Abstract
BACKGROUND: The development of therapies for sickle cell disease has received special attention, particularly those that reduce the polymerization of hemoglobin S. Hydroxyurea is a commonly used medication because it has the ability to raise levels of fetal hemoglobin, decrease the frequency of vaso-occlusive episodes and thus improve the clinical course of sickle cell disease patients. OBJECTIVE: To study hematological data and the clinical profile of sickle cell disease patients taking hydroxyurea in a regional blood center. METHODS: From the charts of 20 patients with sickle cell anemia, the clinical outcomes and a number of hematological variables were analyzed before and during treatment with hydroxyurea. RESULTS: The patients' ages ranged from 6 to 41 years old, most were dark skinned and there was a predominance of women. The main symptom that defined whether patients were prescribed hydroxyurea was painful crises followed by hospitalizations. During treatment with hydroxyurea there were significant increases in hemoglobin, fetal hemoglobin, mean corpuscular volume and mean corpuscular hemoglobin. The reticulocyte and white blood cell counts dropped significantly with treatment. A positive correlation was found between fetal hemoglobin and mean corpuscular volume before and during treatment. Additionally, a correlation was found between the white blood cell and reticulocyte counts before treatment with hydroxyurea. CONCLUSION: Most patients showed improvements with treatment as demonstrated by increases in hemoglobin, fetal hemoglobin and mean corpuscular volume, as well as by reductions in the reticulocyte and white blood cell counts. Clinically, more than 50% of patients had a significant reduction of events.
Anemia, sickle cell; Hydroxyurea; Fetal hemoglobin
ORIGINAL ARTICLE
Patients with sickle cell disease taking hydroxyurea in the Hemocentro Regional de Montes Claros
Fernanda Kelle de Souza SantosI; Caroline Nogueira MaiaII
IUniversidade Estadual de Montes Claros, Montes Claros, MG, Brazil
IIHemocentro Regional de Montes Claros - Fundação Hemominas, Montes Claros, MG, Brazil
Corresponding author Corresponding author: Fernanda Kelle de Souza Santos Rua Dr. Tupiniquins, 447 - Morrinhos 39400-456 - Montes Claros, MG, Brazil Phone: 55 38 3212-4869 fernandakss@yahoo.com.br
ABSTRACT
BACKGROUND: The development of therapies for sickle cell disease has received special attention, particularly those that reduce the polymerization of hemoglobin S. Hydroxyurea is a commonly used medication because it has the ability to raise levels of fetal hemoglobin, decrease the frequency of vaso-occlusive episodes and thus improve the clinical course of sickle cell disease patients.
OBJECTIVE: To study hematological data and the clinical profile of sickle cell disease patients taking hydroxyurea in a regional blood center.
METHODS: From the charts of 20 patients with sickle cell anemia, the clinical outcomes and a number of hematological variables were analyzed before and during treatment with hydroxyurea.
RESULTS: The patients' ages ranged from 6 to 41 years old, most were dark skinned and there was a predominance of women. The main symptom that defined whether patients were prescribed hydroxyurea was painful crises followed by hospitalizations. During treatment with hydroxyurea there were significant increases in hemoglobin, fetal hemoglobin, mean corpuscular volume and mean corpuscular hemoglobin. The reticulocyte and white blood cell counts dropped significantly with treatment. A positive correlation was found between fetal hemoglobin and mean corpuscular volume before and during treatment. Additionally, a correlation was found between the white blood cell and reticulocyte counts before treatment with hydroxyurea.
CONCLUSION: Most patients showed improvements with treatment as demonstrated by increases in hemoglobin, fetal hemoglobin and mean corpuscular volume, as well as by reductions in the reticulocyte and white blood cell counts. Clinically, more than 50% of patients had a significant reduction of events.
