Resumos
O conceito da cascata da coagulação descreve as interações bioquímicas dos fatores da coagulação, entretanto, tem falhado como um modelo do processo hemostático in vivo. A hemostasia requer a formação de um tampão de plaquetas e fibrina no local da lesão vascular, bem como a permanência de substâncias procoagulantes ativadas nesse processo no sítio da lesão. O controle da coagulação sanguínea é realizado por meio de reações procoagulantes em superfícies celulares específicas e localizadas, evitando a propagação da coagulação no sistema vascular. Uma análise crítica do papel das células no processo hemostático permite a construção de um modelo da coagulação que melhor explica hemorragias e tromboses in vivo. O modelo da coagulação baseado em superfícies celulares substitui a tradicional hipótese da "cascata" e propõe a ativação do processo de coagulação sobre diferentes superfícies celulares em quatro fases que se sobrepõem: iniciação, amplificação, propagação e finalização. O modelo baseado em superfícies celulares permite um maior entendimento de como a hemostasia funciona in vivo e esclarece o mecanismo fisiopatológico de certos distúrbios da coagulação.
Coagulação sanguínea; Fatores de coagulação sanguínea; Transtornos da coagulação sanguínea; Plaquetas; Hemostasia; Tromboplastina; Proteína C; Proteína S; Antitrombinas; Anticoagulantes
The concept of a coagulation cascade describes the biochemical interactions of the coagulation factors, but it is flawed as a model of the in vivo hemostatic process. Hemostasis requires both platelet and fibrin plug formation at the site of vessel injury and that the procoagulant substances activated in this process remain at the site of injury. This control of blood coagulation is accomplished as the procoagulant reactions only exist on specific cell surfaces to keep coagulation from spreading throughout the vascular system. A model of coagulation that better explains bleeding and thrombosis in vivo created after considering the critical role of cells. The cellbased model of hemostasis replaces the traditional "cascade" hypothesis, and proposes that coagulation takes place on different cell surfaces in four overlapping steps: initiation, amplification, propagation and termination. The cell-based model allows a more thorough understanding of how hemostasis works in vivo, and sheds light on the pathophysiological mechanism for certain coagulation disorder.
Blood coagulation; Blood coagulation factors; Blood coagulation disorders; Blood platelets; Hemostasis; Thromboplastin; Protein C; Protein S; Antithrombins; Anticoagulants
Atualização/ Update
O novo modelo da cascata de coagulação baseado nas superfícies celulares e suas implicações
Cláudia Natália FerreiraI; Marinez de Oliveira SousaII; Luci Maria Sant'Ana DusseII; Maria das Graças CarvalhoII
IFundação Hospitalar do Estado de Minas Gerais, Maternidade Odete Valadares, e Prefeitura Municipal de Belo Horizonte, Laboratório Distrital do PAM Padre Eustáquio - Belo Horizonte (MG), Brasil
IIDepartamento de Análises Clínicas e Toxicológicas, Faculdade de Farmácia da Universidade Federal de Minas Gerais - UFMG - Belo Horizonte (MG), Brasil
Correspondência Correspondência: Maria das Graças Carvalho Laboratório de Hematologia, Departamento de Análises Clínicas e Toxicológicas Faculdade de Farmácia da Universidade Federal de Minas Gerais Av. Antônio Carlos, 6627 - Pampulha 31270-010 - Belo Horizonte (MG), Brasil Tel: (55 31) 3409-6881; Fax: (55 31) 3409-6895 E-mail: mgcarvalho@farmacia.ufmg.br
RESUMO
O conceito da cascata da coagulação descreve as interações bioquímicas dos fatores da coagulação, entretanto, tem falhado como um modelo do processo hemostático in vivo. A hemostasia requer a formação de um tampão de plaquetas e fibrina no local da lesão vascular, bem como a permanência de substâncias procoagulantes ativadas nesse processo no sítio da lesão. O controle da coagulação sanguínea é realizado por meio de reações procoagulantes em superfícies celulares específicas e localizadas, evitando a propagação da coagulação no sistema vascular. Uma análise crítica do papel das células no processo hemostático permite a construção de um modelo da coagulação que melhor explica hemorragias e tromboses in vivo. O modelo da coagulação baseado em superfícies celulares substitui a tradicional hipótese da "cascata" e propõe a ativação do processo de coagulação sobre diferentes superfícies celulares em quatro fases que se sobrepõem: iniciação, amplificação, propagação e finalização. O modelo baseado em superfícies celulares permite um maior entendimento de como a hemostasia funciona in vivo e esclarece o mecanismo fisiopatológico de certos distúrbios da coagulação.
