Resumos
A terapêutica farmacológica em recém-nascidos confronta-se, por um lado, com um organismo sujeito a marcadas alterações biológicas, resultantes da composição orgânica e da maturação funcional, que decorre a diferentes graus em crianças com a mesma idade, determinando modificações no perfil farmacocinético e farmacodinâmico e, por outro lado, com a necessidade efetiva da utilização de fármacos. Para dar resposta à necessidade de tratamento destes doentes, recorre-se à utilização de medicamentos "off label", sendo esta uma prática com um elevado risco de segurança e de eficácia, na ausência de informação acerca da estabilidade, solubilidade e biodisponibilidade. Considerou-se, assim, que a utilização de derivados das ciclodextrinas altamente solúveis em água seria uma alternativa para a formulação de preparações líquidas aquosas de fármacos fracamente solúveis, aliada à melhoria de biodisponibilidade e de segurança. Esta revisão pretende fundamentar a possibilidade de recurso à complexação de indometacina com hidroxipropil-beta-ciclodextrina, com o objetivo de melhorar as características de biodisponibilidade e de segurança e permitir a administração por via oral para o tratamento farmacológico do fechamento do canal arterial em prematuros ou em recém-nascidos com esta patologia.
Indometacina; Hidroxipropil-beta-ciclodextrina; Recém-nascidos; Canal arterial
Pharmacological therapy for newborns is faced on one hand, with an organism characterized by biological differences and functional immaturity with various grades of evolution for the same age, implying changes on the pharmacokinetic and pharmacodinamic medicine profiles. On the other hand, there is the effective need for pharmacotherapy. The "off label" use of medicines is therefore the only thing left to do, having in mind the risk of using therapeutic agents not studied for this special group of people. On this context it has been considered the use of cyclodextrin derivatives like hydroxypropyl-beta-cyclodextrin as an alternative to prepare oral formulations. With this review we intend to evaluate the rational for using indomethacin's complexation with hydroxypropyl-beta-cyclodextrin, to enhance bioavailability and reduce gastric toxicity characteristics, allowing its oral administration to treat patent ductus arteriosus on preterm and full-term newborns.
Indomethacin; Hydroxypropyl-beta-cyclodextrin; Newborn; Patent ductus arteriosus
TRABALHOS DE REVISÃO
Aspectos biofarmacêuticos da formulação de medicamentos para neonatos. Fundamentos da complexação de indometacina com hidroxipropil-b-ciclodextrina para tratamento oral do fechamento do canal arterial
Biopharmaceutical aspects of drug formulation for neonatology. Rational for indomethacin's complexation with hydroxypropyl-b-cyclodextrin to treat patent ductus arteriosus
Ana Cristina Ribeiro Rama; Francisco Veiga* * Correspondência: F. Veiga Faculdade de Farmácia de Coimbra. Rua do Norte, 3000 Coimbra - Portugal E-mail: fveiga@ci.uc.pt ; Isabel Vitória Figueiredo; Adriano Sousa; Margarida Caramona
Faculdade de Farmácia, Universidade de Coimbra, Coimbra, Portugal
RESUMO
A terapêutica farmacológica em recém-nascidos confronta-se, por um lado, com um organismo sujeito a marcadas alterações biológicas, resultantes da composição orgânica e da maturação funcional, que decorre a diferentes graus em crianças com a mesma idade, determinando modificações no perfil farmacocinético e farmacodinâmico e, por outro lado, com a necessidade efetiva da utilização de fármacos. Para dar resposta à necessidade de tratamento destes doentes, recorre-se à utilização de medicamentos "off label", sendo esta uma prática com um elevado risco de segurança e de eficácia, na ausência de informação acerca da estabilidade, solubilidade e biodisponibilidade. Considerou-se, assim, que a utilização de derivados das ciclodextrinas altamente solúveis em água seria uma alternativa para a formulação de preparações líquidas aquosas de fármacos fracamente solúveis, aliada à melhoria de biodisponibilidade e de segurança. Esta revisão pretende fundamentar a possibilidade de recurso à complexação de indometacina com hidroxipropil-b-ciclodextrina, com o objetivo de melhorar as características de biodisponibilidade e de segurança e permitir a administração por via oral para o tratamento farmacológico do fechamento do canal arterial em prematuros ou em recém-nascidos com esta patologia.
Unitermos: Indometacina. Hidroxipropil-b-ciclodextrina. Recém-nascidos. Canal arterial.
ABSTRACT
Pharmacological therapy for newborns is faced on one hand, with an organism characterized by biological differences and functional immaturity with various grades of evolution for the same age, implying changes on the pharmacokinetic and pharmacodinamic medicine profiles. On the other hand, there is the effective need for pharmacotherapy. The "off label" use of medicines is therefore the only thing left to do, having in mind the risk of using therapeutic agents not studied for this special group of people. On this context it has been considered the use of cyclodextrin derivatives like hydroxypropyl-b-cyclodextrin as an alternative to prepare oral formulations. With this review we intend to evaluate the rational for using indomethacin's complexation with hydroxypropyl-b-cyclodextrin, to enhance bioavailability and reduce gastric toxicity characteristics, allowing its oral administration to treat patent ductus arteriosus on preterm and full-term newborns.
Uniterms: Indomethacin. Hydroxypropyl-b-cyclodextrin. Newborn. Patent ductus arteriosus.
INTRODUÇÃO
A investigação farmacêutica tem como intuito o desenvolvimento de novos agentes terapêuticos direcionados à resolução de situações clínicas, integrando conhecimentos pluridisciplinares.
A prática de cuidados farmacêuticos possibilita a identificação de situações clínicas para as quais os recursos terapêuticos existentes não são os adequados. Fazer parte da equipe de saúde de uma unidade de cuidados intensivos de neonatologia é um desafio para o farmacêutico clínico, confrontado com a necessidade de dar resposta aos problemas biofarmacêuticos decorrentes da anatomofisiologia e das patologias inerentes àquele grupo etário, que abrange prematuros (indivíduo que nasce antes das 37 semanas de gestação) e recém-nascidos (indivíduo com tempo de gestação de 37 a 42 semanas, com idade inferior a 28 dias) (Richardson, 2001; Stewart, Hampton, 1987).
A terapêutica farmacológica confronta-se, por um lado, com um organismo sujeito a marcadas alterações biológicas, resultantes da composição orgânica e da maturação funcional, que decorre de diferentes graus em crianças com a mesma idade, determinando modificações nos perfis farmacocinético e farmacodinâmico e, por outro lado, com a necessidade efetiva da utilização de fármacos (Leff, Roberts, 1987; Pellegrin, Lesne, 1980). Verifica-se, na prática, a falta de adequação das especialidades farmacêuticas a este grupo, quer com relação à formulação, dosagem, forma farmacêutica, osmolalidade, vias de administração ou excipiente utilizado, quer, ainda, com relação às características biofarmacêuticas de eficácia e de segurança.
Para dar resposta à necessidade de tratamento destes doentes, recorre-se à utilização de medicamentos "off label", isto é, medicamentos utilizados fora das indicações terapêuticas aprovadas, sendo esta uma prática com elevado risco de segurança e de eficácia, na ausência de informação acerca da estabilidade, solubilidade e biodisponibilidade.
Nas formulações líquidas, para ultrapassar a fraca solubilidade aquosa de vários fármacos, são usados como agentes suspensores grandes quantidades de co-solventes orgânicos ou xaropes com alta osmolalidade. Ambas as alternativas acarretam riscos potenciais para o recém-nascido, quer devido à imaturidade metabólica, quer devido aos efeitos locais no intestino (Leff, Roberts, 1987). Na ausência de formulações líquidas adequadas, preparam-se, a partir das fórmulas sólidas orais, diluições sólidas em solventes tais como lactose ou amido. A administração de pós misturados com um pequeno volume de água (1 a 2 mL) resulta na perda de parte variável de dose e obstrução da sonda nasogástrica.
Considerou-se, assim, que a utilização de derivados das ciclodextrinas altamente solúveis em água seria uma alternativa para a formulação de preparações líquidas aquosas de fármacos fracamente solúveis, aliada à melhoria de biodisponibilidade e de segurança (Kaukonen et al., 1998; Tötterman et al., 1997).
Esta revisão pretende fundamentar a possibilidade de utilização de sistemas terapêuticos, dos quais é exemplo a complexação de fármacos com ciclodextrinas, especificamente utilizando a hidroxipropil-b-ciclodextrina, com o objetivo de melhorar as características de biodisponibilidade e de segurança da indo metacina e permitir a sua administração por via oral para o tratamento farmacológico do fechamento do canal arterial em prematuros ou em recém-nascidos com esta patologia.
A manutenção do canal arterial em recém-nascidos e implicações na biodisponibilidade de fármacos
Na circulação fetal, o canal arterial (CA) é um canal vascular normal, que liga a artéria pulmonar esquerda, com a aorta descendente. A função do CA é a de permitir que a maior parte do fluxo sanguíneo (90%), que sai do ventrículo direito, flua diretamente para a aorta descendente, evitando a circulação pulmonar altamente resistente. Esta circulação direciona o fluxo de sangue não oxigenado para a placenta, a qual é a fonte fetal de reoxigenação (Bhatt, Nahata, 1989; Gal, Gilman, 1993; Hammerman, Kaplan, 2001; Yeh, Carr, 1991).
Durante os primeiros minutos após o nascimento, a constrição do CA e o seu fecho funcional iniciam-se normalmente de modo espontâneo, redirecionando o fluxo sanguíneo para os pulmões, que assumem a função de oxigenação. A evolução do fechamento funcional do CA em recém-nascidos de termo ocorre durante as primeiras horas de vida: 20% encontram-se fechados às 24 h, 82% às 48 h e 100% às 96 h. O potencial risco de reabertura do CA mantém-se até ocorrer o fecho completo.
Os fatores determinantes do fecho desta veia são a tensão de oxigênio, as características específicas da massa muscular disponível no CA e a concentração de prostaglandinas (PGs) vasodilatadoras circulantes, principalmente a PGE2, a prostaciclina PGI2 e, segundo alguns autores, também o tromboxano A2, tendo-se recentemente investigado o efeito vasoconstritor da angiotensina II (Baptista et al., 1999; Barst, Gersony, 1989; Gal, Gilman, 1993; Hammerman, 1995; Hammerman, Kaplan, 2001; Treszl et al., 2003).
A incidência da manutenção do CA de acordo com a bibliografia é, aproximadamente, de 20 a 25% em recém-nascidos de baixo peso (1500 a 2500 g), podendo atingir 70% nos de muito baixo peso (inferior a 1500 g), ou valores ainda superiores naqueles que são tratados com tensoativo exógeno. O não fechamento tem implicações hemodinâmicas clinicamente significativas (Connuck et al., 2002; Hammerman, 1995; Hammerman, Kaplan, 2001; Richardson, 2001; Romagnoli, 1997; Yeh, Carr, 1991) e que se refletem na biodisponibilidade dos fármacos (Tabela I).
O fato do CA não fechar após o nascimento leva à mistura do sangue oxigenado com o não-oxigenado, conduzindo, freqüentemente, à síndrome de insuficiência respiratória (Bhatt, Nahata, 1989; Yeh, Carr, 1991).
