiothreitol (DTT) and protease inhibitors. The suspension was kept on ice for 15 min and was later centrifuged at 250 × g for 5 min at 4°C. The supernatant was discarded, and the pellet was resuspended in two volumes of cold cytoplasmic lysis buffer. The suspension was homogenized using a small-gauge needle syringe and centrifuged at 8,000 × g for 20 min at 4°C. The resulting pellet contained the nuclear portion of the cell lysate. The supernatant containing the cytosolic fraction was transferred to another tube and stored at -80°C until the BDNF protein level assay was performed.
The pellet was resuspended in a nuclear extraction buffer containing DTT and protease inhibitors, and the suspension was homogenized with a small-gauge needle syringe. The resulting sample was kept in slow agitation for 30-60 min in an orbital shaker at 4°C. Later, the nuclear suspension was centrifuged at 16,000 × g for 5 min at 4°C, and the nuclear extract-containing supernatant was transferred to a new tube and stored at -80°C until further analysis.
HDAC activity
Nuclear extracts from the PFC and PBMCs were subjected to an HDAC activity assay with the HDAC Assay kit (Fluorometric Detection) according to manufacturer instructions (Upstate, USA). Briefly, 5 µL of nuclear extract were mixed with 5 µL of HDAC Assay Buffer and 5 µL of HDAC Assay Substrate in a 384-well plate and incubated at 30°C for 45 min. Concomitantly, a standard curve was performed with serial dilutions of deacetylated substrate and positive and negative controls were added to the plate. Afterwards, 10 µL of activator solution were added to the wells, and the plate was incubated at room temperature for 15 min. A fluorescence reading was obtained in a fluorescence plate reader, with 360 nm for excitation and 460 nm for emission. HDAC activity was calculated on the basis of the standard curve, and values are presented as µM/µg protein. Total protein was measured by a modified Lowry's method2525. Peterson GL. A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal Biochem. 1977;83:346-56. using bovine serum albumin as the standard. HDAC activity was calculated as the micromolar concentration of deacetylated standard substrate per microgram of protein.
BDNF protein levels
Whole blood was collected by intracardiac puncture into a Vacutainer containing ethylenediamine tetraacetic acid (EDTA) to prevent clotting. Plasma samples were obtained by centrifugation (4000 rpm for 10 min) and frozen at -80°C until further analysis. BDNF levels in the cytosolic fraction from PFC and plasma samples were measured using an anti-BDNF sandwich enzyme-linked immunosorbent assay (ELISA) (CYT306), according to manufacturer instructions (Millipore, USA). Briefly, microtiter plates (96-well flat-bottom) were coated for 24 h with the samples, diluted 1:2 in sample diluent, and the standard curve ranged from 7.8 to 500 pg/ml of BDNF. The plates were then washed four times with wash buffer, and a biotinylated monoclonal anti-BDNF rabbit antibody (ChemiKine, USA), diluted 1:1000 in sample diluent, was added to each well and incubated for 3 h at room temperature. After washing, peroxidase-conjugated streptavidin (ChemiKine, USA) (diluted 1: 1000) was added to each well and incubated at room temperature for 1 h. After the addition of substrate and stop solution, the amount of BDNF was determined by absorbance at 450 nm. The standard curve demonstrated a direct relationship between optical density (OD) and BDNF concentration. Total protein was measured by Lowry's method using bovine serum albumin (Sigma, St. Louis, USA) as standard.
RNA isolation
Tissues were subjected to an RNA isolation protocol with the use of TRIzol (Invitrogen, Carlsbad, CA, USA), according to manufacturer instructions. Briefly, tissues (50 to 100 mg) were homogenized in 1 mL TRIzol and incubated at room temperature for 5 min. Afterwards, chloroform was added to the samples and the suspension was vigorously mixed for 15 s and incubated at room temperature for 5 min, followed by centrifugation at 12,000 × g for 15 min at 4°C. The aqueous phase was then placed in another tube to which isopropanol was added, followed by incubation at room temperature for 10 min and centrifugation at 12,000 × g for 10 min at 4°C. The supernatant was then discarded and the pellet was washed with 75% ethanol, followed by another centrifugation at 7,500 × g for 5 min at 4°C. After drying the pellet at room temperature, RNA was resuspended in 30 µL of DEPC-treated water. RNA samples were then quantitated with the Quant-iT RNA assay (Molecular Probe; Invitrogen, Carlsbad, CA, USA), treated with DNAse I, Amp Grade (Invitrogen, Carlsbad, CA, USA), and stored at -80°C until further analysis.
