Resumos
As gamopatias monoclonais constituem um grupo de desordens caracterizado pela proliferação monoclonal de plasmócitos, que produzem e secretam imunoglobulina ou fragmento de imunoglobulina monoclonal (proteína M) . Este artigo propõe uma revisão dos critérios diagnósticos das principais gamopatias monoclonais e diagnósticos diferenciais, uma vez que é comum a sobreposição de muitas características clínicas entre suas variantes. A gamopatia monoclonal de significado indeterminado (MGUS) é definida pela presença de proteína M sérica < 3,0 g/dL e/ou urinária < 1g/24h, infiltração plasmocitária medular menor que 10% e ausência de danos aos órgãos e tecidos. O mieloma múltiplo (MM) assintomático caracteriza-se pela presença de proteína M, infiltração plasmocitária na medula óssea ou em tecido biopsiado e ausência de critérios para MGUS, MM sintomático e plasmocitoma solitário. O MM sintomático é uma neoplasia plasmocitária associada à proteína M sérica e/ou urinária, infiltração medular por plasmócitos e presença de dano orgânico relacionado: hipercalcemia, insuficiência renal, anemia e lesões ósseas. Se a proteína M não é detectada (MM não secretor), a plasmocitose medular precisa ser > 30% ou plasmocitoma documentado por biópsia. Se a lesão óssea decorre de plasmocitoma solitário ou somente osteoporose, sem fratura, a plasmocitose medular também precisa ser > 30%, para preencher critérios de MM. As gamopatias monoclonais podem estar associadas a diversas doenças, incluindo desordens linfoproliferativas, reumatológicas, neurológicas, dermatológicas e infecciosas. A definição das características clínicas e laboratoriais de cada entidade, maligna ou benigna, facilita o diagnóstico das gamopatias monoclonais e, como conseqüência, seu manejo clínico pelos médicos assistentes.
Gamopatias monoclonais; diagnóstico; diagnósticos diferenciais
Monoclonal gammopathies are a group of disorders characterized by proliferation of monoclonal plasma cells, which produce and secrete monoclonal immunoglobulin or fragments of monoclonal immunoglobulin (M protein). This paper proposes to review diagnostic criteria of the most important monoclonal gammopathies and their differential diagnosis, because superposition of many clinical characteristics is common between variants. The monoclonal gammopathy of undetermined significance (MGUS) is defined by the presence of serum M protein < 3g/dL and/or urinary M protein < 1g/24h, bone marrow plasma cell < 10%, and absence of organ and tissue damage. Asymptomatic multiple myeloma (MM) is characterized by the presence of M protein, bone marrow or tissue biopsy plasma cell infiltration, and non-compliance of the criteria for MGUS, symptomatic MM and solitary plasmacytoma. Symptomatic MM is a plasma cell neoplasm associated with serum or urinary M protein, bone marrow or tissue biopsy plasma cell infiltration and related organ or tissue damage: elevated calcium levels, renal insufficiency, anemia and bone lesions. If no M protein is detected (nonsecretory MM), then at least 30% monoclonal bone marrow plasma cell infiltration and/or a biopsy-proven plasmacytoma is required for MM diagnosis. If a solitary (biopsy-proven) plasmacytoma or osteoporosis (without fractures) are the sole defining criteria, then at least 30% plasma cells are required in the bone marrow for MM diagnosis. Monoclonal gammopathies may be associated with many different diseases, including lymphoproliferative disorders, connective tissue disorders, neurologic, dermatologic and infectious diseases. The clinical and laboratorial characteristics should be very well defined in each variant, malign or benign, easily determining the diagnosis of monoclonal gammopathies and then their clinical management.