Keywords: Anemia, sickle cell; Hydroxyurea/therapeutic use; Fetal hemoglobin
Introduction
A point mutation in the sixth position of the β-globin polypeptide chain in chromosome 11 leads to the substitution of the amino acid, glutamic acid, for the amino acid valine, thereby forming hemoglobin S (Hb S). This is the factor that determines sickle cell disease (SCD).(1-3) Sickle cell anemia is a homozygous structural variant of the β chain. Additionally, abnormalities in the two β chains can occur causing double heterozygosis, which culminates in the production of two different modified β chains, as occurs in Hb SC and Hb SD. Finally, double heterozygous β-thalassemia and a β chain variant (sickle β-thalassemia: Hb Sβ+-thalassemia and Hb Sβº-thalassemia) can arise. Thus, sickle cell disease is the symptomatic form of the S gene. Despite the peculiarities that distinguish the different presentations and the varying degrees of severity, all these diseases have overlapping clinical and hematological manifestations in a epidemiologic spectrum.(3,4)
The homozygous form of this mutant hemoglobin under conditions of low oxygen tension undergoes a change in its molecular conformation triggering the formation of polymers that transform the classical shape of red blood cells to a new cellular structure in the shape of a sickle. As a result, there is cellular dehydration, loss of flexibility and greater adhesion to the endothelium. All these changes lead to vaso-occlusive episodes. These changes cause most of the signs and symptoms experienced by sickle cell anemia patients, such as painful crises, hemolytic episodes, lower limb ulcers, acute chest syndrome, splenic sequestration, priapism, aseptic necrosis of the femur, retinopathy, chronic renal failure, autosplenectomy and stroke.(5,6)
One available therapy is hydroxyurea (HU), an antineoplastic drug that increases the production of fetal hemoglobin (α2γ2).(7,8) A high level of fetal hemoglobin (Hb F) results in a compensatory reduction in Hb S to maintain the total concentration of intracellular hemoglobin. Moreover, Hb F tetramers dissociate into dimers that can form mixed hybrids with dimers of Hb S (α2βSγ), preventing polymerization, as neither the mixed hybrids, nor the tetramers of Hb F have affinity to the desoxi-Hb S polymer. Thus, there is greater survival of erythrocytes, resulting in a decrease in the frequency and severity of vaso-occlusive episodes suggesting a less severe course of disease during the administration of this drug.(9,10) There is evidence that the beneficial effect of HU is not only limited to inducing increases in Hb F, as many patients demonstrate clinical improvement before any significant increase in Hb F appears.(11,12)
This survey was conducted with the aim of studying the hematological data and the clinical profile of sickle cell disease patients taking HU in the Hemocentro Regional de Montes Claros.
Methods
This study was approved by the Research Ethics Committee of Hemominas (CEP Registration Nº 249). This is a retrospective study that analyzed the medical records of 20 sickle cell disease patients registered in the Hemocentro Regional de Montes Claros and who were prescribed HU. Data of the pre-medication period and after 2, 4, 8, 12 and 24 weeks of treatment, which corresponds to the period established by the Hemominas HU protocol to monitor therapeutic response, were collected from the patients' records. The inclusion criteria for this study were that during the 24 weeks of the HU protocol, no patient discontinued treatment due to myelotoxicity or for any other reason.
Variables such as type of sickle cell disease, gender, race and age were analyzed. The age of the patients corresponded to the time at start of medication.
One of the criteria for the prescription of HU, as defined in the protocol of the institution, is to present with specific disease-related clinical complications within the preceding year.
The presence or absence of clinical events was recorded during treatment. This evaluation of the patient's clinical status was made on days of consultations (2, 4, 8, 12 and 24 weeks).
The hematologic variables that were analyzed are: hemoglobin (Hb), red blood cell count (RBC), mean corpuscular volume (MCV), mean corpuscular hemoglobin (MCH), white blood cell count (WBC), platelet count, reticulocyte count and Hb F. The mean of three blood tests preceding the start of treatment was used for the premedication value. The WBC and the clinical status of patients were assessed before and during treatment.
Analysis of descriptive data was by measurement of the position and spread of variables before and during treatment. Analysis of variance (ANOVA) was used to evaluate whether there were significant differences between the means of each variable between study periods (before and during treatment). For Hb F, the time analyzed was before drug use and at 24 weeks of treatment because the figures for this variable were not reported in the patients' records for other periods.
The identification of significant differences between means compared two by two was achieved using the Tukey's test.
The Pearson correlation coefficient was used to investigate the relationship between continuous variables in the premedication period and during the 24-week treatment period. The Statistica 7.0 statistics program was used for statistical analysis with the level of significance being set at 5%.
Results
Of the 20 patients enrolled in this study, 11 (55%) were female and 9 (45%) were male. Six female and 12 male patients were aged 18 years or less. Four patients (20%) were white, 13 (65%) were mulatto, two (10%) were black and one (5%) did not state the color. The average age of the patients was 21.2 ± 9.7 years old (range: 6 to 41 years). All 20 patients had sickle cell anemia.
