Abstract
The glucose-1-phosphatase encoding gene (agp) of Pantoea agglomerans was sequenced and heterologously expressed in Escherichia coli. The enzyme showed very high homology to periplasmatic glucose-1-phosphatases of other members of the Enterobacteriaceae family. It was isolated from transformed Escherichia coli cells in a single step in high yields (32.3 ± 1.2 mg per litre of culture) by Ni-NT agarose affinity chromatography to >95% purity as calculated from specific activity determinations. The purified glucose-1-phosphatase was entrapped in alginate beads with an entrapment efficiency of >80%. Temperature stability was enhanced as a consequence of entrapment, whereas pH dependence of enzyme activity was not affected. Maximum catalytic activity of entrapped glucose-1-phosphatase was found at 70°C, whereas the free enzyme exhibited maximal activity at 60°C. A single pH optimum at pH 4.5 was determined for the free and the entrapped enzyme. Kinetic parameters for the hydrolysis of sodium phytate were found to be affected by entrapment. They were determined to be K M = 0.84 mmol l-1 and k cat = 8 s-1 at pH 4.5 and 37°C for the entrapped glucose-1-phosphatase and K M = 0.35 mmol l-1 and k cat = 20.5 s-1 for the free enzyme. Complete conversion of phytate into one single myo-inositol pentakisphosphate isomer, identified as D-myo-inositol(1,2,4,5,6)pentakis-phosphate, was shown to be feasible by using the enzyme-loaded alginate beads in batch operations. The entrapped enzyme showed a high operational stability by retaining almost full activity even after ten uses.
Entrapment; glucose-1-phosphatase; myo-inositol pentakisphosphate Pantoea agglomerans; phytase
AGRICULTURE, AGRIBUSINESS AND BIOTECHNOLOGY
Production of D-myo-inositol(1,2,4,5,6)pentakisphosphate using alginate-entrapped recombinant Pantoea agglomerans glucose-1-phosphatase
Ralf GreinerI, * * Author for correspondence ; SajidanII
ICentre for Molecular Biology; Federal Research Centre for Nutrition and Food; Haid-und-Neu-Strasse 9; ralf.greiner@bfel.de; D-76131 Karlsruhe - Germany
IIDepartment of Biology; Sebelas Maret University Surakarta; Jl. Ir. Sutami 36A, 57126 Solo - Indonesia
ABSTRACT
The glucose-1-phosphatase encoding gene (agp) of Pantoea agglomerans was sequenced and heterologously expressed in Escherichia coli. The enzyme showed very high homology to periplasmatic glucose-1-phosphatases of other members of the Enterobacteriaceae family. It was isolated from transformed Escherichia coli cells in a single step in high yields (32.3 ± 1.2 mg per litre of culture) by Ni-NT agarose affinity chromatography to >95% purity as calculated from specific activity determinations. The purified glucose-1-phosphatase was entrapped in alginate beads with an entrapment efficiency of >80%. Temperature stability was enhanced as a consequence of entrapment, whereas pH dependence of enzyme activity was not affected. Maximum catalytic activity of entrapped glucose-1-phosphatase was found at 70°C, whereas the free enzyme exhibited maximal activity at 60°C. A single pH optimum at pH 4.5 was determined for the free and the entrapped enzyme. Kinetic parameters for the hydrolysis of sodium phytate were found to be affected by entrapment. They were determined to be KM = 0.84 mmol l-1 and kcat = 8 s-1 at pH 4.5 and 37°C for the entrapped glucose-1-phosphatase and KM = 0.35 mmol l-1 and kcat = 20.5 s-1 for the free enzyme. Complete conversion of phytate into one single myo-inositol pentakisphosphate isomer, identified as D-myo-inositol(1,2,4,5,6)pentakis-phosphate, was shown to be feasible by using the enzyme-loaded alginate beads in batch operations. The entrapped enzyme showed a high operational stability by retaining almost full activity even after ten uses.
Key words: Entrapment, glucose-1-phosphatase, myo-inositol pentakisphosphate Pantoea agglomerans, phytase
INTRODUCTION
The major interest in individual dephosphorylation products of phytate [ myo-inositol(1,2,3,4,5,6)hexakisphosphate] results from physiological effects which have been attributed to their action. About 35 of the 63 possible myo-inositol phosphates were identified in different types of cells (Shears, 1998). Depending on cell type, myo-inositol phosphates were linked with a range of cellular function, among others secretion, contraction, cell division, cell differentiation and cell death. It has been recently reported that highly negatively charged myo-inositol polyphosphates can cross the plasma membrane and be internalised by cells (Ferry et al., 2002; Maffucci et al., 2005). Therefore, every extracellular myo-inositol phosphate may possibly affect cellular functions. D-myo-inositol(1,2,6)trisphosphate, for example, has been studied in respect to prevention of diabetes complications and treatment of chronic inflammations (Carrington et al., 1993; Claxon et al., 1990). Because of its antiangiogenic and antitumour effects myo-inositol(1,3,4,5,6)pentakisphosphate was suggested as a promising compound for anticancer therapeutic strategies (Maffucci et al., 2005). In addition, myo-inositol phosphates containing the 1,2,3-trisphosphate grouping may reduce the likelihood for iron-catalysed lipid peroxidation (Phillippy and Graf, 1997), because iron was demonstrated to bind to this grouping in such a way that it cannot catalyse the formation of hydroxyl free radicals (Hawkins et al., 1993).