Monoclonal gammopathies; diagnosis; differential diagnosis
ARTIGO ARTICLE
Gamopatias monoclonais - critérios diagnósticos e diagnósticos diferenciais
Monoclonal gammopathies - diagnosis criteria and differential diagnosis
Rosa Malena D. FariaI; Roberta O. Paula e SilvaII
IProfa. adjunta do Departamento de Propedêutica Complementar da Faculdade de Medicina da UFMG; Médica coordenadora do Atendimento ao Paciente Portador de Mieloma Múltiplo do Hospital das Clínicas da UFMG; Coordenadora do Curso de Medicina da Faculdade de Ciências Médicas da Unifenas-BH
IIPós-graduanda do Programa de Patologia Geral da UFMG
Correspondência Correspondência: Rosa Malena D. Faria Departamento de Propedêutica Complementar, Faculdade de Medicina - UFMG Av. Prof. Alfredo Balena, 190, sala 6000, Santa Efigênia 30130-100 - Belo Horizonte-MG - Brasil Tel: (31)3248-9774; (31)3497-4302; (31)8759-7929; Fax:(31)3497-4314 E-mail: rosa.malena@ufmg.br - rosa.malena@unifenas.br
RESUMO
As gamopatias monoclonais constituem um grupo de desordens caracterizado pela proliferação monoclonal de plasmócitos, que produzem e secretam imunoglobulina ou fragmento de imunoglobulina monoclonal (proteína M) . Este artigo propõe uma revisão dos critérios diagnósticos das principais gamopatias monoclonais e diagnósticos diferenciais, uma vez que é comum a sobreposição de muitas características clínicas entre suas variantes. A gamopatia monoclonal de significado indeterminado (MGUS) é definida pela presença de proteína M sérica < 3,0 g/dL e/ou urinária < 1g/24h, infiltração plasmocitária medular menor que 10% e ausência de danos aos órgãos e tecidos. O mieloma múltiplo (MM) assintomático caracteriza-se pela presença de proteína M, infiltração plasmocitária na medula óssea ou em tecido biopsiado e ausência de critérios para MGUS, MM sintomático e plasmocitoma solitário. O MM sintomático é uma neoplasia plasmocitária associada à proteína M sérica e/ou urinária, infiltração medular por plasmócitos e presença de dano orgânico relacionado: hipercalcemia, insuficiência renal, anemia e lesões ósseas. Se a proteína M não é detectada (MM não secretor), a plasmocitose medular precisa ser > 30% ou plasmocitoma documentado por biópsia. Se a lesão óssea decorre de plasmocitoma solitário ou somente osteoporose, sem fratura, a plasmocitose medular também precisa ser > 30%, para preencher critérios de MM. As gamopatias monoclonais podem estar associadas a diversas doenças, incluindo desordens linfoproliferativas, reumatológicas, neurológicas, dermatológicas e infecciosas. A definição das características clínicas e laboratoriais de cada entidade, maligna ou benigna, facilita o diagnóstico das gamopatias monoclonais e, como conseqüência, seu manejo clínico pelos médicos assistentes.
Palavras-chave: Gamopatias monoclonais; diagnóstico; diagnósticos diferenciais.
ABSTRACT
Monoclonal gammopathies are a group of disorders characterized by proliferation of monoclonal plasma cells, which produce and secrete monoclonal immunoglobulin or fragments of monoclonal immunoglobulin (M protein). This paper proposes to review diagnostic criteria of the most important monoclonal gammopathies and their differential diagnosis, because superposition of many clinical characteristics is common between variants. The monoclonal gammopathy of undetermined significance (MGUS) is defined by the presence of serum M protein < 3g/dL and/or urinary M protein < 1g/24h, bone marrow plasma cell < 10%, and absence of organ and tissue damage. Asymptomatic multiple myeloma (MM) is characterized by the presence of M protein, bone marrow or tissue biopsy plasma cell infiltration, and non-compliance of the criteria for MGUS, symptomatic MM and solitary plasmacytoma. Symptomatic MM is a plasma cell neoplasm associated with serum or urinary M protein, bone marrow or tissue biopsy plasma cell infiltration and related organ or tissue damage: elevated calcium levels, renal insufficiency, anemia and bone lesions. If no M protein is detected (nonsecretory MM), then at least 30% monoclonal bone marrow plasma cell infiltration and/or a biopsy-proven plasmacytoma is required for MM diagnosis. If a solitary (biopsy-proven) plasmacytoma or osteoporosis (without fractures) are the sole defining criteria, then at least 30% plasma cells are required in the bone marrow for MM diagnosis. Monoclonal gammopathies may be associated with many different diseases, including lymphoproliferative disorders, connective tissue disorders, neurologic, dermatologic and infectious diseases. The clinical and laboratorial characteristics should be very well defined in each variant, malign or benign, easily determining the diagnosis of monoclonal gammopathies and then their clinical management.