Primer design and quantitative real-time PCR
FAM-labeled TaqMan primers and a probe (Applied Biosystems, Foster City, CA, USA) specific for the rat BDNF gene were designed with the use of Primer Express software (Applied Biosystems, Foster City, CA, USA) based on the BDNF mRNA sequence obtained from NCBI GenBank (GenBank ID: 122427415, NM_012513). Primers and probe specificities were confirmed through comparison with other sequences available at the GenBank with the use of BLAST. The sequences used were as follows: forward primer, 5′-CTGACACTTTTGAGCACGTGATC-3′; reverse primer, 5′-CGTTGGGCCGAACCTTCT-3′.
Real-time RT-PCR reactions were performed in the 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) with the use of the TaqMan One-Step RT-PCR kit (Applied Biosystems, Foster City, CA, USA) according to manufacturer instructions. Reactions were performed in a final volume of 12 µL with 2 µL of total RNA (5 ng), 6 µL of Master Mix 2x, 0.3 µL of RT enzyme 40x, 0.6 µL of TaqMan Custom Gene expression assay, 0.6 µL of endogenous TaqMan gene expression assay, and 2.5 µL of DEPC-treated water. Expression values were normalized to beta-actin endogenous control expression using a TaqMan VIC/MGB endogenous control inventoried assay (Applied Biosystems, Foster City, CA, USA, 4352340E).
All reactions were performed in triplicate. Relative expression levels (RQ) were determined by the ΔΔCt method.2626. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402-8.
Statistical analysis
Statistical analyses were performed using PASW Statistics 18.0 for Windows. Behavioral data (number of crossings and rearings), HDAC activity, and BDNF protein levels in the PFC fit a standard distribution curve and were therefore subjected to parametric analyses. For comparisons between groups, a one-way analysis of variance (ANOVA) test was performed, followed by the Tukey post-hoc test when the ANOVA was significant. P-values < 0.05 for a two-tailed distribution were considered statistically significant. BDNF mRNA and protein plasma levels did not fit a standard distribution curve and were thus subjected to nonparametric analyses. A Kruskal-Wallis test was performed to compare groups and p-values < 0.05 for two-tailed distributions were considered statistically significant. All biochemical data are expressed as % of control.
Results
Open-field test
In the open-field test (Figure 1), one-way ANOVA followed by Tukey's post-hoc test was performed to compare all groups. AMPH increased horizontal and vertical locomotor activities in saline-treated rats (ANOVA, crossings-F7-82 = 6.673, p < 0.0001, Figure 1A; rearings-F7-82 = 7.980, p < 0.0001, Figure 1B), whereas treatment with Li, VPT, and SB partially reversed this AMPH-induced hyperactivity. The administration of Li, VPT, and SB per se did not significantly alter the behavioral activity, indicating that their effects on AMPH-treated rats were not due to sedation.
Schematic representation of treatment and open-field test. A) Number of crossings. B) Number of rearings. Rats were treated for 7 days with AMPH + 7 days of AMPH and Li, VPT, or SB. One-way ANOVA and Tukey's post-hoc test; * p < 0.0001 (different from the SAL + SAL group). The results are presented as mean ± standard error of the mean (n=11-21). AMPH = D-amphetamine; Li = lithium; SAL = saline; SB = sodium butyrate; VPT = sodium valproate.
HDAC activity
Figure 2 shows the amount of HDAC activity in the PFC and in PBMCs. In the PFC, AMPH treatment (AMPH + saline) significantly increased HDAC activity as compared with the control group (saline + saline) (ANOVA, F7-79 = 2.173, p = 0.01, Figure 2A), and treatment with Li, VPT, and SB in AMPH-treated rats partially reversed this effect. In PBMCs, no differences in HDAC activity were found between groups (p > 0.05 for all comparisons, Figure 2B).
HDAC activity in the prefrontal cortex and PBMCs. A) Prefrontal cortex (n=8-18 per group). B) PBMCs (n=7-19 per group). One-way ANOVA and Tukey's post-hoc test; * p = 0.001. The results are presented as mean ± standard error of the mean. AMPH = D-amphetamine; HDAC = histone deacetylase; Li = lithium; PBMC = peripheral blood mononuclear cell; PFC = prefrontal cortex; SAL = saline; SB = sodium butyrate; VPT = sodium valproate.