The use of non-enzymatic phytate hydrolysis or chemical synthesis to provide access to individual myo-inositol phosphate isomers is often associated with high expense and is not very efficient. The direct production of the desired isomer using enzymes proves to be far more effective. Phytate was reported to be dephosphorylated by a special class of phosphatases regio- and stereo-selective in a stepwise manner by producing, in general, only one myo-inositol pentakis-, tetrakis-, tris-, bis-, and mono-phosphate isomer (Konietzny and Greiner, 2002). Separation of the different partially phosphorylated myo-inositol phosphate esters was shown to be easily feasible by anion-exchange chromatography (Greiner et al., 2002). Recently a unique myo-inositol phosphate phosphatase activity was reported for agp-encoded acid glucose-1-phosphatases of the Gram-negative Enterobacteriaceae family members Escherichia coli (Cottrill et al., 2002), Pantoea agglomerans (Greiner, 2004a), and Enterobacter cloacae (Herter et al., 2006). These enzymes cleave only the D-3 phosphate from phytate and no further hydrolysis takes place. This provides access to sufficient amount of D-myo-inositol(1,2,4,5,6)pentakisphosphate for physiological studies or as a substrate for the enzymatic production of further partially phosphorylated myo-inositol phosphate isomers.
In this communication we report on the production of D-myo-inositol(1,2,4,5,6)pentakisphosphate using alginate-entrapped recombinant Pantoea agglomerans glucose-1-phosphatase. Furthermore, this paper describes the nucleotide sequence of the glucose-1-phosphatase encoding gene, the amino acid sequence of the enzyme and its heterologous expression in Escherichia coli.
MATERIALS AND METHODS
Chemicals
Pantoea agglomerans was isolated from an Indonesian rice field (Oryza sativa var. IR64) and identified by biochemical and phylogenetic analysis (Sajidan, unpublished). Phytic acid, as a dodecasodium salt, was purchased from Aldrich (Steinheim, Germany), Ultrasep ES 100 RP18 from Bischoff (Leonberg, Germany) and high performance ion chromatography Carbo-Pac PA-100 column from Dionex (Sunnyvale, CA, USA).
Cloning of the glucose-1-phosphatase encoding gene and nucleotide sequence analysis
The glucose-1-phosphatase encoding gene without signal peptide was amplified from Pantoea agglomerans DNA using the forward NdeI-linked primer IP5f (5'-CATATGCAAGAGACGCCGGA AGGG-3') and the reverse HindIII-linked primer IP5r (5'-AAGCTTCTTCGCCGCGTTATT-3') by PCR. Both primers were designed according to the nucleotide sequence of the glucose-1-phosphatase of E. coli and Enterobacter cloacae, two further members of the Enterobacteriaceae family. The amplicon was cloned into pGEM-T (Promega). A DNA fragment harbouring the glucose-1-phosphatase encoding gene was obtained by digestion of the resulting plasmid with NdeI and HindIII, and purification of the fragment from an agarose gel was perfomed using the QIAquick Gel Extraction Kit (Qiagen). The purified DNA fragment was inserted between the NdeI and HindIII sites of pET-22b(+) (Novagen). The resulting plasmid, designated pIP5, was transferred into E. coli BL21 (DE3) (Invitrogen). The insert was sequenced in both directions using standard primers (T7 promoter primer (Novagen): 5'-TAAT ACGACTCACTATAGGG-3'; T7 terminator primer (Novagen): 5'-GCTAGTTATTGCTCAGC GG-3') and specific primers (SEQ1, forward 367-384: 5'-ATGGATCCGACCTTCAAT-3'; SEQ2, forward 805-822: 5'-ATCGATAAAACCCTGGT-3'; SEQ3, reverse 429-412: 5'-AATCGCTTGTTC ACGGAA-3'; SEQ4, reverse 903-886: 5'-ATCCA GCGCCGTGAGCAG-3') with an automatic sequencing system (ALF, Pharmacia).
Recombinant enzyme production
E. coli BL21 (DE3) cells harbouring pIP5 were cultured at 37°C in LB medium containing ampicillin (100 µ g ml-1). At OD600=0.6-0.7, glucose-1-phosphatase expression was induced by addition of isopropyl-b-D-thiogalactopyranoside (IPTG, final concentration 1 mM). After further 6 h at 37°C, the cells were harvested by centrifugation at 7,000g and 4°C for 15 min and resuspended in 50 mM sodium acetate buffer, pH 4.5. Bacteria were lysed by the following procedure: 1. Cells were frozen at -80°C for 10 min and thawed at room temperature for 20 min; freezing and thawing was repeated twice. 2. Sonication for 1 min; sonication was repeated twice. Cell debris was removed by centrifugation at 10,000g and 4°C for 30 min and glucose-1-phosphatase was purified from the clear supernatant by affinity chromatography using Ni-NT agarose (Qiagen). To determine the enzymatic properties of the recombinant glucose-1-phosphatase, the enzyme was purified from the enzyme preparation obtained from Ni-NT agarose affinity chromatography to apparent homogeneity as described previously (Greiner, 2004b).