Descritores: Coagulação sanguínea; Fatores de coagulação sanguínea; Transtornos da coagulação sanguínea/fisiopatologia; Plaquetas/metabolismo; Hemostasia; Tromboplastina; Proteína C; Proteína S; Antitrombinas; Anticoagulantes
ABSTRACT
The concept of a coagulation cascade describes the biochemical interactions of the coagulation factors, but it is flawed as a model of the in vivo hemostatic process. Hemostasis requires both platelet and fibrin plug formation at the site of vessel injury and that the procoagulant substances activated in this process remain at the site of injury. This control of blood coagulation is accomplished as the procoagulant reactions only exist on specific cell surfaces to keep coagulation from spreading throughout the vascular system. A model of coagulation that better explains bleeding and thrombosis in vivo created after considering the critical role of cells. The cellbased model of hemostasis replaces the traditional "cascade" hypothesis, and proposes that coagulation takes place on different cell surfaces in four overlapping steps: initiation, amplification, propagation and termination. The cell-based model allows a more thorough understanding of how hemostasis works in vivo, and sheds light on the pathophysiological mechanism for certain coagulation disorder.
Keywords: Blood coagulation; Blood coagulation factors; Blood coagulation disorders/physiopathology; Blood platelets/metabolism; Hemostasis; Thromboplastin; Protein C; Protein S; Antithrombins; Anticoagulants
Introdução
A clássica cascata da coagulação, proposta em 1964, por Macfarlane,(1) Davie e Ratnoff(2) está documentada em numerosos artigos e compêndios. Apesar deste modelo possuir limitações e não conseguir explicar satisfatoriamente todos os fenômenos ligados à hemostasia in vivo, foi aceito por quase cinquenta anos. Este modelo convencional referido como "cascata" foi proposto para explicar a fisiologia da coagulação do sangue, segundo o qual a coagulação ocorre por meio de ativação proteolítica sequencial de pró-enzimas por proteases do plasma, resultando na formação de trombina que, então, quebra a molécula de fibrinogênio em monômeros de fibrina. Tal proposta divide a coagulação em uma via extrínseca (envolvendo elementos do sangue e também elementos que usualmente não estão presentes no espaço intravascular) e uma via intrínseca (iniciada por componentes presentes no espaço intravascular), que convergem para uma via comum, a partir da ativação do fator X (FX). Na via extrínseca, o fator VII plasmático é ativado na presença de seu cofator, o fator tecidual (FT), formando o complexo fator VII ativado/FT (FVIIa/FT), responsável pela ativação do fator X. Na via intrínseca, a ativação do fator XII ocorre quando o sangue entra em contato com uma superfície contendo cargas elétricas negativas. Tal processo é denominado "ativação por contato" e requer ainda a presença de outros componentes do plasma: pré-calicreína (uma serinoprotease) e cininogênio de alto peso molecular (um cofator não enzimático). O fator XII ativado ativa o fator XI que, por sua vez, ativa o fator IX. O fator IX ativado, na presença de fator VIII ativado por traços de trombina, e em presença e íons cálcio (complexo tenase), ativa o fator X da coagulação, desencadeando a geração de trombina e, subsequentemente, formação de fibrina.(3-5)
Embora o conceito da "cascata" da coagulação tenha representado um modelo bem sucedido e um avanço significativo no entendimento da coagulação, observações experimentais e clínicas mais recentes demonstram que a hipótese da cascata não reflete completamente os eventos da hemostasia in vivo.(6)
Nos últimos anos, deficiências neste esquema clássico têm se tornado evidentes. Por exemplo, deficiências de fator XII, precalicreína ou cininogênio de alto peso molecular prolongam o tempo de tromboplastina parcial ativado (TTPa) mas não causam sangramento.(7) Ao contrário, a deficiência do fator IX causa hemofilia B e um sangramento clínico grave. O modelo da "cascata" não explica porque a ativação do fator X pela via extrínseca não é capaz de compensar o comprometimento da via intrínseca pela falta de fator VIII (hemofilia A) ou fator IX (hemofilia B).(8) Além disso, o grau de prolongamento do TTPa em pacientes hemofílicos não necessariamente prediz a extensão da tendência ao sangramento.