Estudos clínicos provam que os níveis elevados de prostaglandinas vasodilatadoras no CA, principalmente da PGE2 e da prostaciclina PGI2, são os principais responsáveis pela manutenção da abertura do CA no recém-nascido, justificando-se, assim, o tratamento farmacológico com inibidores da síntese das PGs (Bhatt, Nahata, 1989; Hammerman, 1995; Hammerman, Kaplan, 2001; Yeh, Carr, 1991). A indometacina (IM) administrada por via intravenosa (IV) é um dos fármacos de eleição para o tratamento farmacológico do fechamento do CA (Malviya et al., 2003).
Absorção gastrointestinal em recém-nascidos
A absorção gastrointestinal é regulada fundamentalmente por um processo de difusão dependente do pH, pelo tempo de esvaziamento gástrico, pelo peristaltismo, pelas condições de desenvolvimento e maturação da membrana intestinal, pelo estado da função biliar e pela flora bacteriana. Há estudos que indicam que o pH gástrico alcalino no nascimento se deve à presença de líquido amniótico e que no recém-nascido de termo a secreção basal de ácido no conteúdo gástrico se inicia alguns minutos após o nascimento, aumentando gradualmente ao longo de algumas horas, sendo, no entanto, inferior ao do adulto (Hyman et al., 1985; Stewart, Hampton, 1987). O pH do conteúdo gástrico é o resultado de vários fatores, os quais incluem, sob condições fisiológicas, a liberação de ácido clorídrico, alimento ingerido, saliva deglutida e regurgitação do conteúdo duodenal (Hyman et al., 1985; Mason, 1962).
No momento do nascimento, o pH gástrico situa-se entre 1 e 3, passando para 6 a 8, nas primeiras 24 horas e alcançando o do adulto aos 3 anos de idade. A relativa acloridria no recém-nascido, juntamente com o esvaziamento gástrico lento, o qual permanece até os 6 meses, dá lugar a diferenças de biodisponibilidade dos fármacos, dependentes das características físico-químicas destes (Besunder, 1988; Kadima, Lesne, 1980). O recém-nascido tem um pH no intestino delgado mais elevado do que o do adulto, o que permite a colonização da flora bacteriana normalmente restrita ao intestino grosso. As alterações da colonização do trato gastrointestinal pela flora bacteriana são dependentes da idade gestacional, do tipo de parto e de alimentação, modificando a hidrólise de fármacos conjugados, que são excretados na bílis (Besunder, 1988; Yoshioka et al., 1983).
Uma vez que a maior parte dos fármacos administrados oralmente são absorvidos no intestino delgado, a rapidez de esvaziamento gástrico é um fator importante na velocidade e extensão de absorção, sendo afetado pela maturidade gestacional, idade pós-natal, efeito da postura, volume, osmolalidade e composição da alimentação. A velocidade de esvaziamento durante o período neonatal é menor do que a do adulto, com valores de 6 a 8 horas. Há autores que defendem a ausência de peristaltismo gástrico durante os primeiros 2 a 4 dias de vida, atribuindo o esvaziamento gástrico ao aumento combinado do tônus, da contração do antrum e da pressão hidrostática (Stewart, Hampton, 1987).
Com relação à atividade enzimática gastrointestinal, o recém-nascido tem baixas concentrações de lipases, as quais, em conjugação com baixas concentrações intraluminais de ácidos biliares, levam a que fármacos lipófilos sejam insolúveis no estômago e, portanto, com absorção reduzida. A baixa concentração intraluminal de ácido biliar (50% em comparação com o adulto) é conseqüência da reabsorção ineficaz de ácidos biliares no íleo, menor velocidade de síntese, tamanho do conjunto de ácidos biliares e maior permeabilidade destes no jejuno. Por outro lado, a atividade da a-glicuronidase no intestino pode ser sete vezes superior à do adulto, o que tem conseqüencias sobre os medicamentos submetidos à circulação entero-hepática (Besunder, 1988; Kadima, Lesne, 1980; Stewart, Hampton, 1987; Watkins, 1973).
Assim, estando ainda cientes dos riscos de aspiração e refluxo que apresenta a via oral ou entérica, devido à imaturidade do trato gastrointestinal, à toxicidade associada a alguns medicamentos e aos múltiplos fatores que imprimem grande variabilidade no comportamento das formas farmacêuticas orais ou entéricas, esta via será a que apresenta melhor balanço benefício/risco para a administração de medicamentos em recém-nascidos.
Indometacina
A indometacina (IM) é um antiinflamatório nãoesteróide (AINE), derivado do ácido indol-acético. O seu mecanismo de ação abrange a inibição da síntese de todas as PGs, por bloqueio reversível da via mediada pelas cicloxigenases (Barst, Gersony, 1989).
Muitos compostos sólidos existem em diversas formas cristalinas modificadas denominadas polimorfos. São compostos cristalinos com a mesma estrutura molecular, que têm um arranjo diferente das moléculas, apresentam a mesma composição química, podem ter diferentes temperaturas de fusão, densidade, solubilidade, estabilidade química e física, velocidade de dissolução e biodisponibilidade (Bettinetti, 1988; Brittain, 1997, 2002; Lin, 1992). O polimorfismo da IM tem sido muito investigado nos últimos anos. Foram primeiro descritas três formas polimórficas a (Forma I), b, e g (Forma II), tendo posteriormente sido identificados outros polimorfos, a Forma III e IV entre outras, não havendo, contudo, consenso quanto à existência de todos os polimorfos descritos. Em relação aos polimorfos Forma I e II, há dados consistentes de ponto de fusão, difração de raios-X e de infravermelho que confirmam a sua existência (Borka, 1974; Lin, 1992; O'Brien et al., 1984).
As tentativas para aumentar a solubilidade da IM através da elevação do pH para valores alcalinos têm sido infrutíferas, pois pode ocorrer decomposição do composto por hidrólise (Bakensfeld et al., 1990; Krasowska, 1974).
Indometacina como fármaco de escolha no tratamento farmacológico do fecho do Canal Arterial
A utilização da IM para o tratamento farmacológico do fecho do CA em recém-nascidos foi descrita pela primeira vez em 1976 (Vert et al., 1980; Wiest et al., 1991), sendo hoje um dos fármacos de eleição efetivo e apropriado, induzindo o fecho do CA em mais de 80% dos recém-nascidos prematuros, quando administrado por via intravenosa (Angerio, Kot, 1998; Archer, 1993; Baptista et al., 1999; Bhatt, Nahata, 1989; Grosfeld et al., 1996; Hammerman, 1995).
É controversa a relação entre a concentração sérica de IM e o fecho do CA, devido à extrema variabilidade que se observa entre recém-nascidos e ao fato da fisiopatologia dessa situação clínica ter reflexos na biodisponibilidade dos fármacos (Gal, Gilman, 1993).
A biodisponibilidade da IM administrada por via oral em recém-nascidos prematuros com CA, numa dose de 0,2 mg/kg, é caracterizada da seguinte forma:
- absorção oral - inferior a 15 a 20% da dose administrada;
- pico sérico - aproximadamente 400 ng/mL, 1 a 6 h após a primeira dose;
- volume de distribuição - 0,36 L/kg;
- ligação às proteínas - 95 a 98%;
- depuração plasmática;
- metabolismo hepático - a limitada atividade enzimática contribui para o aumento percentual de fármaco livre;
- meia-vida plasmática - entre 11 a 20 horas;
- excreção renal - diminuída.
A administração de 0,2 mg/kg por via oral, repetida até 3 administrações de 24 em 24 h, contribuiu para o fechamento do CA na proporção de 57%, nos grupos de doentes estudados (Habib, 1995; Vert et al., 1980; Yeh, Carr, 1991).
O protocolo terapêutico utilizado na prática clínica para o fechamento farmacológico do CA em recém-nascidos, tendo em consideração a variabilidade referida, é de 0,2 mg de IM por quilograma de peso, administrada de 24 em 24 h durante três dias consecutivos por via intravenosa, para alcançar a concentração sérica de 0,37 µg/mL (Renfro et al., 1993; Romagnoli, 1997; Yeh, Carr, 1991).
Toxicidade Gastrointestinal da Indometacina
Existem duas características fundamentais que determinam a ulcerogenicidade dos AINEs:
- A cinética de absorção gastrointestinal dos fármacos;
- O fato de exibirem ataque múltiplo a alguns ou a todos os mecanismos de defesa da mucosa.
O grau de ulcerogenicidade depende destes mecanismos de defesa da mucosa celular ou dos processos bioquímicos afectados e da potência do efeito dos fármacos nesses processos. Para que um fármaco exerça efeito ulcerogênico, deve estar presente nas células da mucosa em concentração e tempo de permanência suficientes para desencadear os mecanismos de toxicidade (Erden, Çelebi, 1988; Liversidge et al., 1989; Menguy, Desbaillets, 1967; Rainsford, 1990; Somasundaram et al., 1997). Em geral, quando os AINEs são administrados por via oral são atingidas grandes concentrações nas células da mucosa gástrica. Quando a administração é parenteral, alguns AINEs sofrem recirculação entero-hepática, podendo produzir lesões na mucosa ou úlceras.
A IM, sendo um AINE, tem capacidade de induzir toxicidade gastrointestinal. O mecanismo responsável pela ulceração induzida pela IM não é claro (Bulbena et al., 1993; Lugea et al., 1993; Mahmud et al., 1998; Parent, 1991; Rainsford, 1990; Somasundaram et al., 1997), contudo, várias têm sido as explicações propostas. Na Figura 1 estão representados esquematicamente os mecanismos que podem estar envolvidos no processo de lesão gastroinstestinal, com base nas principais teorias cientificamente aceitas.
Os primeiros episódios conducentes à indução da lesão incluem uma fase de contato concomitante com a inibição das cicloxigenases nas células da mucosa, causando redução das PGs vasodilatadoras (PGE2 e PGI2) e induzindo o ácido araquidônico a produzir, por um lado, através da via da lipoxigenase, excesso de peptidoleucotrienos (LTC4 e LTD4), que são potentes vasoconstritores e, por outro lado, a gerar radicais peróxido livres, resultantes da clivagem peroxidativa de ácidos hidroxieicosatetranóicos.
Os AINEs também inibem a produção de trifosfato de adenosina (ATP) nas células da mucosa, elevando, assim, o monofosfato de adenosina (AMP), que ativa a forma desidrogenase da xantina, passando esta à forma oxidase e gerando mais radicais superóxido. Este processo conduz, também, à inibição ou diminuição da produção de muco e a alterações da sua composição em hidratos de carbono.
A diminuição da vasodilatação, o aumento da vasoconstrição e da quantidade de radicais livres de oxigênio promovem o desenvolvimento de isquemia e conseqüente lesão vascular.
A fase seguinte é constituída por episódios resultantes da conjugação de vários fatores, que contribuem para o desenvolvimento da inflamação e da ulceração. O condicionamento desses fatores, que são parte integrante dos mecanismos de protecção da mucosa, tais como a permeabilidade intestinal, o conteúdo luminal, os neutrófilos e a microcirculação, poderão estar igualmente na origem da lesão gastrointestinal (Rainsford, 1990; Somasundaram et al., 1997).