Assay of enzyme activity and protein estimation
Enzyme activity measurements were carried out at 37°C. The enzymatic reactions were started by the addition of 10 µ l enzyme to the assay mixtures. The incubation mixture consisted of 350 µ l 0.1 M sodium acetate, pH 4.5 containing either 1.5 µ mol sodium phytate or 1.5 µ mol glucose-1-phosphate. After incubating for 30 min at 37°C, the liberated phosphate was measured according to the ammonium molybdate method (Heinonen and Lahti, 1981) with some modifications. 1.5 ml of a freshly prepared solution of acetone/5 N H2SO4/10 mM ammonium molybdate (2:1:1 v/v) and then 100 µ L 1.0 M citric acid were added to the assay mixture for enzyme activity determination or to 400 µ l of the incubation mixtures of the alginate reactor. Any cloudiness was removed by centrifugation prior to measurement of absorbance at 355 nm. To calculate enzyme activity, a calibration curve was produced over the range of 5-600 nmol phosphate ( = 8.7 cm2 nmol-1). Activity (units) was expressed as 1 µ mol phosphate liberated per min. Blanks were run by addition of the ammonium molybdate solution prior to adding the enzyme to the assay mixture.
Total protein concentration was determined by the Coomassie blue G-250 dye-binding assay using bovine serum albumin as a standard (Bradford, 1976).
Preparation of enzyme-loaded alginate beads and production of D-myo-inositol(1,2,4,5,6)pentakis-phosphate
20 mg of the purified glucose-1-phosphatase were dissolved in 20 ml of a 2% (w/v) Na-alginate solution. The beads were prepared by dropping the sodium alginate solution containing glucose-1-phosphatase from a syringe with a 0.5 inch flat-tip needle to a magnetically stirred 0.05 M calcium chloride solution (200 ml) at a rate of 2 ml min-1. The beads were collected by decanting the calcium chloride solution, washed thoroughly with deionised water and transferred into 50 mM sodium acetate buffer, pH 4.5 (180 ml). The mixtures were incubated in a shaking water bath at 40°C for 30 min. Thereafter, 20 ml of a sodium phytate solution (100-500 mM) in the same buffer pre-heated to 40°C were added and mixed thoroughly. The reactor was run at 40°C in batch operations. From the incubation mixtures, 1 ml samples were removed periodically and assayed for myo-inositol phosphates and phosphate.
Entrapment efficiency was determined by dissolving the enzyme-loaded beads in 20 mM Tris-HCl buffer, pH 8.0 within 2 h. The resulting solution was cleared by centrifugation at 5.000g for 20 min and the supernatant was assayed for phytase and glucose-1-phosphatase activity. Control experiments were run to quantify the effect of pH on enzyme stability. Entrapment efficiency was defined as the ratio of total enzyme activity after dissolving the beads and total enzyme activity used to prepare the beads.
To study the effect of pH on phytate degradation by the free and the entrapped enzyme, the following buffers were used in the above described standard assays: pH 2.0-3.0, 50 mM glycine-HCl; pH 3.5-6.0, 50 mM sodium acetate-HCl; pH 6.0-7.0, 50 mM Tris-acetic acid; pH 8.0, 50 mM Tris-HCl. The temperature dependence of phytate degradation by the free and the entrapped enzyme was determined in the range from 25 to 80°C using the above described standard assays at the given temperature.
In order to check thermal stability the alginate reactor was run in 20 mM sodium acetate buffer, pH 4.5 containing 10 mM sodium phytate at different temperatures for 30 min. The beads were collected by decanting the buffer, washed twice with deionised water and transferred into 200 ml of 50 mM sodium acetate buffer, pH 4.5 containing 10 mM sodium phytate pre-heated to 37°C. The mixtures were incubated at 37°C for another hour.
Kinetic experiments were carried out at 37°C in 50 mM sodium acetate buffer, pH 4.5 containing sodium phytate in a serial dilution of a concentrated stock solution (10 mM). The kinetic constants (Km, vmax) were calculated from Lineweaver-Burk plots of the data. For calculation of kcat a molecular mass of 42 kDa for the Pantoea agglomerans glucose-1-phosphatase was used (Greiner, 2004b).