Conforme salientado por Hoffman,(6,9) pacientes hemofílicos apresentam atividade da via extrínseca normal, avaliada pelo tempo de protrombina (TP), apesar de um TTPa prolongado e uma pronunciada tendência a sangramento. Este fato tem levantado a seguinte indagação: por que a via extrínseca falha na compensação da disfunção da via intrínseca, ou, em outras palavras, por que os hemofílicos sangram? Muitos investigadores reconhecem que o modelo da cascata possui sérias falhas em relação ao modelo fisiológico da coagulação e que as vias extrínseca e intrínseca podem não operar como vias independentes e redundantes, como empregado neste modelo.(6,10,11) Foi reconhecido também, em estudos prévios da coagulação, que as células têm participação importante neste processo e que a hemostasia normal não é possível na ausência do fator tecidual associado às células e plaquetas. Portanto, parece lógico que, substituindo o papel das células nos testes da coagulação in vitro por vesículas de fosfolípides nos testes TP e TTPa, fica negligenciado o papel ativo de tais células na condição in vivo.(10)
Diante dos questionamentos expostos acima e de algumas observações chaves, surgiu a necessidade de uma revisão do modelo clássico da coagulação, já que o mesmo não conseguia responder várias importantes indagações relacionadas à clinica de pacientes portadores de certos distúrbios hemostáticos. Dessa forma, foi desenvolvido um modelo para a hemostasia baseado em superfícies celulares que substitui o modelo clássico da cascata da coagulação. Esse modelo enfatiza a interação dos fatores da coagulação com superfícies celulares específicas e parece ser capaz de explicar muitas questões até então não entendidas, valendo-se apenas da tradicional cascata da coagulação.
Modelo da cascata de coagulação baseado em superfícies celulares
Grandes avanços ocorreram nos últimos 15 anos no campo da hemostasia, à luz de importantes descobertas relacionadas à coagulação sanguínea in vivo, cujo início depende de células que expressam FT em sua superfície. O FT é uma proteína transmembrânica que age como receptor e cofator para o fator VII, estando normalmente expresso em células fora da vasculatura.(12,13) Este entendimento resultou em questionamento do verdadeiro papel da via intrínseca na hemostasia in vivo. Com relação ao fator XII, evidências sugerem que, embora sua deficiência não resulte em problemas de sangramento, a ausência desse não protege contra a trombose.(7,9)
Recentemente foi proposto o modelo baseado em superfícies celulares, no qual a hemostasia requer substâncias procoagulantes ativadas que permaneçam localizadas no sítio da lesão para a formação de tampão plaquetário e de fibrina neste local. Neste novo modelo, o processo de coagulação sanguínea é iniciado pela exposição de FT na corrente sanguínea. O FT não é expresso constitutivamente nas células endoteliais, mas está presente nas membranas das células ao redor do leito vascular, como células do músculo liso e fibroblastos. Dessa forma, o FT é exposto na circulação sanguínea pela lesão endotelial e de células vizinhas ou pela ativação de células endoteliais ou monócitos.(12) Muitas evidências sugerem que o FT está também presente no sangue em micropartículas celulares provenientes de membranas fragmentadas de vários tipos de células, como leucócitos e células endoteliais, bem como de plaquetas. Estas micropartículas podem desempenhar importante papel nos processos trombóticos. Sabe-se que o complexo FVIIa/FT ativa não somente o fator X, mas também o fator IX. Além disso, estudos mostram que esse complexo é fundamental para iniciar a coagulação in vivo.(10)
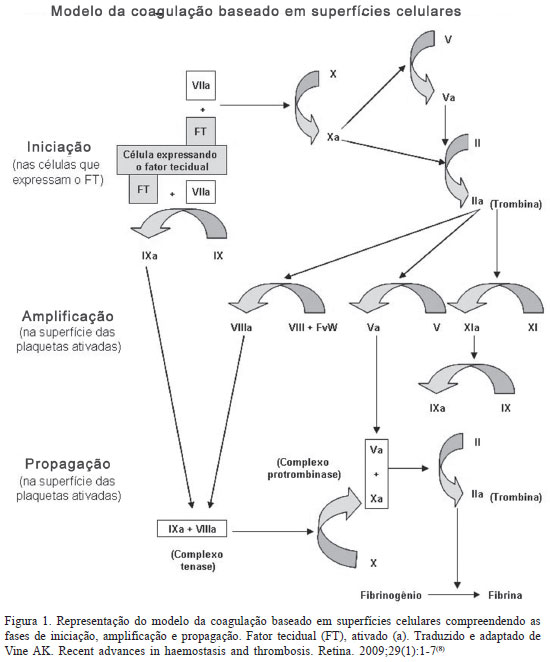
O entendimento atual do processo hemostático considera a interrelação dos processos físicos, celulares e bioquímicos que atuam em uma série de estágios ou fases, e não em duas vias (intrínseca e extrínseca) como antes. As fases de iniciação, amplificação, propagação e finalização ilustram o intrigante processo que garante a circulação do sangue na forma líquida, restrita ao leito vascular. Estas quatro fases, resumidas no Quadro 1, compreendem a atual teoria da coagulação baseada em superfícies celulares.