Key words: Monoclonal gammopathies; diagnosis; differential diagnosis.
Introdução
As gamopatias monoclonais são um grupo de desordens associadas com proliferação monoclonal de plasmócitos, também conhecidas como paraproteinemias, disproteinemias ou imunoglobulinopatias. Caracterizam-se pela produção e secreção de uma proteína monoclonal - imunoglobulina (Ig) ou um fragmento de Ig. A Ig é composta por duas cadeias polipeptídicas pesadas da mesma classe e subclasse (IgG, IgA, IgD, IgE e IgM) e duas cadeias polipeptídicas leves do mesmo tipo (kappa ou lambda). A confirmação da presença da proteína monoclonal é essencial para diferenciar as gamopatias monoclonais das gamopatias policlonais, uma vez que as primeiras são entidades neoplásicas ou potencialmente neoplásicas enquanto as últimas resultam de processos inflamatórios ou infecciosos.
O diagnóstico das gamopatias monoclonais requer a detecção e quantificação do componente monoclonal, o exame da medula óssea para verificação de infiltração plasmocitária e a pesquisa de dano orgânico relacionado (hipercalcemia, anemia, insuficiência renal e lesões ósseas). A eletroforese de proteínas em gel de agarose deve ser utilizada para detecção da proteína monoclonal e a imunofixação para a caracterização das cadeias pesada e leve da imunoglobulina.1
A importância de se estabelecerem critérios diagnósticos bem definidos, com base nas manifestações clínicas e na propedêutica laboratorial, tem como principais objetivos: i) contribuir para o diagnóstico e diagnósticos diferenciais das gamopatias monoclonais; ii) fornecer informações sobre o prognóstico e guiar a terapêutica; e iii) monitorar a eficácia do tratamento.2 As manifestações clínicas variam amplamente, desde um indivíduo assintomático até a associação de múltiplos achados, como fraqueza, dor óssea, fraturas patológicas, hipercalcemia, emagrecimento, hiperviscosidade, insuficiência renal e infecções recorrentes. A caracterização de cada variante é uma das principais dificuldades encontradas na prática clínica. A definição de cada uma delas com ênfase nas suas principais características clínicas e laboratoriais são essenciais para uma melhor abordagem do paciente.1,3,4
Critérios diagnósticos das gamopatias monoclonais
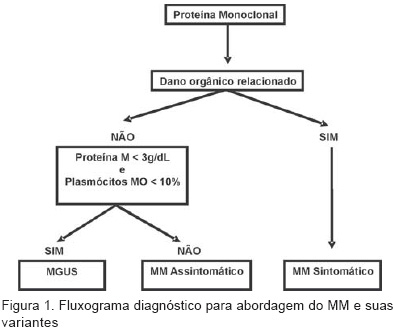
Partiremos do ponto comum, a proteína monoclonal, e a partir daí abordaremos as diversas variantes, privilegiando as de maior prevalência e relevância clínica em nosso meio, como a MGUS e o MM, que nem sempre são de simples diagnóstico (Figura 1).
MGUS
A MGUS é a gamopatia monoclonal mais comum, presente em aproximadamente 3% da população acima de 50 anos, tendo sua prevalência aumentada com a idade (1,7% em pacientes entre 50-59 anos e acima de 5% a partir dos 70 anos).