Myo-Inositol phosphate analysis
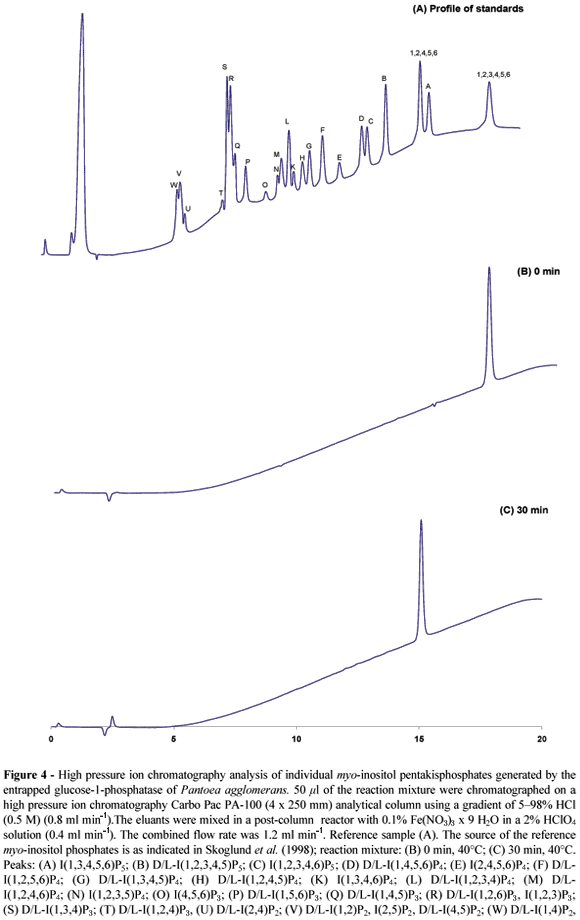
20 µ l of samples were chromatographed on a high performance liquid chromatography Ultrasep ES 100 RP18 (2 x 250 mm) column. The column was run at 45°C and 0.2 ml min-1 with an eluant consisting of formic acid:methanol:water:TBAH (tetrabutylammonium hydroxide) (44:56:5:1.5 v/v), pH 4.25 (Sandberg and Ahderinne, 1986). A mixture of the individual myo-inositol phosphate esters (myo-inositol trisphosphate myo-inositol hexakisphosphate) was used as a standard. To identify the enzymatically formed phytate degradation products, 50 µ l of the samples were chromatographed on a high performance ion chromatography system using a Carbo Pac PA-100 (4 x 250 mm) analytical column and a gradient of 598% HCl (0.5 M, 0.8 ml min-1) (Skoglund et al., 1998). The eluants were mixed in a post-column reactor with 0.1% Fe(NO3)3 x 9 H2O in a 2% HClO4 solution (0.4 ml min-1) (Phillippy and Bland, 1988). The combined flow rate was 1.2 ml min-1.
RESULTS AND DISCUSSION
Nucleotide sequence analysis of the Pantoea agglomerans glucose-1-phosphatase encoding gene (agp) and its heterologous expression in Escherichia coli
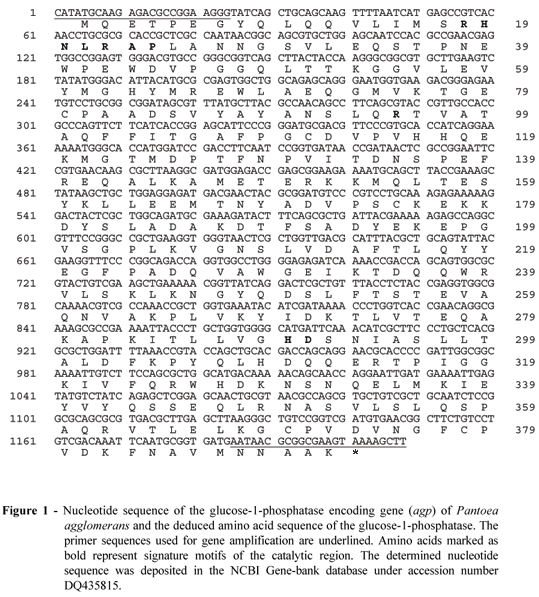
Sequence analysis of the Pantoea agglomerans glucose-1-phosphatase encoding gene (Fig. 1) revealed an open reading frame with 92% nucleotide identity and 96% amino acid identity to Enterobacter cloacae glucose-1-phosphatase. Furthermore, high homology was found to periplasmatic glucose-1-phosphatases of other members of the Enterobacteriaceae family, such as Shigella sonnei (81% nucleotide identity and 82% amino acid identity), Shigella flexneri (81% nucleotide identity and 82% amino acid identity), Escherichia coli (81% nucleotide identity and 82% amino acid identity), Shigella dysenteriae (81% nucleotide identity and 81% amino acid identity), and Salmonella enterica (78% nucleotide identity and 80% amino acid identity). The mature Pantoea agglomerans glucose-1-phosphatase consists of 391 amino acids and its molecular mass was calculated to be 43,871 Da. This is in excellent agreement with the molecular mass of 43,000 ± 1,000 Da estimated by gel filtration and 42,500 ± 2,000 Da by SDS gel electrophoresis (data not shown).
Pantoea agglomerans glucose-1-phosphatase belongs to the family of histidine acid phosphatases. The enzyme contains an N-terminal RHNXRXP sequence motif (residues 18-24) as well as a R (residue 95) and a C-terminal HD motif (residues 289-290) (Fig. 1), which are common for glucose-1-phosphatases from various members of the Enterobacteriaceae family (Herter et al., 2006; Lee et al., 2003). The catalytic region of these glucose-1-phosphatases differs slightly from other histidine acid phosphatases (van Etten et al., 1991), since an Asn replaces the Gly in RHGXRXP. However, the replacement is thought to be of minimal influence because the side chain of Asn-20 points to the opposite direction of the active site and the position of the Asn-20 peptide oxygen is still conserved (Lee The enzyme was primarily produced as an intracellular enzyme and it could be isolated from the crude extract in a single step in high yields (32.3 ± 1.2 mg per litre of culture) by Ni-NT agarose affinity chromatography to >95% purity as calculated from specific activity determinations. Because the recombinant glucose 1-phosphatase was shown to exhibit the same molecular mass (43,000 ± 1,000 Da estimated by gel filtration and 42,500 ± 2,000 Da by SDS gel electrophoresis) compared to the wild-type enzyme (42,000 ± 1,500 Da and 41,500 ± 2,500 Da respectively (Greiner, 2004b)), it could be concluded that the full-length et al., 2003).