Fase de iniciação
A fase de iniciação do processo da coagulação ocorre quando células que expressam o FT em sua superfície são expostas aos componentes do sangue no sítio da lesão.(4,8) O FT, uma vez ligado ao FVII presente no sangue, rapidamente o ativa em FVIIa formando o complexo FVIIa/FT, responsável pela ativação de pequenas quantidades de FIX e FX.(11,14) O FXa associado com o seu cofator, FVa, forma um complexo denominado protrombinase na superfície da célula que expressa o FT. O FV pode ser ativado pelo FXa ou por proteases não coagulantes, resultando em FVa necessário para o complexo protrombinase. Esse complexo transforma pequenas quantidades de protrombina (Fator II) em trombina, que são insuficientes para completar o processo de formação do coágulo de fibrina, mas são de fundamental importância para a fase de amplificação da coagulação (Figura 1).(6,8,14)
Acredita-se que as reações responsáveis pela iniciação da coagulação ocorram constantemente fora do espaço vascular em indivíduos saudáveis. Atualmente, está comprovado que fatores da coagulação, incluindo FVII, FX e protrombina, são capazes de percorrer espaços entre os tecidos, ou seja, podem deixar o espaço vascular. Estes fatores foram detectados na linfa e a quantidade deles fora dos vasos depende especialmente do tamanho da molécula.(6) Com base nestas observações foi proposto que a via de iniciação permanece continuamente ativa, gerando pequenas quantidades de fatores ativados no estado basal. Assim, pequenas quantidades de trombina são produzidas continuamente fora do espaço vascular, independente de lesão vascular.(11) Portanto, admite-se que pequena atividade da via do FT ocorre todo o tempo no espaço extravascular. O processo da coagulação segue para a fase de amplificação somente quando há dano vascular, permitindo que plaquetas e FVIII (ligado ao fator de von Willebrand) entrem em contato com o tecido extravascular onde se aderem às células que expressam FT.(9,10)
Fase de amplificação
Devido ao grande tamanho das plaquetas e do FVIII ligado ao fator de von Willebrand (FvW), esses somente passam para o compartimento extravascular quando há lesão vascular. Quando um vaso é lesado, plaquetas escapam de dentro dos vasos, se ligam ao colágeno e a outros componentes da matrix extracelular no sítio da lesão, onde são parcialmente ativadas, resultando em um tampão plaquetário responsável pela hemostasia primária.(5,15) Neste ponto, pequenas quantidades de trombina produzidas pelas células que expressam o FT podem interagir com as plaquetas e o complexo FVIII/FvW. Dessa forma, inicia-se o processo hemostático culminando na formação de fibrina estável, que consolida o tampão plaquetário inicial. Este processo resulta na hemostasia secundária.(4,11,15)
Esta pequena quantidade de trombina gerada pelas células que expressam o FT possui várias funções importantes, sendo a principal a ativação máxima de plaquetas, que expõem receptores e sítios de ligação para os fatores da coagulação ativados. Como resultado dessa ativação, as plaquetas alteram a permeabilidade de suas membranas, permitindo a entrada de íons cálcio e saída de substâncias quimiotáticas que atraem os fatores da coagulação para sua superfície, além de liberarem FV parcialmente ativados.(4,14) Outra função da trombina formada durante a fase de iniciação é a ativação de cofatores FV e FVIII na superfície das plaquetas ativadas. O complexo FVIII/FvW é dissociado, permitindo o FvW mediar a adesão e agregação plaquetárias no sítio da lesão. Além disso, pequenas quantidades de trombina ativam o FXI a FXIa na superfície da plaqueta durante essa fase. A ativação do FXI pela trombina na superfície das plaquetas explica porque o FXII não é necessário para a hemostasia normal. Simultaneamente, por mecanismos quimiotáticos, os fatores mencionados são atraídos à superfície das plaquetas onde se inícia rapidamente a fase de propagação (Figura 1).(8,9,11,14)