A taxa de progressão da MGUS para MM é de 1% ao ano. Aproximadamente 25% dos pacientes portadores de MGUS desenvolvem mieloma múltiplo, amiloidose, macroglobulinemia ou outras doenças linfoproliferativas, com taxa atuarial de 16% até 10 anos, 33% até 20 anos e 45% até 25 anos.1,3 A proteína M predominante é a IgG (55,5%), seguida da IgM (20%), IgA (10%), biclonal (8%), cadeia leve (6%) e IgD (<0,5%).
A concentração da proteína M, o tipo de imunoglobulina, se IgM ou IgA, infiltração plasmocitária na medula óssea maior que 5% e presença de cadeia leve monoclonal na urina podem ser considerados fatores de risco de progressão para MM. Esses dados podem ser utilizados como estratificação de risco na MGUS.1,4 Os critérios diagnósticos para MGUS são apresentados no quadro 1, de acordo com o Consenso do Grupo de Trabalho Internacional sobre Mieloma.5
Um dos grandes desafios da prática clínica é determinar a diferenciação entre MM e MGUS em pacientes com comprometimento dos órgãos por outras patologias, como, por exemplo: lesão óssea por osteoporose, lesão renal por hipertensão arterial ou diabetes e hipercalcemia por hiperparatireoidismo.
MM assintomático
Condição pré-clínica que representa aproximadamente 15% dos casos de MM recém-diagnosticados. Predomina no sexo masculino e na raça negra, sendo a idade mediana dos pacientes, ao diagnóstico, 65 anos. A maioria dos pacientes progride para MM sintomático em torno de 2-4 anos. Sabe-se que aqueles pacientes com infiltração plasmocitária da MO >10% apresentam progressão ainda mais rápida, em torno de 2-3 anos. O risco de progressão para MM sintomático é substancialmente maior que o da MGUS, apresentando uma taxa de progressão anual de 10%-20%. A presença de plasmócitos circulantes na corrente sangüínea foi demonstrada como principal fator de risco de progressão desta entidade. A concentração de proteína M e o tipo da Ig também são considerados fatores importantes para progressão.1,3,6
O quadro 2 mostra os critérios utilizados para diagnóstico de MM assintomático, de acordo com o Consenso do Grupo de Trabalho Internacional sobre Mieloma.5
MM Sintomático
MM é definido como desordem incurável caracterizada pela proliferação de plasmócitos clonais, que produzem e secretam Ig monoclonal ou fragmento de Ig monoclonal, marcada por destruição óssea, falência renal, anemia e hipercalcemia, freqüentemente precedida por MGUS. A etiologia é desconhecida, porém a exposição a radiações, benzeno e outros solventes orgânicos, inseticidas e herbicidas pode ter importância. A incidência anual do MM nos Estados Unidos é de 4 para 100.000 habitantes, correspondendo a 1% de todas as neoplasias malignas e 10% das neoplasias hematológicas. Ocorre em todas as raças e localizações geográficas, sendo mais freqüente em homens que mulheres e em negros que em brancos. A idade mediana ao diagnóstico é de 66 anos, e apenas 2% dos pacientes possuem idade inferior a 40 anos. O quadro 3 apresenta os critérios utilizados para diagnóstico do MM sintomático, de acordo com o Consenso do Grupo de Trabalho Internacional sobre Mieloma.5