O comportamento farmacocinético da IM possibilita a existência de três vias possíveis pelas quais pode entrar em contato com a mucosa do trato gastrointestinal (TGI) (Mahmud et al., 1998; Somasundaram et al., 1997):
- Fase tópica: após ingestão ao nível gástrico e durante a absorção no intestino delgado e no intestino grosso;
- Via sistêmica: ocorre à medida que o fármaco entra no compartimento vascular e se distribui pelo organismo, podendo atingir as células apicais da mucosa gástrica;
- Excreção através da via biliar: torna a expor o intestino delgado e o estômago a uma fase de contato, por refluxo do conteúdo duodenal (Somasundaram et al., 1997).
Toxicidade gastrointestinal em Recém-nascidos
As características físico-químicas da IM, a reduzida absorção oral e a toxicidade gastrointestinal fazem com que a forma farmacêutica injetável seja a única disponível com ação farmacológica efetiva para o fechamento do canal arterial (Coombs et al., 1990; Habib, 1995).
A toxicidade gastrointestinal observada com a via intravenosa, intramuscular, oral e retal, inclui, entre as situações mais graves, a ocorrência de ulceração, hemorragia intestinal, perfuração e enterocolite necrosante. Em todo este complexo processo, que leva ao aparecimento de toxicidade, deve-se ainda considerar que os efeitos vasculares sistêmicos da IM poderão causar vasoconstrição das veias intestinais, originando situação de isquemia com evidência de desenvolvimento de enterocolite necrosante, a qual é um fator de risco adicional em recém-nascidos cujo CA não fechou (Coombs et al., 1990; Grosfeld et al., 1996; Krasna, Lee, 1993; Schmidt et al., 2001).
Ciclodextrinas
Características gerais
As ciclodextrinas (CDs) são um grupo de sacarídeos estruturalmente relacionados, que são produzidos pela ciclização enzimática do amido, catalizada pela enzima ciclodextrina-glicosil-transferase, formando uma espiral helicoidal de unidades de glicose unidas por ligações ±-(1,4) (Biwer et al., 1988; Szejtli, 1988). Devido à falta de rotação livre à volta das pontes de ligação das unidades de glicose, as CDs não são moléculas cilíndricas, tomando a forma tronco-cônica (Figura 2).
Em conseqüência da conformação em cadeira das unidades de glicopiranose, todos os grupos de hidroxila estão orientados para o exterior da molécula, com os grupos hidroxila primários localizados no lado mais estreito e os secundários no lado mais largo da estrutura, conferindo caráter hidrófilo (Loftsson, Brewster, 1996; Saltão, Veiga, 2001).
A cavidade apresenta características hidrófobas devido ao caráter apolar determinado pelos dois anéis dos grupos C-H e pelo anel de átomos de oxigênio incluídos nas ligações glicosídicas. Esta estrutura molecular invulgar confere às CDs propriedades únicas.
As CDs naturais mais comuns são a a-ciclodextrina (ciclomalto-hexanose), b-ciclodextrina (ciclomalto-heptanose) e g-ciclodextrina (ciclomalto-octanose), contendo respectivamente 6, 7 e 8 unidades de a-1,4 de glicopiranose ligadas. Destes três derivados, a b-ciclodextrina (b-CD) parece ser a mais vantajosa para utilização farmacêutica como agente complexante, devido, entre outras propriedades, ao tamanho da sua cavidade, disponibilidade e baixo custo.
O tamanho da cavidade na parte mais larga, para entrada da molécula hóspede varia de 4,7 a 5,3 Å, 6,0 a 6,5 Å e 7,5 a 8,3 Å, respectivamente para a CD a, b e g (Figura 3) (Cabral-Marques, Morais, 1991; Szejtli, 1990; Thompson, 1997).
Mecanismo de complexação, conseqüências e limitações dos complexos de inclusão com ciclodextrinas
As CDs são capazes de formar complexos de inclusão com muitos fármacos, com tamanho e forma apropriados, encapsulando total ou parcialmente as moléculas-hóspedes. A cavidade das CDs tem afinidade preferencial para a forma neutra de um determinado substrato, sendo capazes de complexar e solubilizar compostos não polares. A complexação pode ser condicionada, entre outros fatores, pela composição do esqueleto do composto, pela fraca solubilidade em água (inferior a 10 mg/mL), pelo estado de ionização, pela temperatura e solventes utilizados, por ponto de fusão inferior a 250 ºC (ponto de fusão superior significa que as forças coesivas entre as moléculas são muito fortes) e por massa molecular compreendida entre 100 e 400 Daltons. Contudo, pode ocorrer a formação de complexos com moléculas muito grandes, desde que contenham cadeias laterais apropriadas para inclusão parcial, originando compostos com solubilidade e estabilidade modificadas (Szejtli, 1991b; Thompson, 1997).
Os mecanismos pelos quais se processa a formação de complexos de inclusão são diversos, contudo todos têm em comum o fato de não ocorrer formação nem quebra de ligações covalentes. Considera-se que a principal força de ligação para a formação de complexo é a liberação de entalpia das moléculas de água da cavidade das CDs, uma vez que estas não conseguem satisfazer a sua necessidade de ligação aos potenciais de hidrogênio, tendo, portanto, elevada entalpia. A energia do sistema diminui quando estas moléculas são substituídas por moléculas-hóspedes, que são menos polares do que a água. O fato de não se formarem ligações covalentes entre a CD e as moléculas-hóspedes faz com que os complexos em condições fisiológicas sejam facilmente dissociáveis (Cabral-Marques, 1994a). Na formação do complexo podem também participar a liberação de forças do anel, interações van der Waals, ligações de hidrogênio, interações hidrofóbicas e alterações na tensão superficial do solvente (Loftsson, Olafsson, 1998).
Em solução aquosa, os complexos dissociam-se, encontrando-se em equilíbrio as moléculas livres com as moléculas ligadas na cavidade da CD. Este é um processo dinâmico, no qual a molécula-hóspede se associa e dissocia constantemente da molécula-hospedeira. Nesta conformidade, o complexo será constituído por uma família de espécies em proporções médias variáveis, sendo a espécie de complexo a que apresenta maior tempo de existência (Loftsson, Brewster, 1996; Stella et al., 1999; Szejtli, 1991a).
Os principais fatores que afetam a extensão e o grau de complexação incluem o tamanho e a geometria da molécula-hóspede, a energia conformacional, a força das interações van der Walls e de outras forças eletrostáticas. Adicionalmente, a solubilidade relativa da molécula-hóspede no ambiente circundante e a solubilidade do complexo contribuem para definir o equilíbrio final. A temperatura, o pH e outras moléculas-hóspedes competidoras podem, também, afetar o equilíbrio (Cabral-Marques, 1994b).
A extensão da complexação em meio aquoso, ou seja, a estabilidade do complexo formado, é caracterizada pela constante de estabilidade, também chamada de associação (Ka) ou dissociação (KD), e depende de como a molécula-hóspede (F) se encaixa na cavidade da CD. A razão entre a constante de velocidade de recombinação (KR) e a constante de velocidade de dissociação traduz a magnitude da estabilidade do complexo, Ke = Ka:b = KR / KD = FaCDb / [F]ª x [CD]b, em que a:b representa a razão molar de F incluído na CD. Quanto maior esta razão, maior é a estabilidade do complexo (Cabral-Marques, 1994a; Stella et al., 1999; Veiga, 1996).
A ação e toxicidade do complexo de inclusão nas condições "in vivo" e o comportamento do produto obtido, tal como o perfil de dissolução, podem constituir fatores limitantes do seu uso. Pode, também, ser fator limitante da aplicabilidade de complexos de inclusão destinados à administração oral, a relação entre a massa molecular do fármaco, a dose necessária para de obter efeito terapêutico e a estequeometria do complexo. Assim, quando a dose a administrar é elevada, requerendo grandes quantidades de ciclodextrina, a porção de CD pode condicionar aspectos de segurança e custo de produção (Cabral-Marques, 1994b; Redenti et al., 2000).
Assim, um modelo de comportamento do complexo de inclusão HP-b-CD + IM no intestino após administração oral e os vários fatores intervenientes no processo de liberação e absorção da IM complexada com a HP-b-CD foi proposto (Figura 4).
Quando se administra por via oral um complexo de inclusão de fármaco em ciclodextrinas hidroxialquiladas, os estudos efetuados levam a prever que:
- no estômago este não sofra qualquer tipo de alteração;
- no intestino delgado haja competição entre as moléculas de sais biliares e a molécula de fármaco pela complexação com a ciclodextrina;
- no intestino grosso haja reduzida hidrólise enzimática da ciclodextrina por parte da flora microbiana e que a maior parte da CD é eliminada nas fezes na forma intacta, ou que, no caso de haver hidrólise, se formem maltodextrinas lineares, maltose e glicose, as quais podem ser absorvidas ou, então, eliminadas sob a forma de dióxido de carbono e água.
Os principais fatores dos quais depende a estabilidade do complexo com CDs hidroxialquiladas ao longo do TGI serão, assim, a constante de formação do complexo e a interação com os sais biliares presentes.
A constante de formação do complexo entre o fármaco e a CD (Ke) é um fator relevante, como já se referiu, dado que se o seu valor for elevado não há liberação da molécula, com conseqüente diminuição da fração livre e da absorção.
Por outro lado, a formação de complexos de inclusão dos sais biliares com as CDs está dependente do efeito de competição com o fármaco-hóspede, originando o deslocamento deste da molécula do complexo, melhorando a sua biodisponibilidade (Nakanishi et al., 1989). Este mecanismo pode ser positivo no caso da IM, dado que promove a sua liberação, ficando disponível para ser absorvida. O processo pelo qual os sais biliares aumentam a dissolução da IM envolve solubilização micelar e efeito molhante (Miyazaki et al., 1981). A IM sob a forma de micelas é inserida na membrana gastrointestinal por interação com os seus componentes, promovendo a absorção do fármaco. Contudo, podem ocorrer potenciais danos na membrana, causados por esta interação.
Em termos farmacêuticos é desejável usar complexos de formação cuja constante se situe entre 100 e 1000 M-1, uma vez que os valores das constantes de formação de complexos com as várias moléculas dos sais biliares a pH 7,2 com a b-CD são, respectivamente 2670 M-1, 1100 M-1, 410 M-1 e 406 M-1 para o desoxicolato, colato, glicocolato e taurocolato de sódio (Miyajima et al., 1986; Nakanishi et al., 1989). Fármacos com constante de formação menor formam complexos instáveis, enquanto que aqueles que têm constante de formação elevada são estáveis demais para que a troca com os sais biliares ocorra. Se a constante se situar nos valores referidos, a competição ocorre para a formação de complexo com os sais biliares no intestino delgado.
A inclusão de moléculas de sais biliares é também benéfica, por diminuir a capacidade das CDs livres de interagir com os fosfolipídeos da membrana intestinal, com formação de complexos de inclusão, que podem provocar a fluidez desta e conseqüente aumento da permeabilidade, podendo esta capacidade ser uma das explicações para a absorção das CDs (Irie, Uekama, 1997).
Estudos metabólicos e farmacocinéticos efetuados em ratos, camundongos e cães, mostraram que a HP-b-CD administrada por via oral pode ser absorvida na forma intacta, numa proporção que varia entre 1 e 3%, sendo o material não absorvido excretado nas fezes. A maior parte da pequena quantidade metabolizada é provavelmente convertida em dissacarídeos no intestino grosso, constituindo, assim, o principal mecanismo de absorção. Contudo, sabe-se que a HP-b-CD é dializável através de membrana de acetato e que os sais biliares podem aumentar a absorção de moléculas intactas (Strattan, 1992).