BDNF mRNA and protein levels in the PFC
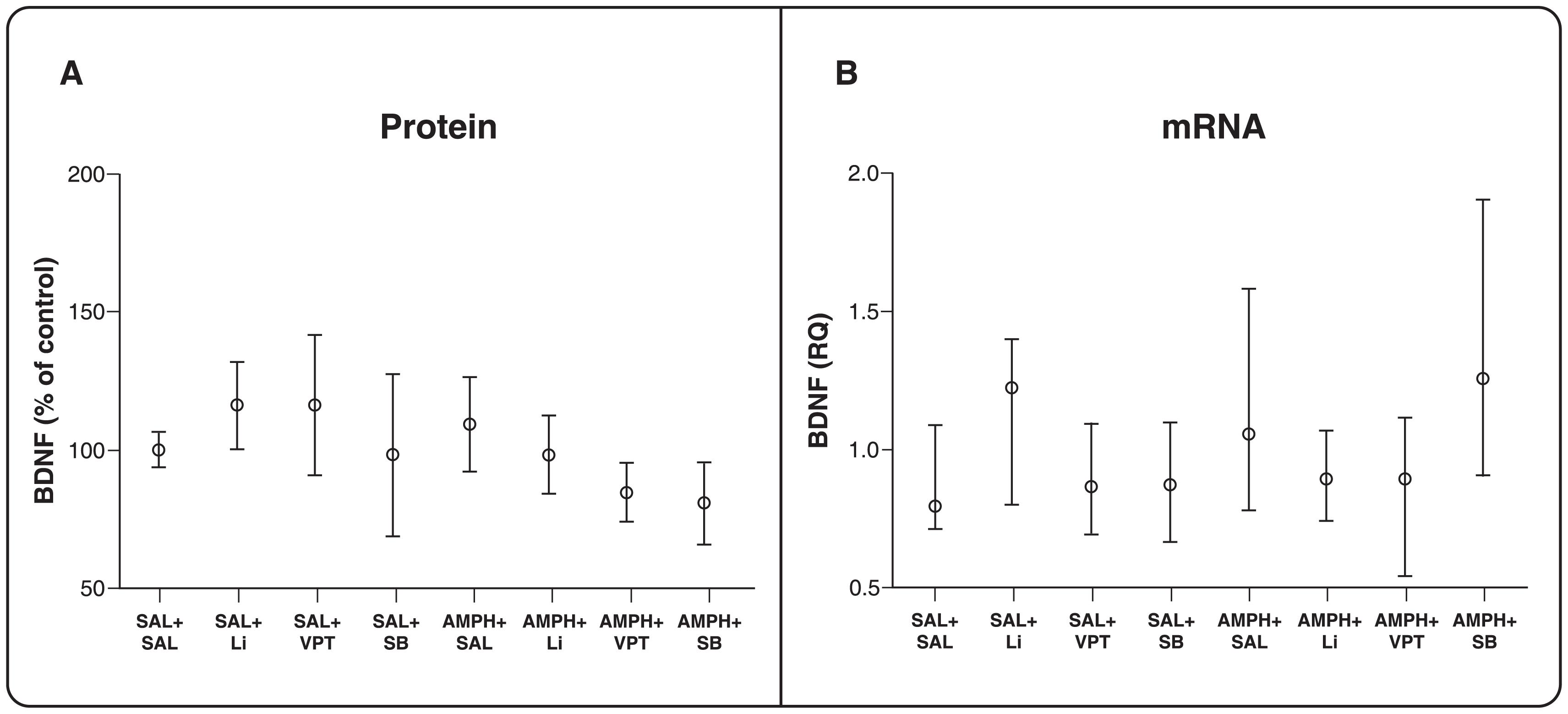
Given that evidence suggests that BDNF expression may be linked to mood-stabilizing effects, it was hypothesized that BDNF levels would increase after treatment with HDACi. However, we did not observe any between-group differences in BDNF protein levels (p > 0.05 for all comparisons, Figure 3A) or mRNA levels (p > 0.05 for all comparisons, Figure 3B) the PFC. This indicates that HDAC modulation in the PFC might alter the expression of genes other than BDNF.
