LETTER TO EDITOR CARTA AO EDITOR
Clinical and laboratory analyses for the identification of Alpha Thalassemia
Análise clínica e laboratorial para a identificação da Talassemia Alfa
Iramaia A. NériI; Gustavo J. LourençoII; Mônica A. FalconiIII; Helia A. S. SouzaIV; Márcia WenningV; Carmen S. P. LimaVI
IBióloga do Laboratório de Rotinas Hematológicas do Hemocentro da Universidade Estadual de Campinas
IIAluno de mestrado do Deptº de Clínica Médica da Faculdade de Ciências Médicas da Universidade Estadual de Campinas
IIIBióloga do Laboratório de Rotinas Hematológicas do Hemocentro da Universidade Estadual de Campinas
IVTécnica do Laboratório de Rotinas Hematológicas do Hemocentro da Universidade Estadual de Campinas
VAluna de mestrado do Deptº de Patologia Clínica da Faculdade de Ciências Médicas da Universidade Estadual de Campinas
VIProfessora Doutora da Disciplina de Oncologia Clínica, do Deptº de Clínica Médica, da Faculdade de Ciências Médicas da Universidade Estadual de Campinas
Correspondence Correspondence: Carmen Silvia Passos Lima Department of Internal Medicine, Faculty of Medical Sciences State University of Campinas Rua Alexander Fleming, nº 181, Cidade Universitária "Zeferino Vaz" Distrito de Barão Geraldo 13083-970 Campinas - SP, Brasil Tel.: (019) 3788-7496; Fax: (019) 3788-7496 E-mail: carmenl@fcm.unicamp.br
RESUMO
A talassemia alfa é uma doença hereditária devida aos genes alfa e sua gravidade depende do número de genes compromtidos. Neste relato apresentamos um caso em que originalmente não havia sido realizado e descrevemos os resultados e os métodos adequados para sua elucidação.
Palavras-chave: Talassemia; hemoglobnopatia; doença hereditária.
To Editor:
Haemoglobin (Hb) is the oxygen-carrying moiety of erythrocytes. It is a polypeptide tetramer consisting of two pairs of unlike globin chains (a plus b, d, or g), which form a shell around a central cavity containing four oxygen-binding haeme groups; each covalently linked to a globin chain. In healthy adults, ~ 95% of the Hb is A (a2b2) with small amounts of HbA2 (a2d) and HbF (a2g2) present. The a-globin gene is encoded in duplicate on chromosome 16, and the non-a chains (b, d, g) are encoded in a cluster on chromosome 11.1
Alpha thalassaemia (a-thal) represents a heterogeneous group of clinical conditions characterised by reduction or suppression in synthesis of the a-globin chain, resulting in the formation of erythrocytes with an inadequate content of Hb and, therefore, microcytic and hypochromic. The imbalance between the synthesis of a- and b-globin chains in adults results in the accumulation of b-globin chains, which are produced in a normal rhythm. The excess b-globin chains are unstable and precipitate intracellularly, causing alterations in the membrane and cellular metabolism, that result, in the destruction of the erythroblasts in the bone marrow (ineffective erythropoiesis) or spleen (haemolysis) and anaemia.2-5
a-thal is caused particularly by an inherited deletion of the a-globin genes and its clinical severity is determined by the number of inherited genes. Patients with two or three alpha genes do not present symptoms and are only identified with certainty, by molecular analysis. Three alpha gene deletions lead to a moderately to severe microcytic, hypochromic anaemia with splenomegaly, which is frequently erroneously treated with iron compounds and red blood cell transfusion. This form of a-thal is known as Hb H disease, since Hb (b4) can be detected in erythrocyte preparations stained with brilliant cresyl blue or electrophoresis of Hb.2-5 The loss of all a-genes leads to death in the uterus (hydrops fetalis).2-4
The diagnostic definition of this illness is, in general, confirmed by molecular studies, where the most usual are the Mediterranean deletions (loss of two alleles of the same chromosome) in Caucasians and 3.7 deletion (loss of one out of two alleles of the same chromosome) in African descendents.6-8
Herein, we present for educational purposes, the case of a patient attended in our clinic and diagnosed with Hb H disease, who had received erroneous treatment for anaemia in other services.