Enzymes used in biotechnological applications should be effective, stable to resist inactivation by processing conditions and storage, and cheap to produce. As a first step to an economically competitive production system for the Pantoea agglomerans glucose-1-phosphatase, the encoding gene was heterologously expressed in Escherichia coli. E. coli BL21 (DE3) cells transformed with plasmid pIP5 showed significant higher glucose-1-phosphatase and phytase activity than those transformed with the empty pET-22b(+) plasmid (Table 1). The difference in both activities has to be attributed to the expression of one single enzyme by E. coli BL21 (DE3) harbouring pIP5 that is the Pantoea agglomerans glucose-1-phosphatase. This enzyme has been demonstrated recently to dephosphorylate glucose-1-phosphate and phytate (Greiner, 2004b). Activity towards glucose-1-phosphate has been shown to be 4.8-fold higher than activity towards phytate. By taking only the enzyme activities that were due to the transformation with pIP5 into account, the same value for the ratio of glucose-1-phosphatase to phytase activity was obtained.
protein has been overexpressed. Specific activities of the Ni-NT agarose purified enzyme towards phytate and glucose-1-phosphate as substrates were determined to 22.1 ± 0.3 U mg-1 and 106.7 ± 1.4 U mg-1, respectively. The corresponding values for the apparently pure recombinant enzyme were determined to be 22.7 U mg-1 and 108.2 U mg-1, respectively (Greiner, 2004b). These values are not significantly different from those of the wild-type enzyme (23.0 U mg-1 and 110.6 U mg-1, respectively (Greiner, 2004b)). In addition, the recombinant glucose-1-phosphatase did not differ significantly from the corresponding wild-type enzyme in respect to enzymatic properties such as pH- and temperature profile for phytate and glucose-1-phosphate hydrolysis, pH- and temperature stability, kinetic constants, substrate specificity, and energy of activation (data not shown). The almost identical enzymatic properties of the recombinant glucose-1-phosphatase and the corresponding wild-type enzyme further confirm the assumption that the full- length enzyme was overexpressed.
Preparation of enzyme-loaded alginate beads and entrapment efficiency
Immobilisation is one possibility to improve processing as well as storage stability of enzymes. The overwhelming majority of enzyme immobilisation methods can be classified into four main categories: matrix entrapment, micro-encapsulation, adsorption and covalent binding. Unlike adsorption and covalent bonding methods, most polymerisation reactions that cause cross-linking and gel formation in entrapment methods do not directly involve the formation of bonds between the support material and the enzyme. There are reports that these bonds change the conformation of the enzyme and modify the enzyme properties (Cao, 2005). Alginate is a biodegradable copolymer of 1,4-linked b-D-mannuronic acid and a-L-guluronic acid, which can be gelled by multivalent cations such as calcium ions. Alginate immobilisation is widely used for micro-organisms and enzymes (Park and Chang, 2000). Enzyme-loaded alginate beads are simple to prepare, cheap and offer good mechanical strength.
The recombinant Pantoea agglomerans glucose-1-phosphatase was entrapped in alginate beads with high entrapment efficiency. Considering that the free enzyme lost less than 5% phosphatase activity within 2 h in 20 mM Tris-HCl buffer, pH 8.0, entrapment efficiency was determined to be 82.3 ± 1.2% (n=3) for phytase activity and 81.7 ± 0.9% (n=3) for glucose-1-phosphatase activity. Similar entrapment efficiencies have been reported for papain (Sankalia et al., 2004) and a-amylase (Dey et al., 2003).
Characterisation of the alginate reactor
Total phytase activity of the enzyme-loaded alginate reactor was determined at various temperatures. As expected, phytate dephosphorylation increased while raising temperature. The maximal activity at 30°C, 40°C, 50°C and 60°C is only 35%, 40%, 46% and 71% of the free-form activity at the given temperature. Thus, compared to the free-form activity, reaction rate of the alginate reactor was markedly reduced (30-75% depending on temperature). The mass transfer consideration indicates that particle shape, particle size, pore size, enzyme loading per particle, and substrate flow rate can affect the reaction rate. The mass transfer limitation arising from entrapment is manifested in an increase in the apparent value of the Michaelis-Menten constant (Km). Thus, the value of this constant as compared to the corresponding intrinsic value of the solubilised enzymes is often recognised as a good indicator of the extent of mass transfer resistance. Km for the entrapped glucose-1-phosphatase was estimated to be 0.84 ± 0.05 mM at pH 4.5 and 37°C with phytate as substrate. Maximal hydrolysis rate and turnover number were computed to be 0.9 ± 0.02 U ml-1 and 8.0 ± 0.2 s-1, respectively. The corresponding values for the free enzyme were reported to be 0.35 mM (Km) and 20.5 s-1 (kcat) (Greiner, 2004b). Therefore it is clearly shown, that transfer limitation is responsible for the reduced reaction rate of the entrapped enzyme. In addition, the catalytic turnover number (kcat) dropped drastically as a consequence of entrapment. Besides the mass transfer resistance to substrates and products, another problem associated with the entrapment method of immobilisation is the leakage of enzymes. Because no significant loss of activity was observed even after 10 repeated batch operations, leakage of glucose-1-phosphatase from the alginate beads is thought to be of minor importance.