BDNF levels in prefrontal cortex. A) Protein levels. One-way ANOVA and Tukey's post-hoc test. The results are presented as mean ± standard error of the mean (n=7-12 per group). B) mRNA levels. A Kruskal-Wallis test was used to compare groups, p > 0.05. The results are presented as median and interquartile range (n=7-12 per group). AMPH = D-amphetamine; BDNF = brain-derived neurotrophic factor; Li = lithium; SAL = saline; SB = sodium butyrate; VPT = sodium valproate.
Plasma BDNF levels
To analyze whether the effects of the tested drugs would be mainly central or could also be systemic, we measured plasma BDNF levels (Figure 4). We did not find any between-group differences in these levels (p > 0.05 for all comparisons), suggesting that there are no systemic effects of AMPH-modulating HDAC in the BDNF gene.
BDNF levels in plasma. A Kruskal-Wallis test was used to compare groups, p > 0.05. The results are presented as median and interquartile range (n=8-19 per group). AMPH = D-amphetamine; BDNF = brain-derived neurotrophic factor; Li = lithium; SAL = saline; SB = sodium butyrate; VPT = sodium valproate.
Discussion
Evidence suggests that HDACi may exert antimanic effects in different treatment regimens and animal models. Accordingly, our results show that treatment with SB, as well as with Li and VPT, was able to partially reverse AMPH-induced hyperactivity after repeated AMPH administration. Moreover, AMPH increased HDAC activity in the PFC, which was partially reversed by SB, VPT, and Li. No between-group differences were found in BDNF protein and mRNA levels. In addition, no differences were found in HDAC activity in PBMCs or in plasma BDNF levels, suggesting that the HDAC-modulating effects of AMPH are mainly central. The present study provides evidence that AMPH-induced behavioral effects may be associated with histone modifications in the PFC and that this can be partially reversed by HDAC inhibitors.
To the best of our knowledge, this is the first study showing that AMPH treatment increases HDAC activity in the PFC of rats. Although there is no evidence of a direct association between AMPH and HDAC, the ability of AMPH to increase dopamine levels in the synaptic cleft may have consequences at a nucleosomal level. Histone deacetylation has been associated with activation of D2 dopamine receptors, leading to the recruitment of a corepressor complex to the promoter of the prolactin gene.2727. Liu JC, Baker RE, Chow W, Sun CK, Elsholtz HP. Epigenetic mechanisms in the dopamine D2 receptor-dependent inhibition of the prolactin gene. Mol Endocrinol. 2005;19:1904-17. Moreover, trichostatin A, which is an HDACi, has been shown to block cocaine-induced behavioral sensitization,2828. Romieu P, Host L, Gobaille S, Sandner G, Aunis D, Zwiller J. Histone deacetylase inhibitors decrease cocaine but not sucrose self-administration in rats. J Neurosci. 2008;28:9342-8. showing that other HDAC inhibitors may also have effects on dopamine-enhancing drugs. More recently, Arent et al. showed different effects of SB treatment in specific brain regions, suggesting that the antimanic effects of SB and of VPT are related to the amygdala, striatum, and, of special interest here, to the PFC, although not to the hippocampus.2929. Arent CO, Valvassori SS, Fries GR, Stertz L, Ferreira CL, Lopes-Borges J, et al. Neuroanatomical profile of antimaniac effects of histone deacetylases inhibitors. Mol Neurobiol. 2011;43:207-14. More studies are warranted to clarify the effects of these HDAC inhibitors in different brain regions. Of note, the lack of between-group differences in HDAC activity in PBMCs in our sample suggests that the modulation of HDAC activity by the tested drugs is mainly central and cannot be observed in the periphery.
Based on our results, we cannot rule out the possibility that AMPH increases the expression of different HDACs, which would ultimately lead to an enhanced activity of this enzyme in our samples. Nevertheless, in this particular experiment, regardless of whether AMPH increased HDAC expression or only its activity, both scenarios would culminate in increased histone deacetylation and, thus, decreased gene expression. Furthermore, we did not assess whether this modulation of HDAC activity is associated with a change in acetylated H3 and H4 levels, which would contribute to the discussion of our results. In this sense, future studies are required to further explore these mechanisms.