Figure 1 shows the different symptoms reported by patients before they started taking HU; 60% of patients were hospitalized for painful crises and in 30% other complications were associated with painful episodes.
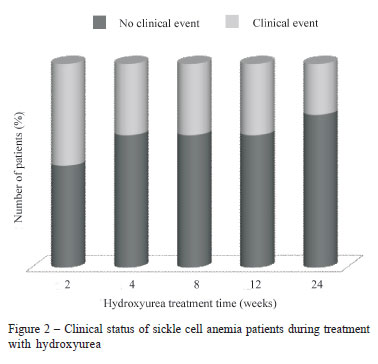
The majority of patients had no further clinical events by the end of the 24-week treatment regimen (50% of patients after two weeks of treatment, 65% after four weeks and 75% of patients after 24 weeks) as illustrated by Figure 2.
Descriptive/comparative analysis of the hematologic variables showed that the Hb, Hb F, MCV and MCH variables were significantly higher in the post-medication period (Table 1). However, in this period the WBC and reticulocyte counts dropped. On comparing before and during treatment, there were no significant differences in the RBC and platelet counts.
There was a significant variation only for Hb at 24 weeks of treatment. The level of MCH differed from the 8th week of treatment. The difference in the WBC count became significant at 12 weeks. The reticulocyte count and MCV differed immediately after the start of HU treatment (two weeks). The measurements of Hb F were significantly higher by 24 weeks of treatment.
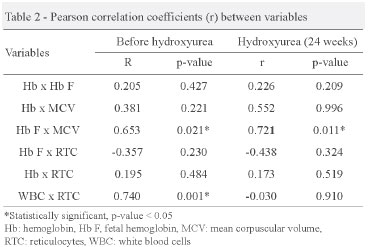
As illustrated in Table 2, there was no correlation between Hb and Hb F, and similarly, between Hb and MCV. The highest concentrations of Hb F are directly related to increased red blood cell volume before and after 24 weeks of HU treatment. The reticulocyte count had no correlation with the concentration of Hb F, suggesting that these are independent events. The highest Hb concentration in patients after HU treatment was not associated with a lower hemolytic process as seen by the reticulocyte count.
The correlation between the WBC and reticulocyte counts was strongly positive before treatment as shown in Figure 3.
As Figure 4 shows, the number of patients with clinical events during medication decreased, coinciding with the decrease in the WBC count.
Discussion
In the state of Minas Gerais in 2001, 87 of 128,326 newborns were carriers of the Hb S, 46 of whom had sickle cell anemia.(13) In another study also in Minas Gerais but in 2000,(14) one in every 1,246 newborns had the disease.
Frequent, but not exclusive to black individuals, sickle cell anemia affects between 0.1% and 0.3% of blacks in Brazil.(15) In our study, most patients with sickle cell anemia were mulattos although the criterion used by the blood center to define skin color is unclear.
Due to its high frequency and because this is an extremely painful and debilitating disease, the treatment of sickle cell anemia has been studied often .(10,16-18) HU is an oral medication that, as there are no reports of carcinogenic effects associated with its use, is the most promising treatment. This is an antineoplastic drug that inhibits enzyme ribonucleotide reductase that incorporates thymine in DNA.(19,20)
With inhibition, an interruption in the DNA synthesis of erythroid precursor cells occurs at later stages of differentiation. This temporary interruption of DNA synthesis would cause a brief suspension of hematopoiesis with the restoration of erythropoiesis occurring under hematopoietic stress. Thus, there is recruitment of young cells of the erythroid lineage which have retained the capacity to synthesize Hb F. These cells are stimulated to undergo rapid differentiation and maturation, causing the resulting erythroblasts to continuously express the γ-globin gene and consequently increase the Hb F expression.(21,22)
Based on evidence that HU is a nitric oxide (NO) donor, there is an increase in Hb F due to activation of the transcription of the γ-globin gene in cells of erythroid lineage and erythrocytes.(6,23) In fact, as reported by some researchers, patients with sickle cell anemia undergoing treatment with HU have elevated blood levels of NO.(22)
This study shows an increase of the concentration of Hb F after treatment with HU supporting the results of other researchers.(10,17)
The higher Hb concentrations after treatment did not result in less hemolysis, since there was no correlation between Hb and reticulocytes. No association between Hb F and Hb levels was demonstrated in this study either. Other researchers also described this finding.(17,24,25)
The increased Hb F level seems to be responsible for the increase in the volume of cells.(17) The MCV of patients in this study after starting treatment with HU increased significantly compared to before treatment. Thus, as there was a significant correlation between MCV and Hb F, it can be stated that in this study the increased Hb F is directly related to the increase of cell volume.