Phytate dephosphorylation by the alginate reactor was analysed from pH 3.0 to 7.0 using a variety of buffers (Fig. 2). The pH dependence of phytase activity was not affected by entrapment. Therefore, immobilised Pantoea agglomerans glucose-1-phosphatase exhibited the same single pH optimum at pH 4.5 as its soluble counterpart and was virtually inactive above pH 7.0 and below pH 3.0.
Temperature dependence of the entrapped phytase activity was conducted from 25 to 80°C (Fig. 3). Maximum catalytic activity of the entrapped glucose-1-phosphatase was found at 70°C, whereas the free enzyme exhibited maximal activity at 60°C. Energy of activation for phytate hydrolysis was calculated from Arrhenius plots to 53.7 kJ mol-1. In comparison to the free recombinant glucose-1-phosphatase, as exemplified by its resistance to heat denaturation at 70 and 80°C. After a period of 1.0 h at 70°C the free recombinant glucose-1-phosphatase showed no remaining activity, whereas the entrapped enzyme did not lose any activity at 70°C and at 80°C only 25% of the initial activity was lost.
Stability of the alginate reactor was investigated at 40°C and pH 4.5.
Even after 10 consecutive batch operations no significant loss of enzymatic activity (<5%) was observed. Storage stability of the entrapped glucose-1-phosphatase was investigated at 4°C. There was no significant loss of activity when the enzyme-loaded alginate beads were stored for a period of three month.
Production of D-myo-inositol(1,2,4,5,6)pentakis-phosphate
In order to assess the catalytic capability of the alginate reactor for production of D-myo-inositol(1,2,4,5,6)pentakisphosphate a buffered phytate solution (10-50 mM) was incubated at 40°C in the presence of the enzyme-loaded alginate beads. Phytate and its hydrolysis products in the. reaction mixture were quantified by high performance ion pair chromatography. This method allows the separation of the individual myo-inositol phosphate species down to myo-inositol trisphosphate. Within 30 minutes at 40°C maximal phytate dephosphorylation was determined to be about 27 µ mol ml-1 without any inhibition by phytate and phosphate (Table 2).
Prolonged incubation times resulted in a complete degradation of phytate. No myo-inositol phosphate esters with less than five phosphate residues were detected in the reaction mixtures (Fig. 4). Thus, complete conversion of phytate into myo-inositol pentakisphosphate was feasible using the alginate reactor. High-pressure ion chromatography analysis established D-myo-inositol(1,2,4,5,6)pentakisphos-phate as the sole product of phytate dephosphorylation (Fig. 4).
Since there is increasing interest in the physiological properties of myo-inositol phosphate isomers, their preparation in pure form and sufficient quantity is required. The developed alginate reactor will contribute to making pure partially phosphorylated myo-inositol phosphate isomers available. Besides D-myo-inositol(1,2,4,5,6)pentakisphosphate, D-myo-inositol(1,2,4,5)tetrakisphosphate and D-myo-inositol(1,2,5,6)tetrakisphosphate could be specifically obtained by using D-myo-inositol(1,2,3,4,5)pentakisphosphate and D-myo-inositol(1,2,3,5,6)pentakisphosphate, respectively, as substrates for the Pantoea agglomerans glucose-1-phosphatase (Greiner, 2004a). To reduced operational costs, the alginate reactor should be operated continuously. The main advantage of a continuous process over a batch process is the ease of automation and control.
Entrapment of Pantoea agglomerans glucose-1-phosphatase was shown to yield longer enzyme lifetime and improved enzyme stability. The entrapped enzyme exhibited the same unique myo-inositol phosphate phosphatase activity as its soluble counterpart. The enzyme hydrolysed only the D-3 phosphate from phytate, producing D-myo-inositol(1,2,4,5,6)pentakisphosphate as the sole product of phytate dephosphorylation. This myo-inositol pentakisphosphate will be used for physiological studies or as a substrate for the enzymatic production of further partially phosphorylated myo-inositol phosphate isomers.
REFERENCES
Bradford, M. (1976), A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry, 72, 248-254.
Cao, L. (2005), Immobilised enzymes: science or art? Current Opinion in Chemical Biology, 9, 217-226.
Claxon, A.; Morris, C.; Blake, D.; Siren, M.; Halliwell, B.; Gustafsson, T.; Löfkvist, B. and Bergelin, I. (1990), The anti-inflammatory effects of D-myo-inositol-1.2,6-trisphosphate (PP56) on animal models of inflammation. Agents Actions, 29, 68-70.
Cottrill, M.A.; Golovan, S.P.; Phillips, J.P. and Forsberg, C.W. (2002), Inositol phosphatase activity of the Escherichia coli agp-encoded acid glucose-1-phosphatase. Canadian Journal of Microbiology, 48, 801-809.
Dey, G.; Singh, B. and Banerjee, R. (2003), Immobilization of a-amylase produced by Bacillus circulans GRS 313. Brazilian Archives of Biology and Technology, 46, 167-176.
Ferry, S.; Matsuda, M.; Yoshida, H. and Hirata, M. (2002), Inositol hexakisphosphate blocks tumor cell growth by activating apoptotic machinery as well as by inhibiting the Akt/NFkB-mediated cell survival pathway. Carcinogenesis, 23, 2031-2041.