Our results show that the reversal of AMPH-induced hyperactivity by Li, VPT, and SB was associated with the ability of these drugs to partially reverse the increase in HDAC activity. However, we found no effects of their administration on HDAC activity per se, suggesting more complex mechanisms regarding HDAC inhibition. Of note, there are two protein families with HDAC activity: the recently discovered SIR2 family of NAD+- dependent HDACs, and the classical HDAC family. Members of the classical HDAC family fall into two different phylogenetic classes, namely class I and class II. Both of these protein families with HDAC activity share an ability to deacetylate histones, but they have different patterns of expression, different pharmacological profiles, different mechanisms of regulation, and different functions.3030. de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737-49. It is possible that different HDAC enzymes are differentially modulated by Li, VPT, and SB in the PFC. For instance, SB has relatively poor HDAC selectivity (disrupting the activity of multiple classes), which also limits our discussion on specific HDAC inhibition taking place in the brain. To address this, an analysis of second-generation compounds (such as hydroxamic acids and benzamides), which would potentially demonstrate the feasibility and importance of dissecting out the roles of particular HDACs in the brain, would be of great interest. In addition, it is also possible that the time elapsed between the last HDACi injection and euthanasia was too long, which would have prevented detection of any enzyme inhibition possibly induced by the drugs. Schroeder et al. showed that histone hyperacetylation in the hippocampus peaks 30 min after i.p. injection of SB in mice, returning to baseline within 1 h after administration.1717. Schroeder FA, Lin CL, Crusio WE, Akbarian S. Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biol Psychiatry. 2007;62:55-64. Further studies are necessary to clarify these issues.
A growing body of evidence suggests a role for histone acetylation in the neurobiology and pharmacology of mood disorders.33. Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci. 2007;8:355-67.,3131. Grayson DR, Kundakovic M, Sharma RP. Is there a future for histone deacetylase inhibitors in the pharmacotherapy of psychiatric disorders? Mol Pharmacol. 2010;77:126-35. Hobaro et al. found that the levels of HDAC2 and HDAC5 mRNA were increased in patients with major depressive disorder (MDD) while in the depressive state, and that HDAC4 mRNA levels were increased in depressive BD patients when compared with controls.3232. Hobara T, Uchida S, Otsuki K, Matsubara T, Funato H, Matsuo K, et al. Altered gene expression of histone deacetylases in mood disorder patients. J Psychiatr Res. 2010;44:263-70. These data suggest that aberrant transcriptional regulation caused by altered HDAC expression may be associated with the pathophysiology of mood disorders. HDAC class I and II inhibitors have shown efficacy in the treatment of neurodegenerative disorders,3333. Kilgore M, Miller CA, Fass DM, Hennig KM, Haggarty SJ, Sweatt JD, et al. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer's disease. Neuropsychopharmacology. 2010;35:870-80. neurodevelopmental disorders, cognitive deficits,3434. Alarcon JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, et al. Chromatin acetylation, memory, and LTP are impaired in CBP+/- mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron. 2004;42:947-59. and psychiatric disorders such as depression and anxiety.1717. Schroeder FA, Lin CL, Crusio WE, Akbarian S. Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biol Psychiatry. 2007;62:55-64.,3535. Covington HE 3rd, Vialou VF, Laplant Q, Ohnishi YN, Nestler EJ. Hippocampal-dependent antidepressant-like activity of histone deacetylase inhibition. Neurosci Lett. 2011;493:122-6. In this same vein, Ferrante et al. showed an attenuation of neuron loss and increased motor function after treatment with SB in an animal model of Huntington's disease.3636. Ferrante RJ, Kubilus JK, Lee J, Ryu H, Beesen A, Zucker B, et al. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington's disease mice. J Neurosci. 2003;23:9418-27. Histone modifications are also associated with memory and learning processes, as shown by the increased formation of contextual fear memory in rats treated with SB.3737. Levenson JM, O'Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD. Regulation of histone acetylation during memory formation in the hippocampus. J Biol Chem. 2004;279:40545-59. In addition, treatment of mice with SB resulted in antidepressant-like effects when administered at a sufficiently high dose that induced global and transitory hyperacetylation of histones in the PFC and in the hippocampus.1717. Schroeder FA, Lin CL, Crusio WE, Akbarian S. Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biol Psychiatry. 2007;62:55-64.