Fase de propagação
A fase de propagação é caracterizada pelo recrutamento de um grande número de plaquetas para o sítio da lesão e pela produção dos complexos tenase e protrombinase na superfície das plaquetas ativadas.(8) Primeiramente, o FIXa ativado durante a fase de iniciação pode agora se ligar ao FVIIIa na superfície das plaquetas formando o complexo tenase. Uma quantidade adicional de FIXa pode também ser produzida pelo FXIa ligado às plaquetas. Como o FXa não pode se mover efetivamente das células que expressam FT para a plaqueta ativada, maior quantidade de FXa deve ser produzida diretamente na superfície da plaqueta pelo complexo FIXa/FVIIIa.(9) Finalmente, o FXa rapidamente se associa ao FVa ligado à plaqueta durante a fase de amplificação, resultando na formação do complexo protrombinase, o qual converte grande quantidade de protrombina em trombina. Esta é responsável pela clivagem do fibrinogênio em monômeros de fibrina, que polimerizam para consolidar o tampão plaquetário inicial (Figura 1).(10)
Fase de finalização
Uma vez formado o coágulo de fibrina sobre a área lesada, o processo de coagulação deve se limitar ao sítio da lesão para se evitar a oclusão trombótica do vaso. Para controlar a disseminação da ativação da coagulação, intervêm quatro anticoagulantes naturais, o inibidor da via do fator tecidual (TFPI), a proteína C (PC), a proteína S (PS), e a antitrombina (AT).
O TFPI é uma proteína secretada pelo endotélio, que forma um complexo quaternário FT/FVIIa/FXa/TFPI inativando os fatores ativados e, portanto, limitando a coagulação.(7) As proteínas C e S são dois outros anticoagulantes naturais, com capacidade de inativar os cofatores procoagulantes FVa e FVIIIa.(16) A proteína C é uma glicoproteína plasmática dependente de vitamina K, cuja síntese, quando ativada, promove a proteólise dos cofatores Va e VIIIa.(17) A proteína C (PC) é ativada pela trombina, que está ligada à proteína transmembrânica trombomodulina (TM) na superfície das células endoteliais intactas.(18) A atividade da PC é aumentada por outro cofator inibidor, também vitamina K dependente, a proteína S (PS). No plasma humano, aproximadamente 30% da PS circula como proteína livre, consistindo na fração que funciona como cofator da PC ativada.(19,20)
Um outro anticoagulante natural é a antitrombina (AT), a qual inibe a atividade da trombina e outras serino proteases, tais como FIXa, FXa, FXIa e FXIIa.(21) As células endoteliais produzem uma variedade de glicosaminoglicanos, que funcionam como sítios de ligação, de alta afinidade, para a AT, que são cruciais para uma rápida inativação da trombina.(8,22)
Vantagens do novo modelo da coagulação
Este novo modelo da hemostasia, baseado em superfícies celulares, é capaz de explicar alguns aspectos clínicos do mecanismo hemostático que o modelo clássico da cascata não permite. Este novo modelo propiciou um melhor entendimento do processo da coagulação in vivo, e apresenta maior consistência com as observações clínicas de vários distúrbios da coagulação.