Greiner, R. (2004a), Degradation of myo-inositol hexakisphosphate by a phytate-degrading enzyme from Pantoea agglomerans. The Protein Journal, 23, 577-585.
Greiner, R. (2004b), Purification and properties of a phytate-degrading enzyme from Pantoea agglomerans. The Protein Journal, 23, 567-576.
Greiner, R.; Larsson Alminger, M.; Carlsson, N.-G.; Muzquiz, M.; Burbano, C.; Cuadrado, C.; Pedrosa, M.M. and Goyoaga, C. (2002), Pathway of dephosphorylation of myo-inositol hexakisphos-phate by phytases from legume seeds. Journal of Agriculture and Food Chemistry, 50, 6865-6870.
Hawkins, P.T.; Poyner, D.R.; Jackson, T.R.; Letcher, A.J.; Lander, D.A. and Irvine, R.F. (1993), Inhibition of iron-catalyzed hydroxyl radical formation by inositol polyphosphate: a possible physiological function for myo-inositol hexakisphosphate. Biochemical Journal, 294, 929-934.
Heinonen, J.K. and Lahti, R.J. (1981), A new and convenient colorimetric determination of inorganic orthophosphate and its application to the assay of inorganic pyrophosphatase. Analytical Biochemistry, 113, 313-317.
Herter, T.; Berezina, O.V.; Zinin, N.V.; Velikodvorskaya, G.A.; Greiner, R. and Borriss, R. (2006), Glucose 1-phosphatase (AgpE) from Enterobacter cloacae displays enhanced phytase activity. Applied Microbiology and Biotechnology, 70, 60-64.
Konietzny, U. and Greiner, R. (2002), Molecular and catalytic properties of phytate-degrading enzymes (phytases). International Journal of Food Science and Technology, 37, 791-812.
Lee, D.C.; Cottrill, M.A.; Forsberg, C.W. and Jia, Z. (2003), Functional insights revealed by the crystal structures of Escherichia coli glucose-1-phosphatase. Journal of Biological Chemistry, 278, 31412-31418.
Maffucci, T.; Piccolo, E.; Cumashi, A.; Iezzi, M.; Riley, A.M.; Saiardi, A.; Godage, H.Y.; Rossi, C.; Broggini, M.; Iacobelli, S.; Potter, B.V.L.; Innocenti, P. and Falasca, M. (2005), Inhibition of the phosphatidylinositol 3-Kinase/Akt pathway by inositol pentakisphosphate results in antiangiogenic and antitumor effects. Cancer Research, 65, 8339-8349.
Park, J.K. and Chang, H.N. (2000), Microencapsulation of microbial cells. Biotechnology Advances, 18, 303-319.
Phillippy, B.Q. and Bland, J.M. (1988), Gradient ion chromatography of inositol phosphates. Analytical Biochemistry, 175, 162-166.
Phillippy, B.Q. and Graf, E. (1997), Antioxidant functions of inositol 1,2,3-trisphosphate and inositol 1,2,3,6-tetrakisphosphate. Free Radical Biology and Medicine, 22, 939-946.
Sandberg, A.-S. and Ahderinne, R. (1986), HPLC method for determination of inositol tri-, tetra-, penta-, and hexaphosphate in foods and intestinal contents. Journal of Food Science, 51, 547-550.
Sankalia, M.G.; Mashru, R.C.; Sankalia, J.M. and Sutariya, V.B. (2005), Papain entrapment in alginate beads for stability improvement and site-specific delivery: Physicochemical characterization and factorial optimization using neural network modelling. AAPS PharmSciTech., 6, E209-E222.
Shears, S.B. (1998), The versatility of inositol phosphates as cellular signals. Biochimica et Biophysica Acta, 1436, 49-67.
Skoglund, E.; Carlsson, N.-G. and Sandberg, A.-S. (1998), High-Performance Chromatographic separation of inositol phosphate isomers on strong anion exchange columns. Journal of Agriculture and Food Chemistry, 46, 1877-1882.
Van Etten, R.L.; Davidson, S.; Stevis, P.E.; MacArthur, H. and Moore, D.L. (1991), Covalent structure, disulfide bonding, and identification of reactive surface and active site residues of human prostatic acids phosphatase. Journal of Biological Chemistry, 266, 2313-2319.
Received: August 24, 2006;
Revised: April 20, 2007;
Accepted: October 23, 2007
- Bradford, M. (1976), A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry, 72, 248-254.
- Cao, L. (2005), Immobilised enzymes: science or art? Current Opinion in Chemical Biology, 9, 217-226.
- Carrington, A.L.; Calcutt, N.A.; Ettlinger, C.B.; Gustafsson, T. and Tomlinson, D.R. (1993), Effects of treatment with myo-inositol or its 1,2,6- trisphosphate (PP56) on nerve conduction in streptozotocin-diabetis. European Journal of Pharmacology, 237, 257-263.
- Claxon, A.; Morris, C.; Blake, D.; Siren, M.; Halliwell, B.; Gustafsson, T.; Löfkvist, B. and Bergelin, I. (1990), The anti-inflammatory effects of D-myo-inositol-1.2,6-trisphosphate (PP56) on animal models of inflammation. Agents Actions, 29, 68-70.