The patient was a 50-year-old medium-brunette female referred to our unit due to weakness, mucosal cutaneous pallor and jaundice over 30 years. She had received treatment with red blood cell transfusion and oral iron compounds sporadically during her life. Mucosal cutaneous pallor, hepatomegaly and splenomegaly were identified during the physical examination.
At this time, the peripheral blood examination showed: RBC: 4.8 x 106/ µL, Hb: 8.5g/dL, MCV: 67.8 fL, MCH: 18.8 pg, MCHC: 27.7g/dL, RDW: 22.5%, reticulocytes: 130.1 x 106/µL, WBC: 4.6 x 103/µL, and Platelets: 132.0 x 103/µl. Hypochromic, microcytic cells with target cells and poikilocytosis were seen on the peripheral blood film (Figure 1). Serum iron: 123.0 µg/dL (normal: 49.0 to 151.0 µg/dL), TIBC: 244.0 (normal: 30.0 to 160.0 mcg/dL) and ferritin: 860.0 ng/mL (normal: 10.0 to 200.0 ng/mL). BT: 0.9 mg/dL (normal: < 1.0 mg/dL), BD: 0.2 mg/dL (normal: < 0.2 mg/dL), BI: 0.70 mg/dL (normal: < 0.8 mg/dL), and haptoglobin: < 5.8 mg/dL (normal: 30.0 to 230.0 mg/dL). Peripheral blood smears stained with brilliant cresyl blue revealed multiple fine, deeply stained deposits caused by the precipitation of b-globin chain aggregates (golf-ball cells) (Figure 2), suggesting the presence of Hb H.9
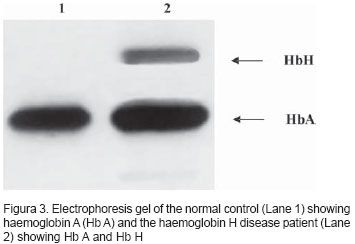
Electrophoresis of Hb showed Hb A2 + Hb A + Hb H. Hb A2: 1.3% (normal: 1.5 to 3.5%), Hb F: 1.1% (normal: < 2.0%) and Hb H: 9.7% (Figure 3). Mediterranean deletion and the 3.7 deletion were demonstrated by molecular studies7,8 (Figure 4).
The patient received the diagnosis of Hb H disease and received 5 mg of folic acid orally per day as therapy. In addition, she was advised to avoid therapies with iron compounds or red blood cell transfusion and to direct members of her family to our service for genetic counselling.
References
1. Clarke GM, Higgins TN. Laboratory investigation of haemo-globinopathies and thalassemias: Review and update. Clin Chem 2000;1284-90.
2. Hoffbrand AV, Pettit JE. Essential Haematology, Fourth edition, ed. Blackwell Science, 2000.
3. Asha S. Thalassemia Syndromes. Indian J Med Sci 2004;58:445-9.
4. Lorey F, Charoenkwan P, Witkowska HE et al. Hb H hydrops foetalis syndrome: a case report and review of literature. Br J Haematol 2001;115:72-8.
5. Chui DHK, Fucharoen S, Chan V. Haemoglobin H disease: not necessarily a benign disorder. Blood 2003;101:791-800.
6. Old JM. Screening and genetic diagnosis of haemoglobin disorders. Blood Rev 2003;17:45-53.
7. Bowden DK, Vickers MA, Higgs DR. A PCR-based strategy to detect the common severe determinants of thalassaemia. Br J Haematol 1992;81:104-8.
8. Dodé C, Krisshnamoorthy R, Lamb J, Rochette, J. Rapid analysis of -a3.7 thalassaemia and aaaanti3.7 triplication by enzymatic amplification analysis. Br J Haematol 1992;82:105-11.
9. Daice JV, Lewis SM. Pratical Haematology, Eighth edition, ed. Churchill Livingstone, 1995.
Avaliação: Editor e dois revisores externos.
Conflito de interesse: não declarado.
Recebido: 27/05/2005
Aceito após modificações: 10/06/2005
Correspondence:
Publication Dates
-
Publication in this collection
11 Jan 2006 -
Date of issue
June 2005