Amphetamine-induced hyperactivity in rats has been consistently put forward as a dopaminergic model of mania with predictive validity.3838. Dencker D, Husum H. Antimanic efficacy of retigabine in a proposed mouse model of bipolar disorder. Behav Brain Res. 2010;207:78-83. Therefore, the finding that AMPH-induced hyperlocomotion is partially blocked by SB treatment, as well as by Li and VPT treatment, suggests that SB may have mood-stabilizing properties.1818. Moretti M, Valvassori SS, Varela RB, Ferreira CL, Rochi N, Benedet J, et al. Behavioral and neurochemical effects of sodium butyrate in an animal model of mania. Behav Pharmacol. 2011;22:766-72.,2929. Arent CO, Valvassori SS, Fries GR, Stertz L, Ferreira CL, Lopes-Borges J, et al. Neuroanatomical profile of antimaniac effects of histone deacetylases inhibitors. Mol Neurobiol. 2011;43:207-14. Based on this pre-established relevance of the AMPH treatment as a valid animal model of mania, we are able to suggest that the results found in our experiments can be of relevance for human patients with BD. Nevertheless, clear extrapolations to the human condition regarding the doses of the tested drugs are less likely to be made, mostly due to differences in the metabolism of rats and humans and, therefore, in the kinetics of the drugs. Moreover, this finding suggests that other HDAC inhibitors should be further investigated as agents with possible therapeutic effects for manic episodes in patients with BD.
Finally, the use of HDAC inhibitors as mood stabilizers has been proposed on the basis of their ability to increase the expression of neuroprotective genes, some of which have already been investigated in patients with BD and other neuropsychiatric disorders.3939. Kim HJ, Rowe M, Ren M, Hong JS, Chen PS, Chuang DM. Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: multiple mechanisms of action. J Pharmacol Exp Ther. 2007;321:892-901. The modulation of neurotrophin levels by HDAC inhibitors has already been shown in vitro,1111. Yasuda S, Liang MH, Marinova Z, Yahyavi A, Chuang DM. The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Mol Psychiatry. 2009;14:51-9. and the differential pattern of HDAC inhibition in different brain regions is in line with previous findings of region-specific expression of neurotrophins.4040. Aid T, Kazantseva A, Piirsoo M, Palm K, Timmusk T. Mouse and rat BDNF gene structure and expression revisited. J Neurosci Res. 2007;85:525-35. We found no differences in BDNF protein or mRNA levels in the PFC of rats, even though AMPH increased HDAC activity in this brain region. Schroeder et al. did not find any change in BDNF levels after SB treatment in vivo either, and no other study has ever been conducted with this aim.1717. Schroeder FA, Lin CL, Crusio WE, Akbarian S. Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biol Psychiatry. 2007;62:55-64. Although a significant body of evidence points to changes in serum BDNF levels in BD patients, there are still discrepancies between studies. Some did not find modulation of this factor in BD phases.4141. Kapczinski F, Dal-Pizzol F, Teixeira AL, Magalhaes PV, Kauer-Sant'Anna M, Klamt F, et al. Peripheral biomarkers and illness activity in bipolar disorder. J Psychiatr Res. 2011;45:156-61. Several factors that may influence BDNF levels could explain these differences, such as medication, comorbidities, and staging of illness. Likewise, in studies with animals, some variables could have a direct impact on the results. In the present study, the absence of alterations in BDNF is likely to be related to the duration of HDACi treatment, which was brief. Fukumoto et al. demonstrated that only chronic Li treatment was able to increase BDNF protein levels in the rat cortex and hippocampus.4242. Fukumoto T, Morinobu S, Okamoto Y, Kagaya A, Yamawaki S. Chronic lithium treatment increases the expression of brain-derived neurotrophic factor in the rat brain. Psychopharmacology (Berl). 2001;158:100-6. Furthermore, as we did not perform a dose-response experiment, one cannot rule out the possibility that higher doses would have stronger effects on BDNF. As far as we can assume, HDAC modulation in the PFC might alter the expression of genes other than BDNF. Further analyses are required to explore the SB-induced effects found in our model as well as in other behavioral tests, including evaluation of the dopaminergic system and of other structures, such as the hippocampus and the amygdala.