The endothelium is abnormal in sickle cell anemia as it has an increased expression of adhesion molecules that interact with the adhesion receptors expressed by sickled red blood cells, reticulocytes and white blood cells.(26)
Research has shown that vaso-occlusion is initiated by reticulocytes that temporarily seize deoxygenated mature red blood cells.(17) The localized ischemia and reperfusion process can cause a chronic state of inflammation. WBC are also involved in this process and high WBC counts have been considered a risk factor in the development of pain and infarctions.(17) Our results support this evidence, since, before medication, the WBC and reticulocyte counts were high and all patients were symptomatic with their main complications being painful crises. The reticulocyte count is positively linked to the WBC count before treatment, even though the same result was not observed after treatment.
A reduction in adhesion would facilitate the movement of RBC in circulation before the sickling process. Thus, the reduction in the number of reticulocytes after starting HU therapy may improve the clinical course of sickle cell anemia patients regardless of induction of the Hb F synthesis.(17) In this study, the reticulocyte count dropped and there was an improvement in clinical status in 50% of patients within the first two weeks of starting HU treatment. However, as the only data available for Hb F refer to after six months of treatment, it is not possible to define the role of Hb F in this discussion. At six months of medication there was no correlation between the reticulocyte count and Hb F.
A drop in the reticulocyte count was not related to Hb or to the white blood cell count after starting treatment with HU. This research cannot define any correlation between these variables however according to some studies,(11,12,27) the increase in Hb F due to HU varies among patients. Many of them show clinical improvement before showing a significant increase in Hb F levels suggesting that HU may benefit patients by other mechanisms that can result from drug-induced myelosuppression and which may explain the decrease in the reticulocyte count in patients in this study.
The majority of patients showed improvement with the use of HU. A fact demonstrated by the increase in concentration of Hb, Hb F and MCV, as well as by a drop in reticulocyte and white blood cell counts and, clinically, the significant reduction in clinical events.
Acknowledgments
The authors wish to thank Leonardo Silva Aguiar Junior for his help in this study.
Submitted: 8/25/2010
Accepted: 10/25/2010
Conflict-of-interest disclosure: The authors declare no competing financial interest
- 1. Shiu Y, Udden MM, Mclntire LV. Perfusion with sickle erytrocytes up-regulates ICAM-1 and VCAM-1 gene expression in cultured human endothelial cells. Blood. 2000;95(10):3232-41.
- 2. Rosse WF, Narla M, Petz LD, Steinberg MH. New views of sickle cell disease, pathophysiology and treatment. Hematology Am Soc Hematol Educ Program. 2000:2-17.
- 3. Sonati MF, Costa FF. Genética das doenças hematológicas: as hemoglobinopatias hereditárias. J Pediatr. 2008;84(Supl 4): S40-51.
- 4. Zago MA, Pinto ACS. Fisiopatologia das doenças falciformes: da mutação genética à insuficiência de múltiplos órgãos. Rev Bras Hematol Hemoter. 2007;29(3):207-14.
- 5. Costa FF. Anemia Falciforme. In: Zago MA, Falcão RP, Paquini R. Hematologia, Fundamentos e prática. São Paulo: Atheneu; 2004. p. 289-308.
- 6. Nahavandi M, Tavakkoli F, Wyche MQ, Perlin E, Winter WP, Castro O. Nitric oxide cyclic GMP levels in sickle cell patients receiving hydroxyurea. Br J Haematol. 2002;119(3):855-7.
- 7. Rodgers GP, Dover GJ, Noguchi CT, Schechter AN, Nienhuis AW. Hematologic responses of patients with sickle cell disease to treatment with hydroxyurea. N Engl J Med. 1990;322(15):1037-45.
- 8. Charache S, Terrin ML, Moore MD, Dover GJ, Barton FB, Eckert SV, et al. Effects of hydroxyurea on the frequency of painful crises in sickle cell anemia. N Engl J Med. 1995;332(20):1317-22.