Dor óssea é a manifestação clínica mais freqüente, presente ao diagnóstico em cerca de 60% dos casos. Anemia está presente em 73% dos pacientes ao diagnóstico, hipercalcemia em 13%, e creatinina maior que 2 mg/dl em 19%. Proteína M pode ser detectada por eletroforese de proteína sérica em 82% dos pacientes e a imunofixação mostra proteína M em 93% dos casos. Proteína monoclonal na urina pode ser identificada em 78% dos casos. O tipo mais freqüente de proteína M é IgG (53%), seguida de IgA (21%), cadeia leve (16%), IgD (2%), biclonal (2%) e IgM (0,5%).7 Alterações radiológicas são detectadas em 79% dos pacientes através de radiografias convencionais. Lesões líticas estão presentes em 67% dos casos, e aproximadamente 20% apresentam osteoporose, fraturas patológicas ou compressão medular. Ao diagnóstico, 25% dos pacientes não apresentam anormalidades radiológicas, porém desenvolvem-nas durante o curso da doença. A tomografia computadorizada e a ressonância nuclear magnética podem ser utilizadas em situações específicas, por possuírem maior sensibilidade e especificidade na detecção das lesões ósseas pequenas e localizadas, uma vez que a radiografia convencional só detecta osteólise quando cerca de 30% de substância óssea trabecular foi perdida.6,8
O aspirado de medula óssea demonstra a presença de plasmócitos atípicos, porém o percentual varia significativamente na dependência do sítio e da qualidade do material aspirado. Em 6% dos casos, somente a biópsia de medula óssea é capaz de demonstrar a infiltração medular.2,9
Formas raras do MM
MM IgD e MM IgE
O subtipo IgD corresponde a 2% dos casos de MM e caracteriza-se por apresentar pequeno pico monoclonal à eletroforese de proteínas séricas. Clinicamente assemelha-se ao MM de cadeia leve, por apresentar alta incidência de insuficiência renal e associação com amiloidose. O envolvimento extramedular está presente em 19%-27% dos casos. Estudos têm mostrado que o MM IgD confere menor sobrevida quando comparado a outros tipos de MM.10 O MM IgE é uma forma muito rara de apresentação do MM e associa-se com maior freqüência à leucemia de células plasmocitárias.11
MM IgM
Tipo raro de MM, responsável por apenas 0,5% dos casos. A dor óssea é o sintoma mais comum, o que o diferencia clinicamente da macroglobulinemia de Waldenström. A medula óssea é infiltrada por plasmócitos pequenos, os níveis de IgG e IgA policlonais são mais baixos que nos outros subtipos e há alta incidência de translocação (11;14).12,13,14
MM não secretor
Corresponde a 3% do total de pacientes com MM. Atualmente testes mais sensíveis para pesquisa de cadeia leve de Ig mostram que muitos casos de MM não secretor são na verdade oligossecretores. A apresentação deste tipo de MM é similar à forma secretora, porém a doença renal não está presente. A sobrevida média destes pacientes parece ser maior especialmente pela ausência do comprometimento renal. A principal dificuldade encontrada neste tipo de MM está na monitorização da resposta terapêutica uma vez que não há proteína M.1,8
Diagnósticos diferenciais das gamopatias monoclonais
Amiloidose
Amiloidose sistêmica primária é uma desordem rara caracterizada pela deposição de fibrila amilóide (fragmento da cadeia leve das Ig) cujo acúmulo geralmente afeta coração, pulmões, rins, nervos periféricos, pele, língua e intestino. Os sintomas são vagos e caracterizam-se por fadiga, edema e perda de peso. A suspeita diagnóstica ocorre quando o paciente apresenta proteína monoclonal no soro ou na urina e mais manifestações de infiltração e comprometimento de outros órgãos tais como albuminúria (amiloidose corresponde a 10% das síndromes nefróticas não diabéticas do adulto), cardiopatia (50% dos pacientes apresentam problemas cardíacos ao diagnóstico), hepatomegalia, neuropatia periférica e medula óssea infiltrada por menos de 10% de plasmócitos. A comprovação histológica do comprometimento é fundamental e é feita utilizando-se a coloração com vermelho Congo, que mostra o aspecto "fluorescente" da proteína amilóide, positiva em 60% dos casos em material obtido através de aspirado de medula óssea e, em 70%-80% dos casos, através do aspirado de gordura subcutânea. A resposta ao tratamento da amiloidose é medida pela redução da cadeia leve no soro e/ou urina.15,16