In conclusion, we demonstrated that repeated AMPH administration increased HDAC activity in the PFC without altering BDNF protein or mRNA levels. Moreover, Li, VPT, and SB administration partially reversed this increase in HDAC activity, which might account for the ability of these agents to reverse AMPH-induced hyperactivity. Further studies are required to explore the cellular consequences of HDAC inhibition in different brain areas, including assessment of the expression of other genes likely to be associated with these treatments. Nonetheless, the available results support the hypothesis that histone modifications are associated with the mechanisms of action of AMPH and mood stabilizers. In addition, the present data highlight the notion that HDAC inhibitors may hold important potential for the development of new drugs for the treatment of BD.
This work was supported by grants from National Science and Technology Institute for Translational Medicine (INCT-TM), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), and Fundo de Incentivo è Pesquisa - Hospital de Clínicas de Porto Alegre (FIPE-HCPA).
References
-
1Wu X, Chen PS, Dallas S, Wilson B, Block ML, Wang CC, et al. Histone deacetylase inhibitors up-regulate astrocyte GDNF and BDNF gene transcription and protect dopaminergic neurons. Int J Neuropsychopharmacol. 2008;11:1123-34.
-
2Caspi A, Moffitt TE. Gene-environment interactions in psychiatry: joining forces with neuroscience. Nat Rev Neurosci. 2006;7:583-90.
-
3Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci. 2007;8:355-67.
-
4Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074-80.
-
5Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem. 2001;276:36734-41.
-
6Bordonaro M, Lazarova DL, Sartorelli AC. The activation of beta-catenin by Wnt signaling mediates the effects of histone deacetylase inhibitors. Exp Cell Res. 2007;313:1652-66.
-
7Kuipers SD, Bramham CR. Brain-derived neurotrophic factor mechanisms and function in adult synaptic plasticity: new insights and implications for therapy. Curr Opin Drug Discov Devel. 2006;9:580-6.
-
8Grande I, Fries GR, Kunz M, Kapczinski F. The role of BDNF as a mediator of neuroplasticity in bipolar disorder. Psychiatry Investig. 2010;7:243-50.
-
9Cunha AB, Frey BN, Andreazza AC, Goi JD, Rosa AR, Goncalves CA, et al. Serum brain-derived neurotrophic factor is decreased in bipolar disorder during depressive and manic episodes. Neurosci Lett. 2006;398:215-9.
-
10Tramontina JF, Andreazza AC, Kauer-Sant'anna M, Stertz L, Goi J, Chiarani F, et al. Brain-derived neurotrophic factor serum levels before and after treatment for acute mania. Neurosci Lett. 2009;452:111-3.
-
11Yasuda S, Liang MH, Marinova Z, Yahyavi A, Chuang DM. The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Mol Psychiatry. 2009;14:51-9.
-
12Frey BN, Andreazza AC, Cereser KM, Martins MR, Valvassori SS, Reus GZ, et al. Effects of mood stabilizers on hippocampus BDNF levels in an animal model of mania. Life Sci. 2006;79:281-6.
-
13Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, et al. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890-3.
-
14Fukuchi M, Nii T, Ishimaru N, Minamino A, Hara D, Takasaki I, et al. Valproic acid induces up- or down-regulation of gene expression responsible for the neuronal excitation and inhibition in rat cortical neurons through its epigenetic actions. Neurosci Res. 2009;65:35-43.
-
15Kwon B, Houpt TA. Phospho-acetylation of histone H3 in the amygdala after acute lithium chloride. Brain Res. 2010;1333:36-47.
-
16Yamawaki Y, Fuchikami M, Morinobu S, Segawa M, Matsumoto T, Yamawaki S. Antidepressant-like effect of sodium butyrate (HDAC inhibitor) and its molecular mechanism of action in the rat hippocampus. World J Biol Psychiatry. 2012;13:458-67.
-
17Schroeder FA, Lin CL, Crusio WE, Akbarian S. Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biol Psychiatry. 2007;62:55-64.
-
18Moretti M, Valvassori SS, Varela RB, Ferreira CL, Rochi N, Benedet J, et al. Behavioral and neurochemical effects of sodium butyrate in an animal model of mania. Behav Pharmacol. 2011;22:766-72.
-
19Machado-Vieira R, Kapczinski F, Soares JC. Perspectives for the development of animal models of bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28:209-24.
-
20de Lima MN, Presti-Torres J, Vedana G, Alcalde LA, Stertz L, Fries GR, et al. Early life stress decreases hippocampal BDNF content and exacerbates recognition memory deficits induced by repeated D-amphetamine exposure. Behav Brain Res. 2012;224:100-6.