- 9. Davies SC, Roberts-Harewood M. Blood transfusion in sickle cell disease. Blood Rev.1997;11(2):57-71.
- 10. Bandeira FM, Peres JC, Carvalho EJ, Bezerra I, Araújo AS, Mello MR, et al. Hidroxiuréia em pacientes com síndromes falciformes acompanhados no Hospital Hemope, Recife-PE. Rev Bras Hematol Hemoter. 2004;26(3):189-94.
- 11. Huang J, Kim-Shapiro DB, King SB. Catalase-mediated nitric oxide formation from hydroxyurea. J Med Chem. 2004;47(14): 3495-501.
- 12. Halsey C, Roberts IA. The role of hydroxyurea in sickle cell disease. Br J Haematol. 2003;120(2):177-86.
- 13. Paixão MC, Cunha-Ferraz MH, Januário JN, Viana MB, Lima JM. Reliability of isoelectrofocusing for the detection of Hb S, Hb C, and Hb D in a pioneering population-based program of the newborn screening in Brazil. Hemoglobin. 2001;25(3):297-303.
- 14. Serjeant GR. Screening for sickle-cell disease in Brazil. Lancet. 2000;356(9224):168-9.
- 15. Ramalho AS, Magna LA, Paiva e Silva RB. A Portaria nş 822/01 do Ministério da Saúde e as peculiaridades das hemoglobinopatias em saúde pública no Brasil. Cad Saúde Pública. 2003;19(4):1195-9.
- 16. Hoppe C, Vichinsky E, Quirolo K, van Warmerdam J, Allen K, Styles L.Use of hydroxyurea in children ages 2 to 5 years with sickle cell disease. J Pediatr Hemat Oncol. 2000;22:330-4.
- 17. Borba R, Lima CS, Grotto HZ. Reticulocyte parameters and hemoglobin F production in sickle cell disease patients undergoing hydroxyurea therapy. J Clin Lab Anal. 2003;17(2):66-72.
- 18. Gambero S, Canalli AA, Traina F, Albuquerque DM, Saad ST, Costa FF, et al. Therapy with hydroxyurea is associated reduced adhesion molecule gene and protein expression in sickle red cells with a concomitant reduction in adhesive properties. Eur J Haematol. 2007;78(4):144-51.
- 19. Charache S, Dover GJ, Moore RD, Eckert S, Ballas SK, Koshy M, et al. Hydroxyurea: effects on hemoglobin F production in patients with sickle cell anemia. Blood. 1992;79(10):2555-65.
- 20. de Lima PD, Cardoso PC, Khayat AS, Bahia Mde O, Burbano RR. Evaluation of the mutagenic activity of hydroxyurea on the G1S-G2 phases of the cell cycle: an in vitro study. Genet Mol Res. 2003;2(3):328-33.
- 21. Kaufman RE. Hydroxyurea: specific therapy for sickle cell anemia? Blood. 1992;79(10):2503-6.
- 22. Gladwin MT, Schechter AN. Nitric oxide therapy in sickle cell disease. Semin Hematol. 2001;38(4):333-42.
- 23. Jiang J, Jordan SJ, Barr DP, Gunther MR, Maeda H, Manson RP. In vivo production of nitric oxide in rats after administration of hydroxyurea. Mol Pharmacol. 1997;52(6):1081-6.
- 24. Silva LB, Gonçalves RP, Martins MF. Estudo da correlação entre os níveis de hemoglobina fetal e o prognóstico dos pacientes com anemia falciforme. Rev Bras Hematol Hemoter. 2009;31(6):417-20.
- 25. Adorno EV, Zanette A, Lyra I, Seixas MO, Reis MG, Gonçalves MS. Clinical and molecular characteristics of sickle cell anemia in the northeast of Brazil. Genet Mol Biol. 2008;31:621-5.
- 26. Chiang EY, Frenette PS. Sickle cell vaso-occlusion. Hematol Oncol Clin North Am. 2005;19(5):771-84.
- 27. De Franceschi L, Corrocher R. Established and experimental treatments for sickle cell disease. Haematologica. 2004;89(3): 348-56.
Corresponding author:
Publication Dates
-
Publication in this collection
13 June 2011 -
Date of issue
2011
History
-
Received
25 Aug 2010 -
Accepted
25 Oct 2010