Implicações do novo modelo da coagulação nos testes laboratoriais
Tradicionalmente, os métodos de triagem para avaliação da coagulação sanguínea compreendem o tempo de tromboplastina parcial ativado (TTPa), que analisa a via intrínseca, e o tempo de protrombina (TP), que avalia a via extrínseca da coagulação.(9,23) O novo modelo da coagulação baseado em superfícies celulares vem mostrar que as vias extrínseca e intrínseca não são redundantes. A via extrínseca opera na superfície das células que expressam FT para iniciar e amplificar o processo de coagulação. Os componentes da via intrínseca operam na superfície das plaquetas ativadas para produzir grande quantidade de trombina que resultará na formação e estabilização do coágulo de fibrina. Assim, o TP avalia os níveis de procoagulantes envolvidos na fase de iniciação da coagulação, enquanto o TTPa avalia os níveis de procoagulantes envolvidos na produção de grande quantidade de trombina na superfície das plaquetas ativadas, gerada durante a fase de propagação.(11)
Cumpre ressaltar que o modelo da cascata da coagulação e os testes de coagulação da clínica comum não refletem a complexidade da hemostasia in vivo. Apesar disso, os testes de coagulação disponíveis possuem sensibilidade para a detecção de deficiência de um ou mais fatores da coagulação, sendo, portanto, eficientes para a definição de alterações de fatores da coagulação em pacientes com tendência asangramento. É importante ressaltar que nenhum ensaio é capaz de fornecer um perfil completo e fidedigno da função hemostática, considerando que o modelo proposto para a hemostasia incorpora participação ativa de estruturas celulares no direcionamento e controle do processo e nenhum dos testes disponíveis inclui componentes celulares. Segundo Monroe & Hoffman,(11) apesar do TP e do TTPa não refletirem o papel desempenhado pelos inibidores e, não necessariamente, o risco de sangramento clínico, não devem ser os mesmos destituídos de valor, porém há necessidade de enfatizar que a interpretação de seus resultados deve ser considerada à luz do conjunto clínico.
Implicações do novo modelo da coagulação nas hemofilias
Quando comparada à tradicional cascata, o modelo baseado em superfícies celulares permite um maior entendimento do mecanismo fisiopatológico envolvido na hemofilia. Por exemplo, o modelo da cascata não explica por que a via extrínseca parece ser incapaz de produzir quantidades suficientes de FX para compensar parcialmente a deficiência de FVIII ou FIX. Em outras palavras, um dos intrigantes questionamentos citado por Hoffman(6) se refere à falta de explicação plausível para o fato de que a ativação do FX pelo complexo FT/VIIa fracassa na substituição do FXa que normalmente é gerado pelo complexo FIXa/FVIIIa. O modelo baseado na superfície celular não sugere que o fator Xa gerado pelo complexo FT/VIIa seja insuficiente na hemofilia, mas que este ocorre "insuficientemente" na superfície das células. O complexo FIXa/FVIIIa ativa o fator X na superfície das plaquetas durante a fase de propagação; entretanto, o FT/FVIIa pode somente produzir o FXa na superfície das células que expressam o FT, sendo incapaz de se mover para a superfície das plaquetas ativadas. Além disso, é importante mencionar que existem dois inibidores muito eficientes de FXa no plasma: TFPI e AT. Os níveis plasmáticos normais de TFPI e AT inibem o FXa tão rápida e efetivamente que a meia vida do FXa é de um minuto ou menos na fase fluida. Portanto, o fator Xa que permanece nas células que expressam o FT é relativamente protegido dos inibidores, já que todo FXa que difunde da superfície celular é rapidamente inibido.(6,9)
O modelo da coagulação baseado em superfícies celulares propõe que a hemofilia seja especificamente uma deficiência de geração de FXa na superfície das plaquetas, resultando na falta de produção de trombina na superfície das mesmas. Pacientes hemofílicos apresentam as fases da coagulação de iniciação e de amplificação relativamente normais, sendo capazes de formar o tampão plaquetário inicial no sítio do sangramento. No entanto, eles são incapazes de gerar uma quantidade de trombina na superfície das plaquetas suficiente para estabilizar o coágulo de fibrina.(6)
Considerações finais
O conceito baseado no modelo de superfícies celulares na hemostasia permite um melhor entendimento dos problemas clínicos observados em alguns distúrbios da coagulação, por enfatizar o papel central de superfícies celulares específicas no controle e direcionamento dos processos hemostáticos. Este modelo fornece uma representação potencialmente mais exata do processo hemostático, bem como facilita a interpretação dos testes da coagulação e dos mecanismos fisiopatológicos dos distúrbios da coagulação, tal como as hemofilias. Enfim, a nova teoria da coagulação baseada em superfícies celulares, pode ser considerada um avanço na avaliação de grandes eventos clínicos ligados à hemostasia. No entanto, investigações adicionais estão sendo realizadas, buscando, cada vez mais, evoluir no entendimento do complexo mecanismo hemostático.
Agradecimentos
Á Fundação Hospitalar do Estado de Minas Gerais - Maternidade Odete Valadares e Prefeitura Municipal de Belo Horizonte - Laboratório Distrital do PAM Padre Eustáquio.