Em estudos de administração IV da HP-b-CD mostram que ela é eliminada intacta na urina após 24 h por filtração glomerular, com cinética de eliminação linear, que implica ausência de nefrotoxicidade (Frijlink et al., 1990b). Autores que estudaram o comportamento da HP-b-CD administrada por via IV concluem que esta se mantém em circulação e que não atravessa membranas lipídicas em razão da sua polaridade, podendo, quando em contato com elas, extrair componentes lipídicos, preferencialmente o colesterol, atuando, assim, também como transportadores de moléculas lipídicas existentes na corrente sanguínea (Irie et al., 1992a e 1992b). Os complexos de colesterol:HP-b-CD dissociam-se no filtrado glomerular, a HP-b-CD passa através do glomérulo sem provocar obstrução e ocorre deposição de colesterol no rim (Fini et al., 1995; Flourié et al., 1993; Frijlink et al., 1990a; Gerloczy et al., 1994; Nakanishi et al., 1992; Shamat, 1993; Tan, Lindenbaum, 1991).
Toxicidade das ciclodextrinas administradas por via oral
A administração oral das CDs naturais não induz qualquer tipo de toxicidade aguda e, mesmo em doses mais elevadas, não origina mortalidade quando administrada a animais. O DL50, em ratos, para a via oral, é descrito como sendo superior a 12,5 g/kg para a a-CD, 18,8 g/kg para a b-CD e 8 g/kg para a g-CD (Duchêne, Wouessidjewe, 1990b; Mosher, Thompson, 2002; Saltão, Veiga, 2001; Szejtli, 1990).
A administração prolongada não promove alterações significativas em qualquer órgão nem nos parâmetros analíticos do sangue. Observa-se, contudo, em alguns dos estudos efetuados, que a administração crônica de ciclodextrinas a ratos pode provocar algumas alterações:
- Reduz o aumento de peso, bem como os triglicerídeos séricos e a deposição de gordura, uma vez que os produtos da hidrólise das CDs incluem glicose e malto-oligosacarídeos, conhecidos por serem rapidamente fermentados pela flora anaeróbia do intestino grosso e originarem, entre outros produtos, ácidos graxos (Flourié et al., 1993).
- O rim e o fígado mostraram ser órgãos alvo da toxicidade da b-CD, provocando alterações morfológicas com aumento do hepatócito centrolobular compatíveis com elevação das enzimas hepáticas (transaminase glutâmico-pirúvica, glutâmico-oxalacética e carbamoil-ornitina). Estas modificações, consideradas moderadas, foram observadas com doses superiores a 1313 mg/kg/dia em ratos macho e 1743 mg/kg/dia em fêmeas, administradas durante 52 semanas (Bellringer et al., 1995).
- Um dos efeitos sistêmicos secundários das CDs é o aumento da eliminação gastrointestinal de certos nutrientes e sais biliares. Este efeito, porém, só é observado para doses superiores a 20% da dose recomendada. O efeito produz-se pela conversão aumentada do colesterol sérico a sais biliares, com conseqüente diminuição dos seus níveis plasmáticos. A capacidade das CDs para complexar o colesterol pode, também, contribuir para desestabilizar a membrana. Estes efeitos podem estar diretamente relacionados com o fato de algumas CDs poderem atuar como promotores da permeabilidade da membrana ou causar irritação dos tecidos, após administração por via injetável (Stella, Rajewski, 1997).
- Não se observou evidência macroscópica da existência de patologias, após um ano de exposição oral de ratos à b-CD, mas a avaliação microscópica dos tecidos revelou alterações. Os órgãos mais afetados pela exposição foram o rim e o fígado. Os efeitos no rim foram considerados como não tendo gravidade toxicológica. Foi observada alguma necrose celular no fígado em percentagens que variam entre os 2,5 a 5% dos animais, bem como o aparecimento de infiltrado inflamatório e aumento do nível sérico das enzimas hepáticas. As observações foram consideradas como representando hepatotoxicidade moderada (Mosher, Thompson, 2002).
O mecanismo pelo qual a b-CD causa alterações hepáticas não está ainda definido, apresentando os estudos em outras espécies de animais, principalmente em cães, ausência de lesão quando expostos durante um ano a dietas contendo até 5% de ciclodextrina, levando a considerar que pode ser um efeito relacionado com a espécie e não refletir hepatotoxicidade geral (Thompson, 1997).
As toxicidades aguda e subcrônica da HP-b-CD são insignificantes após a sua administração intraperitonial, intravenosa, intramuscular, intracraniana ou tópica e, mesmo quando administrada por via intravenosa na dose de 10 g/kg, não se observa o acúmulo renal observado com outras ciclodextrinas (Brewster et al., 1990, 1992; Carpenter et al., 1995; Loftsson, Brewster, 1997; Pitha, Pitha, 1985; Pitha et al., 1986, 1994; Stella, Rajewski, 1997; Strattan, 1992; Yoshida et al., 1988).
Pesquisa efetuada em ratos, por administração intravenosa de 400 mg de HP-b-CD durante 90 dias, resultou em toxicidade moderada, evidenciada pela diminuição do aumento de peso corporal, alterações dos parâmetros séricos, aumento da atividade das células fagocitárias mononucleares dos pulmões e fígado e, ainda, alterações do pâncreas (Mosher, Thompson, 2002).
Os estudos clínicos evidenciam que a HP-b-CD é considerada não tóxica numa dose diária inferior a 16 g/kg. Não apresenta potencial mutagênico, não tem efeitos adversos na fertilidade nem no desenvolvimento pré e pós-natal e não é nem embriogênica nem teratogênica (Irie, Uekama, 1997).
Vários são os estudos que foram realizados em ratos com a finalidade de avaliar a segurança da administração oral da HP-b-CD. Algumas das investigações revelaram que a toxicidade a longo prazo não induziu mortalidade, nem sinais visíveis de doença, apesar do grupo experimental ter apresentado ligeiro aumento de peso comparativamente com o grupo controle. O aumento do tamanho do fígado após administração crônica durante 112 dias, sem alterações morfológicas, nem diferença significativa do colesterol sérico, pode ser atribuída à formação de complexos de inclusão com os ácidos biliares no intestino. A inibição da captação destes poderá levar ao aumento da sua síntese e possivelmente ao aumento do tamanho do fígado (Duchêne, Wouessidjewe, 1990b; Gerloczy et al., 1994; Pitha, Pitha, 1985). Estudos realizados em células humanas de epitélio intestinal (Caco-2) para testar a resistência transepitelial demonstraram que a HP-b-CD não afeta a sua integridade e não evidencia toxicidade na atividade enzimática intracelular da desidrogenase, parecendo, assim, ser segura no que respeita aos efeitos locais no intestino (Tötterman et al., 1997). Avaliou-se, também, a segurança da HP-b-CD em ratos por um período de 2 anos. As doses atingiram os 5000 mg/kg/dia. Não foram detectados efeitos adversos, exceto o efeito laxativo considerado não-relevante (Mosher, Thompson, 2002).
Utilização terapêutica de complexos de inclusão para administração por via oral
Os complexos de inclusão com CDs abrem novas perspectivas à utilização dos fármacos (Brewster et al., 1997; Duchêne, Wouessidjewe, 1990a,b; Duchêne et al., 1989; Fujioka et al., 1983; Miyaji et al., 1992; Rajeweski, Stella, 1996; Szejtli, 1990, 1991a; Thompson, 1997; Veiga et al., 1996).
Os principais objetivos a atingir com a utilização por via oral destes novos sistemas incluem:
- Aumento da solubilidade aquosa e melhoria da biodisponibilidade através do aumento da velocidade aparente e extensão da dissolução;
- Melhoria das características físicas para melhor homogeneização e compressão do fármaco sob a forma de comprimidos;
- Aumento da estabilidade ou do tempo de liberação, durante o trânsito gastrointestinal, pela modificação do local e/ou do perfil de tempo de liberação do fármaco;
- Modificação dos parâmetros farmacocinéticos, implicando na melhoria da absorção e em aumento dos níveis sanguíneos de fármacos pouco solúveis na água;
- Diminuição da irritação dos tecidos, dos efeitos colaterais e das reações hemolíticas;
- Mascaramento do cheiro e sabor desagradável;
- Redução de incompatibilidades entre fármacos, se pelo menos um deles estiver complexado;
- Obtenção de soluções aquosas estáveis de fármacos pouco solúveis em água, evitando, assim, o recurso à adição de solventes orgânicos.
Centenas de CDs modificadas têm sido preparadas e mostraram ter aplicação em investigação. Contudo, só alguns derivados têm potencial utilização terapêutica.
O aumento da biodisponibilidade é, assim, um dos efeitos mais significativos da complexação com CDs. A biodisponibilidade de uma especialidade farmacêutica, administrada por via oral, depende de vários fatores, entre os quais a velocidade de dissolução, a solubilidade e a velocidade de absorção intestinal. Doses idênticas de uma mesma especialidade farmacêutica administradas por via oral podem resultar, dependendo da formulação, em diferentes curvas concentração/tempo, podendo ocorrer em muitos casos absorção incompleta.
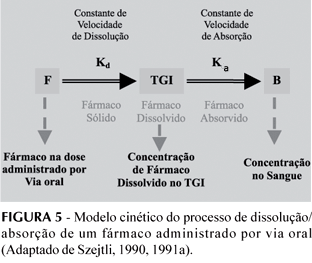
O processo de dissolução e absorção de um fármaco administrado por via oral pode, em síntese, ser expresso pelo modelo cinético representado na Figura 5.
- Se Kd > Ka - o fármaco é muito solúvel e a absorção é o fator limitante. Nestes casos, a complexação com CD não promove a absorção, podendo eventualmente diminuí-la.
- Se Kd < Ka - o fármaco é pouco solúvel e a dissolução é o fator limitante. Nestes casos, Kd tem que ser aumentado quer por micronização das partículas do fármaco, preparando soluções sólidas em matrizes hidrofílicas, quer adicionando agentes tensoativos, ou, ainda, formando complexos menos hidrofóbicos, por exemplo com ciclodextrinas. Sendo reconhecido que a velocidade de dissolução e solubilidade de fármacos hidrofóbicos aumenta consideravelmente quando são incluídos na cavidade das CDs, se Ka se mantiver constante, a concentração sanguínea aumentará (Szejtli, 1990, 1991a).
A inclusão de fármacos hidrofóbicos dentro da cavidade da CD pode, assim, melhorar a sua solubilidade e estabilidade em água, o grau e extensão da dissolução do complexo F:CD e a biodisponibilidade do fármaco, quando a dissolução e a solubilidade são fatores limitantes da disponibilidade (Duchêne et al., 1985; Mosher, Thompson, 2002).
A liberação de um fármaco no TGI, do ponto de vista da otimização farmacoterapêutica, deve ser controlada de acordo com a finalidade terapêutica e as propriedades farmacológicas da substância ativa, sendo possíveis diferentes formas de liberação (Figura 6).
As propriedades de biodegradação das CD são particularmente úteis, quando estas são utilizadas como transportadoras de fármacos para os vários locais de absorção ao longo do TGI.