- Cottrill, M.A.; Golovan, S.P.; Phillips, J.P. and Forsberg, C.W. (2002), Inositol phosphatase activity of the Escherichia coli agp-encoded acid glucose-1-phosphatase. Canadian Journal of Microbiology, 48, 801-809.
- Dey, G.; Singh, B. and Banerjee, R. (2003), Immobilization of a-amylase produced by Bacillus circulans GRS 313. Brazilian Archives of Biology and Technology, 46, 167-176.
- Ferry, S.; Matsuda, M.; Yoshida, H. and Hirata, M. (2002), Inositol hexakisphosphate blocks tumor cell growth by activating apoptotic machinery as well as by inhibiting the Akt/NFkB-mediated cell survival pathway. Carcinogenesis, 23, 2031-2041.
- Greiner, R. (2004a), Degradation of myo-inositol hexakisphosphate by a phytate-degrading enzyme from Pantoea agglomerans The Protein Journal, 23, 577-585.
- Greiner, R. (2004b), Purification and properties of a phytate-degrading enzyme from Pantoea agglomerans The Protein Journal, 23, 567-576.
- Greiner, R.; Larsson Alminger, M.; Carlsson, N.-G.; Muzquiz, M.; Burbano, C.; Cuadrado, C.; Pedrosa, M.M. and Goyoaga, C. (2002), Pathway of dephosphorylation of myo-inositol hexakisphos-phate by phytases from legume seeds. Journal of Agriculture and Food Chemistry, 50, 6865-6870.
- Hawkins, P.T.; Poyner, D.R.; Jackson, T.R.; Letcher, A.J.; Lander, D.A. and Irvine, R.F. (1993), Inhibition of iron-catalyzed hydroxyl radical formation by inositol polyphosphate: a possible physiological function for myo-inositol hexakisphosphate. Biochemical Journal, 294, 929-934.
- Heinonen, J.K. and Lahti, R.J. (1981), A new and convenient colorimetric determination of inorganic orthophosphate and its application to the assay of inorganic pyrophosphatase. Analytical Biochemistry, 113, 313-317.
- Herter, T.; Berezina, O.V.; Zinin, N.V.; Velikodvorskaya, G.A.; Greiner, R. and Borriss, R. (2006), Glucose 1-phosphatase (AgpE) from Enterobacter cloacae displays enhanced phytase activity. Applied Microbiology and Biotechnology, 70, 60-64.
- Konietzny, U. and Greiner, R. (2002), Molecular and catalytic properties of phytate-degrading enzymes (phytases). International Journal of Food Science and Technology, 37, 791-812.
- Lee, D.C.; Cottrill, M.A.; Forsberg, C.W. and Jia, Z. (2003), Functional insights revealed by the crystal structures of Escherichia coli glucose-1-phosphatase. Journal of Biological Chemistry, 278, 31412-31418.
- Maffucci, T.; Piccolo, E.; Cumashi, A.; Iezzi, M.; Riley, A.M.; Saiardi, A.; Godage, H.Y.; Rossi, C.; Broggini, M.; Iacobelli, S.; Potter, B.V.L.; Innocenti, P. and Falasca, M. (2005), Inhibition of the phosphatidylinositol 3-Kinase/Akt pathway by inositol pentakisphosphate results in antiangiogenic and antitumor effects. Cancer Research, 65, 8339-8349.
- Park, J.K. and Chang, H.N. (2000), Microencapsulation of microbial cells. Biotechnology Advances, 18, 303-319.
- Phillippy, B.Q. and Bland, J.M. (1988), Gradient ion chromatography of inositol phosphates. Analytical Biochemistry, 175, 162-166.
- Phillippy, B.Q. and Graf, E. (1997), Antioxidant functions of inositol 1,2,3-trisphosphate and inositol 1,2,3,6-tetrakisphosphate. Free Radical Biology and Medicine, 22, 939-946.
- Sandberg, A.-S. and Ahderinne, R. (1986), HPLC method for determination of inositol tri-, tetra-, penta-, and hexaphosphate in foods and intestinal contents. Journal of Food Science, 51, 547-550.
- Sankalia, M.G.; Mashru, R.C.; Sankalia, J.M. and Sutariya, V.B. (2005), Papain entrapment in alginate beads for stability improvement and site-specific delivery: Physicochemical characterization and factorial optimization using neural network modelling. AAPS PharmSciTech, 6, E209-E222.
- Shears, S.B. (1998), The versatility of inositol phosphates as cellular signals. Biochimica et Biophysica Acta, 1436, 49-67.
- Skoglund, E.; Carlsson, N.-G. and Sandberg, A.-S. (1998), High-Performance Chromatographic separation of inositol phosphate isomers on strong anion exchange columns. Journal of Agriculture and Food Chemistry, 46, 1877-1882.
- Van Etten, R.L.; Davidson, S.; Stevis, P.E.; MacArthur, H. and Moore, D.L. (1991), Covalent structure, disulfide bonding, and identification of reactive surface and active site residues of human prostatic acids phosphatase. Journal of Biological Chemistry, 266, 2313-2319.
Publication Dates
-
Publication in this collection
28 May 2008 -
Date of issue
Apr 2008
History
-
Accepted
23 Oct 2007 -
Reviewed
20 Apr 2007 -
Received
24 Aug 2006