-
21Pinheiro RM, de Lima MN, Fries GR, Garcia VA, Presti-Torres J, Hallmenschlager LH, et al. Early life stress exacerbates cognitive dysfunction induced by d-amphetamine: amelioration by valproic acid. J Neural Transm. 2012;119:627-37.
-
22Brambilla P, Stanley JA, Nicoletti MA, Sassi RB, Mallinger AG, Frank E, et al. 1H magnetic resonance spectroscopy investigation of the dorsolateral prefrontal cortex in bipolar disorder patients. J Affect Disord. 2005;86:61-7.
-
23Yurgelun-Todd DA, Ross AJ. Functional magnetic resonance imaging studies in bipolar disorder. CNS Spectr. 2006;11:287-97.
-
24Paxinos G, Watson C. The rat brain in stereotaxic coordinates. 6th ed. San Diego: Academic Press; 1986.
-
25Peterson GL. A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal Biochem. 1977;83:346-56.
-
26Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402-8.
-
27Liu JC, Baker RE, Chow W, Sun CK, Elsholtz HP. Epigenetic mechanisms in the dopamine D2 receptor-dependent inhibition of the prolactin gene. Mol Endocrinol. 2005;19:1904-17.
-
28Romieu P, Host L, Gobaille S, Sandner G, Aunis D, Zwiller J. Histone deacetylase inhibitors decrease cocaine but not sucrose self-administration in rats. J Neurosci. 2008;28:9342-8.
-
29Arent CO, Valvassori SS, Fries GR, Stertz L, Ferreira CL, Lopes-Borges J, et al. Neuroanatomical profile of antimaniac effects of histone deacetylases inhibitors. Mol Neurobiol. 2011;43:207-14.
-
30de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737-49.
-
31Grayson DR, Kundakovic M, Sharma RP. Is there a future for histone deacetylase inhibitors in the pharmacotherapy of psychiatric disorders? Mol Pharmacol. 2010;77:126-35.
-
32Hobara T, Uchida S, Otsuki K, Matsubara T, Funato H, Matsuo K, et al. Altered gene expression of histone deacetylases in mood disorder patients. J Psychiatr Res. 2010;44:263-70.
-
33Kilgore M, Miller CA, Fass DM, Hennig KM, Haggarty SJ, Sweatt JD, et al. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer's disease. Neuropsychopharmacology. 2010;35:870-80.
-
34Alarcon JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, et al. Chromatin acetylation, memory, and LTP are impaired in CBP+/- mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron. 2004;42:947-59.
-
35Covington HE 3rd, Vialou VF, Laplant Q, Ohnishi YN, Nestler EJ. Hippocampal-dependent antidepressant-like activity of histone deacetylase inhibition. Neurosci Lett. 2011;493:122-6.
-
36Ferrante RJ, Kubilus JK, Lee J, Ryu H, Beesen A, Zucker B, et al. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington's disease mice. J Neurosci. 2003;23:9418-27.
-
37Levenson JM, O'Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD. Regulation of histone acetylation during memory formation in the hippocampus. J Biol Chem. 2004;279:40545-59.
-
38Dencker D, Husum H. Antimanic efficacy of retigabine in a proposed mouse model of bipolar disorder. Behav Brain Res. 2010;207:78-83.
-
39Kim HJ, Rowe M, Ren M, Hong JS, Chen PS, Chuang DM. Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: multiple mechanisms of action. J Pharmacol Exp Ther. 2007;321:892-901.
-
40Aid T, Kazantseva A, Piirsoo M, Palm K, Timmusk T. Mouse and rat BDNF gene structure and expression revisited. J Neurosci Res. 2007;85:525-35.
-
41Kapczinski F, Dal-Pizzol F, Teixeira AL, Magalhaes PV, Kauer-Sant'Anna M, Klamt F, et al. Peripheral biomarkers and illness activity in bipolar disorder. J Psychiatr Res. 2011;45:156-61.
-
42Fukumoto T, Morinobu S, Okamoto Y, Kagaya A, Yamawaki S. Chronic lithium treatment increases the expression of brain-derived neurotrophic factor in the rat brain. Psychopharmacology (Berl). 2001;158:100-6.
Publication Dates
-
Publication in this collection
10 Dec 2013 -
Date of issue
Jan-Mar 2014
History
-
Received
21 Jan 2013 -
Accepted
7 Mar 2013