Plasmocitoma Solitário - Ósseo (PSO) e Extramedular (PSE)
Trata-se de lesão única constituída de plasmócitos monoclonais, localizada no osso ou menos comumente como uma massa extramedular. A maioria dos pacientes com PSO evolui para MM em 2-4 anos e cerca de 25% dos pacientes, quando submetidos à ressonância nuclear magnética, apresentam outros focos de acometimento ósseo. Em contraste, o PSE corresponde a uma lesão geralmente localizada e possui uma alta chance de cura com tratamento local. O PSO tem maior prevalência em homens que mulheres, com idade mediana de 55 anos e localização geralmente no esqueleto axial. O sintoma mais comum é a dor óssea localizada, seguido de compressão medular. A melhor forma para avaliar a extensão da lesão é através da tomografia computadorizada ou ressonância magnética. A biópsia guiada por TC é fundamental para a comprovação diagnóstica. A presença da proteína monoclonal pode ser detectada em 27%-72% dos pacientes e os níveis são geralmente baixos. O PSE também acomete mais homens que mulheres na faixa etária de 55 anos, sendo que a maioria das lesões (90%) se localiza nas regiões do pescoço e da cabeça, especialmente acometendo trato respiratório. A proteína M é detectada em apenas 25% dos pacientes com PSE. A tomografia computadorizada ou ressonância magnética é necessária para delinear a extensão da lesão e a biópsia é importante para definir o diagnóstico.17 O quadro 4 apresenta os critérios utilizados no diagnóstico do plasmocitoma, de acordo com o Consenso do Grupo de Trabalho Internacional sobre mieloma.5
Macroglobulinemia de Waldenström (MW)
Doença linfoproliferativa caracterizada por infiltração linfoplasmocitária da medula óssea e pela síntese de IgM monoclonal. Responsável por 2% das neoplasias hematológicas. Acomete pacientes idosos, com idade mediana de 63 anos, mais prevalente em homens que mulheres. A sobrevida média dos pacientes com MV está entre 5-7 anos. Apesar de possuírem alguns aspectos semelhantes, a MW não deve ser confundida com MM IgM.8,12,15 A tabela 1 descreve as principais características diferenciais entre MW e MM IgM.
Síndrome de POEMS
Definida pela presença de polineuropatia (P), organomegalia (O), endocrinopatia (E), proteína monoclonal (M) e alterações da pele (S), seguida por outras manifestações menos freqüentes, tais como: edema, ascite e trombocitose. A presença de lesões osteoescleróticas é uma característica importante presente em 95% dos pacientes. Os pacientes usualmente apresentam MO com < de 5% de infiltração plasmocitária e raramente apresentam hipercalcemia ou insuficiência renal.8,18
Leucemia plasmocitária
Variante rara do MM, definida pela presença de plasmócitos circulantes em número superior a 2.000/mm3 e plasmocitose maior que 20% do total de glóbulos brancos. A forma primária ocorre quando o diagnóstico inicial é feito em fase leucêmica, já a secundária corresponde à transformação leucêmica de um MM previamente diagnosticado. As manifestações clínicas mais comuns são astenia, insuficiência renal, dor óssea, esplenomegalia e hepatomegalia. Em ambas as formas o prognóstico é ruim, sobrevida média de um ano.10,19
Doença de Depósito de Cadeia Leve