As CD atuam essencialmente como transportadoras e protetoras do fármaco enquanto este atravessa o TGI, tendo sido aplicadas com sucesso quando o passo limitante é a solubilidade, a dissolução, o estado físico ou a degradação da molécula e não a sua absorção.
Quando um fármaco é administrado por via oral, inicia rapidamente a dissociação no fluido gastrointestinal, estando esta dependente da magnitude da constante de estabilidade do complexo e das características da CD utilizada relacionadas à degradação enzimática, observando-se, de um modo geral, que o aumento máximo de absorção é conseguido quando quantidade suficiente de CD é utilizada para solubilizar o fármaco. A adição de mais CD à solução de fármaco diminui a fração livre deste e, conseqüentemente, reduz a biodisponibilidade (Hirayama, Uekama, 1999).
Fundamentação para a utilização de complexos de inclusão de indometacina com hidroxipropil-b-ciclodextrina para administração por via oral a recém-nascidos
As principais vias de administração de medicamentos em recém-nascidos são a via intravenosa (IV) e a via oral/enteral.
O problema decorrente das técnicas de administração é o aparecimento de respostas não esperadas à terapêutica medicamentosa e desta dependente, as quais podem ser incorretamente atribuídas a outras causas. Foram identificados vários fatores que afetam a exatidão e previsibilidade do comportamento de medicamentos administrados por via IV.
Entre os fatores intrínsecos aos sistemas de administração de medicamentos, chama-se a atenção para o comprimento do sistema utilizado para a perfusão, uma vez que, quanto maior, mais prolongado é o tempo necessário para que o medicamento entre na corrente sanguínea e, ainda, superior, a hipótese de interações quer entre medicamentos, quer entre estes e o material de sistema. O diâmetro intraluminal do tubo do sistema é igualmente importante, porque influencia o volume, o ritmo de infusão e o tempo necessário à administração da dose. Em crianças muito pequenas, o tempo necessário ao início de infusão pode ser tão longo como 4 h e o necessário para terminar 6 h. Outro problema é inerente à bomba de perfusão utilizada em medicamentos de estreita margem terapêutica e doses da ordem das microgramas, uma vez que os seus impulsos alteram os picos de concentração, cujo ritmo de infusão deve ser contínuo para evitar flutuações, especialmente quando a duração de ação do medicamento é curta e o ritmo de infusão lento.
Os fatores extrínsecos que podem causar problemas na administração IV de medicamentos são a administração múltipla no que respeita à compatibilidade físico-química e hiperosmolalidade da solução final, bem como o volume hídrico que não deve ultrapassar os 60 mL nas 24 horas. Em relação às vias de acesso IV, elas são escassas no recém-nascido, podendo ser ao nível periférico - na mão, no pé, na flexura do braço, na cabeça, na veia e artéria umbilical - e central - introdução de catéter pela flexura até à veia cava - com evidentes problemas de fragilidade e diâmetro capilar, com a conseqüente dificuldade de punção e o risco de extravasamento, com necrose dos tecidos. O local de administração é, também, crítico, dado que podem sequestrar ou aprisionar parte da dose administrada.
Pelo que foi exposto e ainda cientes dos riscos de aspiração e refluxo que apresenta a via oral/enteral, devido à imaturidade do trato gastrointestinal e à toxicidade associada a alguns medicamentos e aos múltiplos fatores que imprimem grande variabilidade no comportamento das formas farmacêuticas orais/enterais, esta é, mesmo assim, a via que apresenta melhor relação benefício/risco para a administração de medicamentos em recém-nascidos.
As características de solubilidade e hidrólise da IM, a reduzida absorção gastrointestinal e potencial toxicidade gástrica em recém-nascidos são fatores que condicionam a elaboração de uma forma farmacêutica líquida para administração oral.
Neste contexto e tendo em consideração as vantagens da complexação de fármacos com ciclodextrinas, surge a idéia deste trabalho, com o objetivo de complexar a IM com HP-b-CD, conseguindo-se a modificação das características físico-químicas e de toxicidade, que até agora impediram a utilização de uma solução para administração por via oral ao recém-nascidos.
A análise das dimensões quer da cavidade da HP-b-CD, quer da molécula de IM inviabilizam a hipótese de inclusão completa. Todavia, estudos efetuados por diversos autores evidenciam que a molécula de IM sofre inclusão parcial na molécula de HP-b-CD, havendo, contudo, divergência quanto à parte da estrutura química da IM que é incluída na ciclodextrina. Na Figura 7 estão representadas as duas principais hipóteses de forma de inclusão: inclusão da parte do anel indol (Djedaïni et al., 1990) e inclusão da parte p-clorobenzeno, sendo esta última a que tem maiores defensores, fundamentada na sua dimensão (7 Å) e comprovada através de técnica de simulação do modelo molecular tipo Stuart e modelagem computacional, permitindo obter informação sobre a geometria e a estrutura dos compostos de inclusão (Backensfeld et al., 1990, 1991; Hamada et al., 1975; Kurozumi et al., 1975; Lin et al., 1991; Redenti et al., 2001). O caráter lipofílico da IM favorece, também, a formação do complexo.
A inclusão do grupo p-clorobenzeno confere proteção contra a hidrólise que ocorre em pH básico, dado que evita a cisão ao nível do grupo N-acil, aumentando a solubilidade e a velocidade de dissolução, prevenindo o contato direto com a mucosa gástrica e diminuindo o tempo de permanência da molécula, com redução da sua toxicidade local.
Uma vez que uma das objeções da administração por via oral da IM a recém-nascidos é a eventualidade de hidrólise devido ao pH alcalino no TGI, a complexação com HP-b-CD poderá, assim, possibilitar a administração por esta via, evitando a hidrólise da IM por proteção estereoquímica do grupo N-acil, uma vez que uma das conseqüências da interação entre a molécula-hóspede e a ciclodextrina é a proteção efetiva contra qualquer tipo de reação, para além daquelas que podem ocorrer com os grupos hidroxila da ciclodextrina (Szejtli, 1990). Por outro lado, a complexação aumenta a solubilidade e, conseqüentemente, a dissolução, o que contribui para a diminuição da toxicidade, não afetando o coeficiente de partilha, o que leva a supor que se manterá a capacidade de difusão transmembranar da IM.
Uma forma farmacêutica pó para preparação de solução oral apresenta diversas vantagens no que concerne à administração de fármacos a recém-nascidos, especialmente a facilidade de preparação extemporânea, a utilização da água como veículo, a não-obstrução das sondas nasogástricas de diâmetro muito reduzido pelo fato de ser uma solução e no, caso da administração oral, não ter sabor desagradável.
CONCLUSÕES
A investigação farmacêutica tem como intuito o desenvolvimento de novos agentes terapêuticos direcionados à resolução de situações clínicas, integrando conhecimentos pluridisciplinares e que a atenção farmacêutica possibilita a identificação de situações clínicas para as quais os recursos terapêuticos existentes não são os adequados.
O problema da carência de formas farmacêuticas adequadas, com que diariamente se debatem os profissionais de saúde que prestam cuidados a recém-nascidos e a pediatria no geral, impõe a atenção premente e a integração dos conhecimentos de todos os envolvidos nos cuidados de saúde, no sentido de dar solução ao dilema de quem prescreve, prepara e administra medicamentos às crianças. Medicamentos esses, que carecem da informação que é legalmente exigida para a introdução de novas formulações no mercado comercial, porque entre não tratar e correr o risco de morte e tratar com riscos de incerteza de eficiência e de segurança, opta-se pela segunda hipótese.
Os objetivos biofarmacêuticos em neonatologia devem, assim, visar à obtenção de agentes adequados ao grupo etário e requerem o estudo de possíveis vias de administração alternativas e avaliação de aspectos biofarmacêuticos, de segurança e eficácia de fármacos "in vivo" e "in vitro".
REFERÊNCIAS BIBLIOGRAFICAS
ANGERIO, A. D.; KOT, P. A. Closure of the ductus arteriosus: new insights for critical care. Crit. Care Nurs. Quarterly, Gaithersburg, v.20, n.4, p.80-85, 1998.
ANTENUCCI, R.N.; PALMER, J.K. Enzymatic degradation of a and b-cyclodextrins by Bacteroides of the human colon. J. Agric. Food Chem., Washington, v.32, n.6, p.1316-1321, 1984.
ARCHER, N. Patent ductus arteriosus in the newborn. Arch. Dis. Child., London, v.69, p. 529-532, 1993.
ARIMA, H.; KONDO, T.; IRIE, T.; UEKAMA, K. Enhanced rectal absorption and reduced local irritation of the anti-inflammatory drug ethyl 4-biphenylylacetate in rats by complexation with water-soluble b-cyclodextrin derivatives and formulation as oleaginous suppository. J. Pharm. Sci., New York, v.81, n.11, p.1119-1125, 1992.
BACKENSFELD, T.; MÜLLER, B. W.; WIESE, M.; SEYDEL, J. K. Effect of cyclodextrin derivatives on indomethacin stability in aqueous solution. Pharm. Res., New York, v.7, n.5, p.484-490, 1990.
BACKENSFELD, T.; MÜLLER, B.W.; KOLTER, K. Interaction of NSA with cyclodextrins and hydroxypropyl cyclodextrin derivatives. Int. J. Pharm., Amsterdam, v.74, p.85-93, 1991.
BAPTISTA, M.J.; CORREIA-PINTO, J.; AREIAS, J.C.; GUIMARÃES, H. Canal arterial patente em cuidados intensivos neonatais. Rev. Port. Cardiol., Lisboa, v.18, n.12, p.1095-1100, 1999.
BARST, R.J.; GERSONY, W.M. The pharmacological treatment of patent ductus arteriosus: a review of the evidence. Drugs, Auckland, v.38, n.2, p.249-266, 1989.
BELLRINGER, M.E.; SMITH, T. G.; READ, R., GOPINATH, C., OLIVIER, P. b-Cyclodextrin: 52-week toxicity studies in the rat and dog. Food. Chem. Toxicol., Oxford, v.33, n.5, p.367-376, 1995.
BESUNDER, J.B.; REED, M. D.; BLUMER, J. L. Principles of drug biodisposition, in the neonate. A critical evaluation of the pharmacokinetic-pharmacodynamic interface (part I). Clin. Pharmacokinet., Auckland, v.14, p.189-216, 1988.
BETTINETTI, G.P. Analisi del polimorfismo di un farmaco. Fármaco, Pavia, v.43, n.3, p.71-99, 1988.
BHATT, V.; NAHATA, M.C. Pharmacologic management of patent ductus arteriosus. Clin. Pharm., Bethesda, v.8, p.17-33, 1989.
BIWER, A.; ANTRANIKIAN, G.; HEINZLE, E. Enzimatic prodution of cyclodextrins. Appl. Microbiol. Biotechnol., Heidelberg, v.59, p.609-617, 2002.
BODOR, N.S.; HUANG, M-J.; WATTS, J.D. Theoretical studies on the structures of natural and alkylated cyclodextrins. J. Pharm. Sci., New York, v.84, n.3, p.330-336, 1995.
BORKA, L. The polymorphism of indomethacin. Acta Pharm. Suec., Stockholm, v.11, p.295-303, 1974.