Recebido: 27/10/2009
Aceito: 23/11/2009
Conflito de interesse: sem conflito de interesse
- 1. Macfarlane RG. An enzyme cascade in the blood clotting mechanism, and its function as a biological amplifier. Nature. 1964;202:498-9.
- 2. Davie EW, Ratnoff OD. Waterfall sequence for intrinsic blood clotting. Science. 1964;145:1310-2.
- 3. Lotspeich-Steininger CA. Introduction to hemostasis. In: Lotspeich-Steininger CA, Stiene-Martin EA, Koepke JA. Clinical hematology. New York: J.B. Lippincott; 1992.
- 4. Handin RI, Lux SE, Stossel TP. Blood: principles and practice of hematology. 2a ed. Philadelphia: Lippincott Williams & Wilkins; 2003. 2304p.
- 5. Zago MA, Falcão RP, Pasquini R. Hematologia: fundamentos e prática. ed rev atual. São Paulo: Atheneu; 2005. 1081p.
- 6. Hoffman M. A cell-base model of coagulation and the role of factor VIIa. Blood Rev. 2003;17(Suppl 1):S1-5.
- 7. Malý MA, Tomasov P, Hájek P, Blasko P, Hrachovinová I, Salaj P, Veselka J. The role of tissue factor in thrombosis and hemostasis. Physiol Res. 2007;56(6):685-95.
- 8. Vine AK. Recent advances in haemostasis and thrombosis. Retina. 2009;29(1):1-7.
- 9. Hoffman M. Remodeling the blood coagulation cascade. J Thromb Thrombolysis. 2003;16(1/2):17-20.
- 10. Riddel Jr JP, Aouizerat BE, Miaskowski C, Lillicrap DP. Theories of blood coagulation. J Pediatr Oncol Nurs. 2007;24(3):123-31.
- 11. Monroe DM, Hoffman M. The coagulation cascade in cirrosis. Clin Liver Dis. 2009;13(1):1-9.
- 12. Nemerson Y. Tissue factor and haemostasis. Blood. 1988;71(1):1-8. Erratum in: Blood. 1988;71(4):1178.
- 13. Edgington TS, Mackman N, Brand K, Ruf W. The structural biology of expression and function of tissue factor. Thromb Haemost. 1991;66(1):67-79.
- 14. Pérez-Gómez F, Bover R. La nueva cascada de la coagulación y su posible influencia en el difícil equilibrio entre trombosis y hemorragia. Rev Esp Cardiol. 2007;60(12):1217-9.
- 15. Boucher BA, Traub O. Achieving hemostasis in the surgical field. Pharmacotherapy. 2009;29(7):2S-7S.
- 16. Valen G, Sigurdardottir O, Vaage J. Systemic release of thrombomodulin, but not from the cardioplegic, reperfused heart during open heart surgery. Thromb Res. 1996;83(4):321-8.
- 17. Shearer MJ. Vitamin K. Lancet. 1995;345(8944):229-34.
- 18. Ohlin AK, Morser J, Ohlin H. Soluble thrombomodulin antigen in plasma is increased in patients with acute myocardial infarction treated with thrombolytic therapy. Thromb Res. 1996;82(4): 313-22.
- 19. Dahlbäck B. Protein S and C4b-binding protein: components involved in regulation of the protein C anticoagulant system. Thromb Haemost. 1991;66(1):49-61.
- 20. Gemmati D, Serino ML, Verzola I, Mari R, Moratelli S, Ballerini G. Resistance to activated protein C and low levels of protein S activity in nine thrombophilic families: a correct diagnosis. Blood Coagul Fibrinolysis. 1997;8(2):118-23.
- 21. Elias A, Bonfils S, Daoud-Elias M, Gauthier B, Sié P, Boccalon H, et al. Influence of long term oral anticoagulants upon prothrombin fragment 1+2, thrombin-antithrombin III complex and D-Dimer levels in patients affected by proximal deep vein thrombosis. Thromb Haemost. 1993;69(4):302-5.
- 22. Franco RF. Fisiologia da coagulação, anticoagulação e fibrinólise. Medicina (Ribeirão Preto). 2001;34(3/4):229-37.
- 23. Lewis SM, Bain BJ, Bates I. Hematologia prática de Dacie e Lewis. 9a ed. Porto Alegre: Artmed; 2006. 572p.
Datas de Publicação
-
Publicação nesta coleção
01 Fev 2012 -
Data do Fascículo
2010
Histórico
-
Recebido
27 Out 2009 -
Aceito
23 Nov 2009