A doença de depósito de cadeia leve não amilóide se caracteriza pela deposição da proteína monoclonal em vários tecidos e órgãos. Diferente da amiloidose, estes depósitos não são corados pelo vermelho Congo e são vistos à microscopia eletrônica como depósitos amorfos nodulares. Esta condição pode ocorrer associada ou não ao MM. Envolvimento renal é o mais comum seguido pelo cardíaco e hepático. O depósito de cadeia leve no rim possui predileção glomerular mais que tubular, o que leva a uma proteinúria inespecífica, com perda de albumina. A expressão clínica habitual é proteinúria e insuficiência renal.8,20
Outras doenças
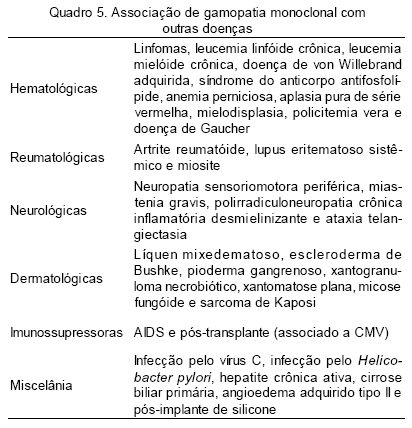
A presença de proteína M sérica tem sido reportada em pacientes portadores de diversas outras doenças, hematológicas ou não, como apresentado no quadro 5. Por isso, diante de uma gamopatia monoclonal, faz-se necessário afastar causas secundárias reacionais.4
Proposta para abordagem diagnóstica das gamopatias monoclonais
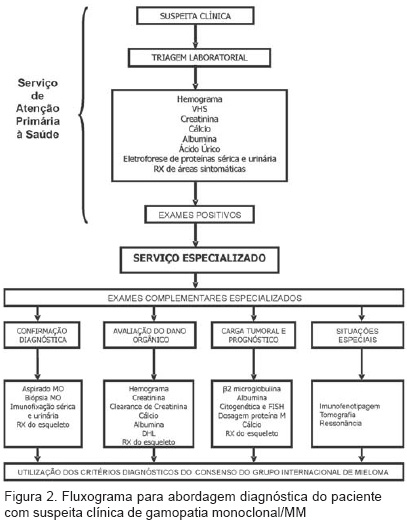
O conhecimento, por parte da população médica, das características clínicas, epidemiológicas e laboratoriais das gamopatias monoclonais é fundamental, entretanto é necessária uma sistematização de condutas para abordagem diagnóstica do paciente com suspeita clínica. Estudos realizados no Brasil mostram que a maioria dos pacientes portadores de MM, ao diagnóstico, apresenta-se em fase avançada da doença, com importante repercussão na sobrevida.21,22,23 Assim, propomos um fluxograma (Figura 2) para abordagem diagnóstica do paciente com suspeita clínica de gamopatia monoclonal/MM, esperando que o instrumento seja capaz de gerar um impacto positivo na sobrevida e qualidade de vida dos pacientes, uma vez que facilitará a condução do diagnóstico aos profissionais que realizam o primeiro atendimento ao paciente, em geral um médico generalista que trabalha em serviços de atenção primária à saúde. A partir do conhecimento clínico, tem-se a base para a realização da propedêutica complementar de triagem. Se os exames de triagem do paciente forem compatíveis com a hipótese de gamopatia monoclonal/MM, o paciente deve ser encaminhado a um serviço especializado, onde será realizada a propedêutica complementar necessária à confirmação diagnóstica, avaliação prognóstica, tratamento e seguimento clínico.
Conclusão
O diagnóstico das gamopatias monoclonais nem sempre é simples e exige do profissional de saúde conhecimentos sobre características clínicas, critérios diagnósticos e prognóstico de cada variante, para uma adequada abordagem do paciente. Portanto, todo esforço deve ser realizado na intenção de se realizar o diagnóstico correto, uma vez que erros diagnósticos possibilitam a progressão da doença, interferem na conduta e resultam em subtratamento ou até supertratamento, implicando prejuízos para a vida humana.
Recebido: 25/11/2006
Aceito: 05/01/2007
O tema apresentado e o convite ao(s) autor(es) consta da pauta elaborada pelo co-editor.
Avaliação: Co-editor e um revisor externo.
Publicado após revisão e concordância do editor.
Conflito de interesse: não declarado.
- 1. Kyle RA, Child JA, Anderson K, et al Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol 2003;121:749-757.
- 2. San Miguel J, Gutiérrez NC, Mateo G, Orfao A. Conventional diagnostics in multiple myeloma. Eur J Cancer 2006;42:1.510-1.519.
- 3. Rajkumar SV. MGUS and smoldering multiple myeloma: Update on pathogenesis, natural history, and management. Hematology Am Soc Hematol Educ Program 2005;340-5.
- 4. Kyle RA, Rajkumar SV. Monoclonal gammopathies of undetermined significance: a review. Immunol Rev 2003;191:112-139.