BREWSTER, M.E.; ESTES, K.S.; BODOR, N. An intravenous toxicity study of 2-hydroxypropyl-b-cyclodextrin, a useful drug solubilizer, in rats and monkeys. Int. J. Pharm., Amsterdam, v.59, p.231-243, 1990.
BREWSTER, M.E.; LOFTSSON, T.; ESTES, K.S.; LIN, J-L.; FRIDRIKSDÓTTIR, H.; BODOR, N. Effect of various cyclodextrins on solution stability and dissolution rate of doxorubicin hydrochloride. Int. J. Pharm., Amsterdam, v.79, p.289-299, 1992.
BREWSTER, M.E.; ESTES, K.S.; BODOR, N. Intravenous and oral pharmacokinetic evaluation of a 2-hydroxypropyl-b-cyclodextrin-based formulation of carbamazepine in the dog: comparison with commercially available tablets and suspensions. J. Pharm. Sci., New York, v.86, n.3, p.335-339, 1997.
BRITTAIN, H.G. Spectral methods for the characterization of polymorphs and solvates. J. Pharm. Sci., New York, v.86, n.4, p.405-412, 1997.
BRITTAIN, H.G. Polymorphism: pharmaceutical aspects. In SWARBRICK, J.; BOYLAN, J.C., ed. Encyclopedia of Pharmaceutical Technology. 2nd ed. v.3. New York; Basel: Marcel Dekker, Inc., 2002. p. 2239-2249.
BULBENA, O., ESCOLAR, G., NAVARRO, C., BRAVO, L., PFEIFFER., C.J. Gastroprotective effect of zinc acexamate against damage induced by nonsteroidal anti-inflammatory drugs: a morphological study. Dig. Dis. Sci., New York, v.38, n.4, p.730-739, 1993.
BURI, P. Préformulation: pKa et coefficient de partage. Labo-Pharma Prob. Techn., Paris, v.307, p.181-186, 1981.
CABRAL-MARQUES, H. Structure and properties of cyclodextrins: inclusion complex formation. Rev. Port. Farm., Lisboa, v.44, n.2, p.77-84, 1994a.
CABRAL-MARQUES, H. Applications of cyclodextrins: thermodynamic aspects of cyclodextrin complexes. Rev. Port. Farm., Lisboa, v.44, n.2, p.85-96, 1994b.
CABRAL-MARQUES, H. Cyclodextrin's derivatives: absorption, toxicity, metabolism and fate. Rev. Port. Farm., Lisboa, v.44, n.4, p.147-156, 1994c.
CABRAL-MARQUES, H.; MORAIS, J.A. Ciclodextrinas e seus complexos de inclusão. Rev. Port. Farm., Lisboa, v.41, n.2, p.5-8, 1991.
CARPENTER, T.O.; GERLOCZY, A.; PITHA, J. Safety of parenteral hydroxypropyl-b-cyclodextrin. J. Pharm. Sci., New York, v.84 n.2, p.222-225, 1995.
CONNUCK, D.; SUN, J.P.; SUPER, D.M.; KIRCHNER, H.L.; FRADLEY, L.G.; HARCAR-SEVCIK, R.A.; SALVATOR, A.M.S.; SINGER, L.; MEHTA, S.K. Incidence of patent ductus arteriosus and patent foramen ovale in normal infants. Am. J. Cardiol., New York, v.89, n.2, p.244-247, 2002.
COOMBS, R.C.; MORGAN, M.E.I.; DURBIN, G.M.; BOOTH, I.W.; MCNEISH, A.S. Gut blood flow velocities in the newborn: effects of patent ductus arteriosus and parenteral indomethacin. Arch. Dis. Child., London, v.65, p.1067-1071, 1990.
DJEDAÏNI, F.; LIN, S.Z.; PERLY, B.; WOUESSIDJEWE, D. High-field nuclear magnetic resonance techniques for the investigation of a b-cyclodextrin:indomethacin inclusion complex. J. Pharm. Sci., New York, v.79, n.7, p.643-646, 1990.
DUCHÊNE, D.; WOUESSIDJEWE, D. Physicochemical characteristics and pharmaceutical uses of cyclodextrin derivatives. Pharm. Tech. Int., Eugene, v.6, p.21-29, 1990a.
DUCHÊNE, D.; WOUESSIDJEWE, D. Physicochemical characteristics and pharmaceutical uses of cyclodextrin derivatives, part II. Pharm. Tech., Eugene, v.14, n.8, p.22-30, 1990b.
DUCHÊNE, D.; VAUTION, C.; GLOMOT, F. La biodisponibilité des principes actifs par inclusion dans les cyclodextrines. S.T.P. Pharma., Paris, v.1, n.4, p.323-332, 1985.
DUCHÊNE, D.; VAUTION, C.; GLOMOT, F. Cyclodextrins, their value in pharmaceutical technology. In RUBINSTEIN, M.H. Pharmaceutical Technology. Drug Stability. New York: John Wiley & Sons, 1989. p.9-23.
ERDEN, N.; ÇELEBI, N. A study of the inclusion complex of naproxen with b-cyclodextrin. Int. J. Pharm., Amsterdam, v.48, p.83-89, 1988.
FINI, A.; FAZIO, G.; FEROCI, G. Solubility and solubilization properties of non-steroidal anti-inflammatory drugs. Int. J. Pharm., Amsterdam, v.126, p.95-102, 1995.
FLOURIÉ, B.; MOLIS, C.; ACHOUR, L.; DUPAS, H.; HATAT, C., RAMBAUD, J.C. Fate of b-cyclodextrin in the human intestine. J. Nutr., Bethesda, v.123, p.676-680, 1993.
FRIJLINK, H.W.; EISSENS, A.C.; SCHOONEN, A.J.M.; LERK, C.F. The effects of cyclodextrins on drug absorption II: in vivo observations. Int. J. Pharm., Amsterdam, v.64, p.195-205, 1990a.
FRIJLINK, H.W.; VISSER, J.; HEFTING, N.R.; OOSTING, R.; MEIJER, D.K.F.; LERK, C.F. The pharmacokinetics of b-cyclodextrin and hydroxypropyl-²-cyclodextrin in the rat. Pharm. Res., New York, v.7, n.12, p.1248-1252, 1990b.
FUJIOKA, K.; KUROSAKI, Y.; SATO, S.; NOGUCHI, T.; NOGUSHI, T.; YAMAHIRA, Y. Biopharmaceutical study of inclusion complexes. I. Pharmaceutical advantages of cyclodextrin complexes of bencyclane fumarate. Chem. Pharm. Bull., Tokyo, v.31, n.7, p.2416-2423, 1983.
GAL, P.; GILMAN, J.T. Drug disposition in neonates with patent ductus arteriosus. Ann. Pharmacother., Cincinnati, v.27, p.1383-1388, 1993.
GERLOCZY, A.; HOSHINO, T.; PITHA, J. Safety of oral cyclodextrins: effects of hydroxypropyl cyclodextrins, cyclodextrin sulfates and cationic cyclodextrins on steroid balance in rats. J. Pharm. Sci., New York, v.83, n.2, p.193-196, 1994.
GROSFELD, J.L.; CHAET, M.; MOLINARI, F.; ENGLE, W.; ENGUM, S.A.; WEST, K.W.; RESCORLA, F.J.; SCHERER, L.R. Increased risk of necrotizing enterocolitis in premature infants with patent ductus arteriosus treated with indomethacin. Ann. Surg., Philadelphia, v.224, n.3, p.350-357, 1996.
HABIB, M.J.; GHOSH, T.K.; AKOGYERAM, C.O.; AHMADI, B. Effects of cyclodextrines and phospholipids in enhancing dissolution of indomethacin. Drug Dev. Ind. Pharm., New York, v.21, n.15, p.1815-1822, 1995.
HAMADA, Y., NAMBU, N., NAGAI, T. Interactions of a and b-cyclodextrin with several non-steroidal anti-inflammatory drugs in aqueous solution. Chem. Pharm. Bull., Tokyo, v.23, n.6, p.1205-1211, 1975.
HAMMERMAN, C. Patent ductus arteriosus: clinical relevance of prostaglandins and prostaglandin inhibitors in PDA pathophysiology and treatment. Clin. Perinatol., Philadelphia, v.22, n.2, p.457-479, 1995.
HAMMERMAN, C.; KAPLAN, M. Comparative tolerability of pharmacological treatments for patent ductus arteriosus. Drug Saf., Auckland, v.24, n.7, p.537-551, 2001.
HIRAYAMA, F.; UEKAMA, K. Cyclodextrin-based controlled drug release system. Adv. Drug Deliv. Rev., Amsterdam, v.36, p.125-141, 1999.
HYMAN, P.E.; CLARKE, D.D.; EVERETT, S.L.; SONNE, B.; STEWART, D.; HARADA, T.; WALSH, J.H.; TAYLOR, I.L. Gastric acid secretory function in preterm infants. J. Pediatr., St. Louis, v.106, n.3, p.467-471, 1985.
IRIE, T.; FUKUNAGA, K.; PITHA, J. Hydroxypropylcyclodextrins in parenteral use. I: lipid dissolution and effects on lipid transfers in vivo. J. Pharm. Sci., New York, v.81, n.6, p.521-523, 1992a.
IRIE, T.; FUKUNAGA, K.; GARWOOD, M.K.; CARPENTER, T.O.; PITHA, J.; PITHA, J. Hydroxypropylcyclodextrins in parenteral use. II: effects on transport and disposition of lipids in rabbits and humans. J. Pharm. Sci., New York, v.81, n.6, p.524-528, 1992b.
IRIE, T.; UEKAMA, K. Pharmaceutical applications of cyclodextrins III: toxicological issues and safety evaluation. J. Pharm. Sci., New York, v.86, n.2, p.147-162, 1997.
JONES, S.P.; GRANT, D.J.W.; HADGRAFT, J.; PARR, G.D. Cyclodextrins in the pharmaceutical sciences. Part I: preparation, structure and properties of cyclodextrins and cyclodextrin inclusion compounds. Acta Pharm. Technol., Stuttgart, v.30, n.3, p.213-223, 1984.
KADIMA, L.N.; LESNE, M. Le métabolisme des médicaments chez le nouveau-né et le nourrisson. J. Pharm. Belg., Bruxelles, v.35, n.4, p.301-310, 1980.
KAUKONEN, A.M.; LENNERNÄS, H.; MANNERMAA, J.P. Water-soluble b-cyclodextrins in paediatric oral solutions of spironolactone: preclinical evaluation of spironolactone bioavailability from solutions of b-cyclodextrin derivatives in rats. J. Pharm. Pharmacol., London, v.50, p.611-619, 1998.
KRASNA, I.H.; LEE, R.T. Allopurinol protects the bowel from necrosis caused by indomethacin and temporary intestinal ischemia in mice. J. Pediatr. Surg., New York, v.28, n.9, p.1175-1177, 1993.
KRASOWSKA, H. Kinetics of indomethacin hydrolysis. Acta Pharm. Jugosl., Zagreb, v.24, p.193-201, 1974.
KUROZUMI, M.; NAMBU, N.; NAGAI, T. Inclusion compounds of non-steroidal anti-inflammatory and other slightly water soluble drugs with a and b-cyclodextrins in powdered form. Chem. Pharm. Bull., Tokyo, v.23, n.12, p.3062-3068, 1975.
LEFF, R.D.; ROBERTS, R.J. Problems in drug therapy for pediatric patients. Am. J. Hosp. Pharm., Bethesda, v.44, p.865-870, Apr, 1987.
LIN, S.Z. Isolation and solid-state characteristics of a new crystal form of indomethacin. J. Pharm. Sci., New York, v.81, n.6, p.572-576, 1992.
LIN, S.Z.; WOUESSIDJEWE, D.; POELMAN, M-C., DUCHÊNE, D. Indomethacin and cyclodextrin complexes. Int. J. Pharm., Amsterdam, v.69, p.211-219, 1991.
LIVERSIDGE, G.G.; DENT, J.; EICKHOFF, W.M. Influence of indomethacin amphoteric gel on gastric ulcerogenicity and absorption of indomethacin in rats. Pharm. Res., New York, v.6, n.1, p.44-48, 1989.
LOFTSSON, T.; BREWSTER, M.E. Pharmaceutical applications of cyclodextrins. 1. Drug solubilization and stabilization. J. Pharm. Sci., New York, v.85, n.10, p.1017-1025, 1996.
LOFTSSON, T.; BREWSTER, M.E. Cyclodextrins as pharmaceutical excipients. Pharm. Tech. Eur., Eugene, v.5, p.26-34, 1997.
LOFTSSON, T.; OLAFSSON, J.H. Cyclodextrins: new drug delivery systems in dermatology. Int. J. Dermatol., Philadelphia, v.37, n.4, p.241-246, 1998.
LUGEA, A.; SALAS, A.; GUARNER, F.; MALAGELADA, J-R. Influence of dietary fat on duodenal resistance to acid. Gut., London, v.34, p.1303-1309, 1993.
MAHMUD, T.; SOMASUNDARAM, S.; SIGTHORSSON, G.; SIMPSON, R.J.; RAFI, S.; FOSTER, R.; JACOB, M.; PACY, J.; BJARNASON, I.; SCOTT, D.L.; TAVARES, I.A.; WRIGGLESWORTH, J.M.; ROSETH, A. Enantiomers of flurbiprofen can distinguish key pathophysiological steps of NSAID enteropathy in the rat. Gut., London, v.43, p.775-82, 1998.
MALVIYA, M.; OHLSSON, A.; SHAH, S. Surgical versus medical treatment with cyclooxygenase inhibitors for symptomatic patent ductus arteriosus in preterm infants. COCHRANE NEONATAL GROUP. In The Cochrane Database of Systematic Reviews. v.3, 2003. Acessível em: http://gateway1.ovid.com/ovidweb.cgi . Consultado em: 21.01.2004.
MASON, S. Some aspects of gastric function in the newborn. Arch. Dis. Child., London, v.37, p.387-391, 1962.
MENGUY, R.; DESBAILLETS, L. Role of inhibition of gastric mucous secretion in the phenomenon of gastric mucosal injury by indomethacin. Am. J. Dig. Dis., Philadelphia, v.12, n.9, p.862-866, 1967.
MIYAJI, T.; INOUE, Y., ACARTÜRK, F.; IMAI, T.; OTAGIRI, M.; UEKAMA, K. Improvement of oral bioavailability of fenbufen by cyclodextrin complexations. Acta Pharm. Nord., Stockholm, v.4, n.1, p.17-22, 1992.
MIYAJIMA, K.; YOKOI, M.; KOMATSU, H.; NAKAGAKI, M. Interaction of b-cyclodextrin with bile salts in aqueous solutions. Chem. Pharm. Bull., Tokyo, v.34, n.3, p.1395-1398, 1986.
MIYAZAKI, S.; YAMAYRA, T.; NADAI, T. Effect of bile flow on indomethacin absorption in rats. Acta Pharm. Suec., Stockholm, v.18, p.135-138, 1981.
MOSHER, G.; THOMPSON, D.O. Complexation and Cyclodextrins. In SWARBRICK, J., BOYLAN, J.C. Encyclopedia of Pharmaceutical Technology. 2nd ed. v.1. New York; Basel: Marcel Dekker, Inc., p.531-558, 2002.
NAKANISHI, K.; MASADA, M.; NADAI, T.; MIYAJIMA, K. Effect of the interaction of drugs-²-cyclodextrin complex with bile salts on the drug absorption from rat small intestinal lumen. Chem. Pharm. Bull., Tokyo, v.37, n.1, p.211-214, 1989.
NAKANISHI, K.; NADA, T.; MASADA, M.; MIYAJIMA, K. Effect of cyclodextrins on biological membrane. I. Mechanism of enhancement on the intestinal absorption of non-absorbable drug by cyclodextrins. Chem. Pharm. Bull., Tokyo, v.40, n.5, p.1252-1256, 1992.
O'BRIEN, M.; McCAULEY, J.; COHEN, E. Indomethacin. In FLOREY, K. (Ed.) Analytical Profiles of Drug Substances. San Diego: Academic Press Inc., v.13, 1984. p.211-238.
PARENT, L.S. NSAID: induced gastropathy. Drug Topics, New York, v.135, n.3, p.63-71, 1991.
PELLEGRIN, P.; LESNE, M. La posologie des médicaments chez le nouveau-né, le nourrisson et l'enfant et leurs contre-indications. J. Pharm. Belg., Bruxelles, v.35, n.4, p.289-300, 1980.
PITHA, J.; PITHA J. Amorphous water-soluble derivatives of cyclodextrins: nontoxic dissolution enhancing excipients. J. Pharm. Sci., New York, v.74, n.9, p.987-990, 1985.
PITHA, J.; MILECKI, J.; FALES, H.; PANNELL, L.; UEKAMA, K. Hydroxypropryl-b-cyclodextrin: preparation and characterization; effects on solubility of drugs. Int. J. Pharm., Amsterdam, v.29, p.73-82, 1986.
RAINSFORD, K.D. NSAID gastropathy: novel physicochemical approaches for reducing gastric mucosal injury by drug complexation with cyclodextrins. Drug Invest., Auckland, v.2, suppl.4, p.3-10, 1990.
RAJEWESKI, R.A.; STELLA, V.J. Pharmaceutical applications of cyclodextrins. 2. In vivo drug delivery. J. Pharm. Sci., New York, v.85, n.11, p.1142-1169, 1996.
REDENTI, E.; SZENTE, L.; SZEJTLI, J. Drug/Cyclodextrin/hydroxyl acid multicomponent systems. Properties and Pharmaceutical applications. J. Pharm. Sci., New York, v.89, n.1, p.1-8, 2000.
REDENTI, E.; SZENTE, L.; SZEJTLI, J. Cyclodextrin complexes of salts of acidic drugs: thermodynamic properties, structural features, and pharmaceutical applications. J. Pharm. Sci., New York, v.90, n.8, p.979-986, 2001.
RENFRO, W.H.; BURR, J.M.; RAWLINGS, D.J. Criteria for use of indomethacin injection in neonates. Clin. Pharm., Bethesda, v.12, p.232-234, 1993.
RICHARDSON, D. A woman with an extremely premature newborn. J. Am. Med. Ass., Chicago, v.286, n.12, p.1498-1505, 2001.
ROMAGNOLI, C.; ZECCA, E.; PAPACCI, P.; CAROLIS, M.P.; GIANNINI, R.; GALLINI, F.; TORTOROLO, G. Furosemide does not prevent indomethacin-induced renal side effects in preterm infants. Clin. Pharmacol. Ther., St. Louis, v.62, n.2, p.181-186, 1997.
SALTÃO, R.; VEIGA, F. Ciclodextrinas em novos sistemas terapêuticos. Rev. Bras. Cien. Farm., São Paulo, v.37, n.1, p.1-17, 2001.
SHARGEL, L.; YU, A. B. C. Biopharmaceutics. In: SWARBRICK, J., BOYLAN, J. C. Enciclopedia of Pharmaceutical Technology, 2nd. ed. v.1. New York; Basel: Marcel Dekker, Inc. 2002. p.156-176.
SCHMIDT, B.; DAVIS, P.; MODDEMANN, D.; OHLSSON, A.; ROBERTS, R.S.; SAIGAL, S.; SOLIMANO, A.; VINCER, M.; WRIGHT, L.L. Long-term effects of indomethacin prophylaxis in extremely-low-birth-weight infants. New Engl. J. Med., Boston, v.344, n.26, p.1966-1972, 2001.
SHAMAT, M.A. The role of the gastrointestinal microflora in the metabolism of drugs. Int. J. Pharm., Amsterdam, v.97, p.1-13, 1993.
SOMASUNDARAM, S.; RAFI, S.; HAYLLAR, J.; SIGTHORSSON, G.; JACOB, M.; PRICE, A.B.; MACPHERSON, A.; MAHMOD, T.; SCOTT, D.; WRIGGLESWORTH, J.M.; BJARNASON, I. Mitochondrial damage: a possible mechanism of the "topical" phase of NSAID induced injury to the rat intestine. Gut, London, v.41, n.3, p.344-353, 1997.
STELLA, V.J.; RAO, V.M.; ZANNOU, E.A.; ZIA, V. Mechanisms of drug release from cyclodextrin complexes. Adv. Drug Deliv. Rev., Amsterdam, v.36, p.3-16, 1999.
STELLA, V.J.; RAJEWSKI, R.A. Cyclodextrins: their future in drug formulation and delivery. Pharm. Res., New York, v.14, n.5, p.556-567, 1997.
STEWART, C.F.; HAMPTON, E.M. Therapy review: effect of maturation on drug disposition in pediatric patients. Clin. Pharm., Bethesda, v.6, p.548-564, 1987.
STRATTAN, C.E. 2-Hydroxypropyl-b-cyclodextrin, part II: safety and manufacturing issues. Pharm. Tech., Eugene, v.2, p.52-58, 1992.
SZEJTLI, J. Cyclodextrin inclusion complexes. In: SZEJTLI, J. Ed. Cyclodextrin Technology. London: Kluwer Academic Publishers, 1988, p. 79-185.
SZEJTLI, J. Cyclodextrins: properties and applications. Drug Invest., Auckland, v.2, suppl.4, p.11-21, 1990.
SZEJTLI, J. Cyclodextrins in drug formulations: part I. Pharm. Tech. Int., Eugene, v.2, p.15-22, 1991a.
SZEJTLI, J. Cyclodextrins in drug formulations: part II. Pharm. Tech. Int., Eugene, v.3, p.16-24, 1991b.
TAN, X.; LINDENBAUM, S. Studies on complexation between b-cyclodextrin and bile salts. Int. J. Pharm., Amsterdam, v.74, p.127-135, 1991.
THOMPSON, D.O. Cyclodextrins - enabling excipients: their present and future use in pharmaceuticals. Crit. Rev. Ther. Drug Carrier Syst., New York, v.14, n.1, p.1-104, 1997.
TöTTERMAN, A.N.; SCHIPPER, N.G.M.; THOMPSON, D.O.; MANNERMAA, J-P. Intestinal safety of water-soluble b-cyclodextrins in paediatric oral solutions of spironolactone: effects on humanintestinal epithelial caco-2 cells. J. Pharm. Pharmacol., London, v.49, p.43-48, 1997.