- 5. Durie BGM, Kyle RA, Belch A, et al Myeloma management guidelines: a consensus report from the Scientific Advisors of the International Myeloma Foundation. The Hematology Journal 2003;4:379-398.
- 6. Smith A, Wisloff F, Samson D. Guidelines on the diagnosis and management of multiple myeloma 2005. Br J Haematol 2005; 132:410-451.
- 7. Kyle RA, Gertz MA, Witzig TE et al Review of 1.027 patients with newly diagnosed multiple mieloma. Mayo Clin Proc 2003;78:21-33.
- 8. Dispenzieri A, Kyle RA. Multiple myeloma: clinical features and indications for therapy. Best Pract Res Clin Haematol 2005;18:553-568.
- 9. Harousseau JL, Shaughnessy J Jr, Richardson P. Multiple myeloma. Hematology Am Soc Hematol Educ Program 2004;237-256.
- 10. Blade J, Kyle RA. Nonsecretory myeloma, immunoglobulin D myeloma and plasma cell leukemia. Hematol Oncol Clin North Am 1999;13:1.259-1.272.
- 11. Blade J, Lust JA, Kyle RA. Immunoglobulin D multiple myeloma: presenting features, response to therapy and survival in a series of 53 cases. J Clin Oncol 1994;12:2.398-2.404.
- 12. Chehal A, Taher A, Shamseddine A. IgM myeloma and Waldenstrom's macroglobulinemia: a distinct clinical feature, histology, immunophenotype, and chromosomal abnormality. Clin Lab Haematol 2003;25:187-190.
- 13. Avet-Loiseau H, Garand R, Lodé L, Robillard N, Bataille R. 14q32 translocations discriminate IgM multiple myeloma from Waldenström's macroglobulinemia. Semin Oncol 2003;30:153-155.
- 14. Avet-Loiseau H, Garand R, Lodé L, Harousseau JL, Bataille R Translocation t(11;14)(q13;q32) is the hallmark of IgM, IgE and nonsecretory multiple myeloma variants. Blood 2003;101:1.570-1.571.
- 15. Gertz MA, Merlini G, Treon SP. Amyloidosis and Waldenstrom's macroglobulinemia. Hematology Am Soc Hematol Educ Program. 2004;257-282.
- 16. Merlini G, Bellotti V. Molecular mechanism of amyloidosis. New Engl J Med 2004;349:583-596.
- 17. Soutar R, Lucraft H, Jackson G, et al Guidelines on the diagnosis and management of solitary plasmacytoma of bone and solitary extramedullary plasmacytoma. Clin Oncol (R Coll Radiol) 2004; 16:405-413.
- 18. Dispenzieri A. POEMS syndrome. Hematology Am Soc Hematol Educ Program 2005;360-7.
- 19. Costello R, Sainty D, Bouabdallah R, et al Primary plasma cell leukaemia: a report of 18 cases. Leuk Res 2001;25:103-107.
- 20. Heilman RL. Long term follow-up and response to chemotherapy in patients with light-chain deposition disease. Am J Kidney Dis 1992;20:34-41.
- 21. Faria RMD, Oliveira JSRO, Faria JR, et al Prognostic value of clinical, laboratory and morphological characteristics in multiple myeloma. Cancer Research Therapy and Control 1999;10:155-161.
- 22. Ortega MM, Faria RMD, Shitara ES, et al N-Ras and K-Ras gene mutations in Brazilian patients with multiple myeloma. Leukemia & Lymphoma 2006;47:285-289.
- 23. Hungria VTM, Maiolino A, Martinez G, et al Multiple myeloma (MM) in Brazil: Clinical and demographic features and the utility of ISS in 1,017 patients, mostly with advanced disease. Haematologica 2006;91 (suppl1):96.
Datas de Publicação
-
Publicação nesta coleção
22 Out 2007 -
Data do Fascículo
Mar 2007
Histórico
-
Recebido
25 Nov 2006 -
Aceito
05 Jan 2005