TRESZL, A.; SZABÓ, M.; DUNAI, G.; NOBILIS, A.; KOCSIS, I.; MACHAY, T.; TULASSAY, T.; VÁSÁRHELYI, B. Angiotensin II type 1 receptor A1166C polymorphism and prophylactic indomethacin treatment induced ductus arteriosus closure in very low birth weight neonates. Pediatr. Res., Baltimore, v.54, n.5, p.753-755, 2003.
VEIGA, F. Complexos de inclusão com ciclodextrinas hidrófilas. Caracterização físico-química e biofarmacêutica. Coimbra, 1996. [Tese de doutoramento. Faculdade de Farmácia. Universidade de Coimbra].
VEIGA, F.; TEIXEIR-DIAS, J.J.C.; KEDZIEREWICZ, F.; SOUSA, A.; MAINCENT, P. Inclusion complexation of tolbutamide wiyh b-cyclodextrin and hydroxypropyl-b-cyclodextrin. Int. J. Pharm., Amsterdam, v.129, n.63-71, 1996.
VERT, P.; BIANCHETTI, G.; MARCHAL, F.; MONIN, P.; MORSELLI, P.L. Effectiveness and pharmacokinetics of indomethacin in premature newborns with patent ductus arteriosus. Eur. J. Clin. Pharmacol., Berlin, v.18, p.83-88, 1980.
WATKINS, J.B.; INGALL, D.; SZCZEPANIK, P.; KLEIN, P.D.; LESTER R. Bile-salt metabolism in the newborn: measurement of pool size and synthesis by stable isotope technique. N. Eng. J. Med., Boston, v.288, n.9, p.431-433, 1973.
WIEST, D.B.; PINSON, J.B.; GAL, P.S.; BRUNDAGE, R.C.; SCHALL, S.; RANSOM, J.L.; WEAVER, R.L.; PUROHIT, D.; BROWN, Y. Population pharmacokinetics of intravenous indomethacin in neonates with symptomatic patent ductus arteriosus. Clin. Pharmacol. Therap., St. Louis, v.49, n.5, p.550-557, 1991.
YEH, T.F.; CARR, I. Pharmacological closure of patent ductus arteriosus. In: YEH, T.F. Neonatal Therapeutics. St. Louis: Mosby Year Book, 1991. p.123-138.
YOSHIDA, A.; ARIMA, H.; UEKAMA, K.; PITHA, J. Pharmaceutical evaluation of hydroxyalkyl ethers of ²-cyclodextrins. Int. J. Pharm., Amsterdam, v.46, p.217-222, 1988.
YOSHIOKA, H.; ISEKI, K.; FUJITA, K. Development and differences of intestinal flora in the neonatal period in breast-fed and bottle-fed infants. Pediatrics, Evaston, v.72, n.3, p.317-321, 1983.
Recebido para publicação em 02 de junho de 2004.
Aceito para publicação em 22 de agosto de 2005.
- ANGERIO, A. D.; KOT, P. A. Closure of the ductus arteriosus: new insights for critical care. Crit. Care Nurs. Quarterly, Gaithersburg, v.20, n.4, p.80-85, 1998.
- ANTENUCCI, R.N.; PALMER, J.K. Enzymatic degradation of a and b-cyclodextrins by Bacteroides of the human colon. J. Agric. Food Chem., Washington, v.32, n.6, p.1316-1321, 1984.
- ARCHER, N. Patent ductus arteriosus in the newborn. Arch. Dis. Child., London, v.69, p. 529-532, 1993.
- ARIMA, H.; KONDO, T.; IRIE, T.; UEKAMA, K. Enhanced rectal absorption and reduced local irritation of the anti-inflammatory drug ethyl 4-biphenylylacetate in rats by complexation with water-soluble b-cyclodextrin derivatives and formulation as oleaginous suppository. J. Pharm. Sci., New York, v.81, n.11, p.1119-1125, 1992.
- BACKENSFELD, T.; MÜLLER, B. W.; WIESE, M.; SEYDEL, J. K. Effect of cyclodextrin derivatives on indomethacin stability in aqueous solution. Pharm. Res, New York, v.7, n.5, p.484-490, 1990.
- BACKENSFELD, T.; MÜLLER, B.W.; KOLTER, K. Interaction of NSA with cyclodextrins and hydroxypropyl cyclodextrin derivatives. Int. J. Pharm, Amsterdam, v.74, p.85-93, 1991.
- BAPTISTA, M.J.; CORREIA-PINTO, J.; AREIAS, J.C.; GUIMARÃES, H. Canal arterial patente em cuidados intensivos neonatais. Rev. Port. Cardiol., Lisboa, v.18, n.12, p.1095-1100, 1999.
- BARST, R.J.; GERSONY, W.M. The pharmacological treatment of patent ductus arteriosus: a review of the evidence. Drugs, Auckland, v.38, n.2, p.249-266, 1989.
- BELLRINGER, M.E.; SMITH, T. G.; READ, R., GOPINATH, C., OLIVIER, P. b-Cyclodextrin: 52-week toxicity studies in the rat and dog. Food. Chem. Toxicol., Oxford, v.33, n.5, p.367-376, 1995.
- BESUNDER, J.B.; REED, M. D.; BLUMER, J. L. Principles of drug biodisposition, in the neonate. A critical evaluation of the pharmacokinetic-pharmacodynamic interface (part I). Clin. Pharmacokinet, Auckland, v.14, p.189-216, 1988.
- BETTINETTI, G.P. Analisi del polimorfismo di un farmaco. Fármaco, Pavia, v.43, n.3, p.71-99, 1988.
- BHATT, V.; NAHATA, M.C. Pharmacologic management of patent ductus arteriosus. Clin. Pharm, Bethesda, v.8, p.17-33, 1989.
- BIWER, A.; ANTRANIKIAN, G.; HEINZLE, E. Enzimatic prodution of cyclodextrins. Appl. Microbiol. Biotechnol, Heidelberg, v.59, p.609-617, 2002.
- BODOR, N.S.; HUANG, M-J.; WATTS, J.D. Theoretical studies on the structures of natural and alkylated cyclodextrins. J. Pharm. Sci., New York, v.84, n.3, p.330-336, 1995.
- BORKA, L. The polymorphism of indomethacin. Acta Pharm. Suec, Stockholm, v.11, p.295-303, 1974.
- BREWSTER, M.E.; ESTES, K.S.; BODOR, N. An intravenous toxicity study of 2-hydroxypropyl-b-cyclodextrin, a useful drug solubilizer, in rats and monkeys. Int. J. Pharm, Amsterdam, v.59, p.231-243, 1990.
- BREWSTER, M.E.; LOFTSSON, T.; ESTES, K.S.; LIN, J-L.; FRIDRIKSDÓTTIR, H.; BODOR, N. Effect of various cyclodextrins on solution stability and dissolution rate of doxorubicin hydrochloride. Int. J. Pharm., Amsterdam, v.79, p.289-299, 1992.
- BREWSTER, M.E.; ESTES, K.S.; BODOR, N. Intravenous and oral pharmacokinetic evaluation of a 2-hydroxypropyl-b-cyclodextrin-based formulation of carbamazepine in the dog: comparison with commercially available tablets and suspensions. J. Pharm. Sci., New York, v.86, n.3, p.335-339, 1997.
- BRITTAIN, H.G. Spectral methods for the characterization of polymorphs and solvates. J. Pharm. Sci, New York, v.86, n.4, p.405-412, 1997.
- BRITTAIN, H.G. Polymorphism: pharmaceutical aspects. In SWARBRICK, J.; BOYLAN, J.C., ed. Encyclopedia of Pharmaceutical Technology 2nd ed. v.3. New York; Basel: Marcel Dekker, Inc., 2002. p. 2239-2249.
- BULBENA, O., ESCOLAR, G., NAVARRO, C., BRAVO, L., PFEIFFER., C.J. Gastroprotective effect of zinc acexamate against damage induced by nonsteroidal anti-inflammatory drugs: a morphological study. Dig. Dis. Sci, New York, v.38, n.4, p.730-739, 1993.
- BURI, P. Préformulation: pKa et coefficient de partage. Labo-Pharma Prob. Techn., Paris, v.307, p.181-186, 1981.
- CABRAL-MARQUES, H. Structure and properties of cyclodextrins: inclusion complex formation. Rev. Port. Farm., Lisboa, v.44, n.2, p.77-84, 1994a.
- CABRAL-MARQUES, H. Applications of cyclodextrins: thermodynamic aspects of cyclodextrin complexes. Rev. Port. Farm., Lisboa, v.44, n.2, p.85-96, 1994b.
- CABRAL-MARQUES, H. Cyclodextrin's derivatives: absorption, toxicity, metabolism and fate. Rev. Port. Farm., Lisboa, v.44, n.4, p.147-156, 1994c.
- CABRAL-MARQUES, H.; MORAIS, J.A. Ciclodextrinas e seus complexos de inclusão. Rev. Port. Farm., Lisboa, v.41, n.2, p.5-8, 1991.
- CARPENTER, T.O.; GERLOCZY, A.; PITHA, J. Safety of parenteral hydroxypropyl-b-cyclodextrin. J. Pharm. Sci., New York, v.84 n.2, p.222-225, 1995.
- CONNUCK, D.; SUN, J.P.; SUPER, D.M.; KIRCHNER, H.L.; FRADLEY, L.G.; HARCAR-SEVCIK, R.A.; SALVATOR, A.M.S.; SINGER, L.; MEHTA, S.K. Incidence of patent ductus arteriosus and patent foramen ovale in normal infants. Am. J. Cardiol., New York, v.89, n.2, p.244-247, 2002.
- COOMBS, R.C.; MORGAN, M.E.I.; DURBIN, G.M.; BOOTH, I.W.; MCNEISH, A.S. Gut blood flow velocities in the newborn: effects of patent ductus arteriosus and parenteral indomethacin. Arch. Dis. Child., London, v.65, p.1067-1071, 1990.
- DJEDAÏNI, F.; LIN, S.Z.; PERLY, B.; WOUESSIDJEWE, D. High-field nuclear magnetic resonance techniques for the investigation of a b-cyclodextrin:indomethacin inclusion complex. J. Pharm. Sci., New York, v.79, n.7, p.643-646, 1990.
- DUCHÊNE, D.; WOUESSIDJEWE, D. Physicochemical characteristics and pharmaceutical uses of cyclodextrin derivatives. Pharm. Tech. Int., Eugene, v.6, p.21-29, 1990a.
- DUCHÊNE, D.; WOUESSIDJEWE, D. Physicochemical characteristics and pharmaceutical uses of cyclodextrin derivatives, part II. Pharm. Tech., Eugene, v.14, n.8, p.22-30, 1990b.
- DUCHÊNE, D.; VAUTION, C.; GLOMOT, F. La biodisponibilité des principes actifs par inclusion dans les cyclodextrines. S.T.P. Pharma., Paris, v.1, n.4, p.323-332, 1985.
- DUCHÊNE, D.; VAUTION, C.; GLOMOT, F. Cyclodextrins, their value in pharmaceutical technology. In RUBINSTEIN, M.H. Pharmaceutical Technology. Drug Stability. New York: John Wiley & Sons, 1989. p.9-23.
- ERDEN, N.; ÇELEBI, N. A study of the inclusion complex of naproxen with b-cyclodextrin. Int. J. Pharm., Amsterdam, v.48, p.83-89, 1988.
- FINI, A.; FAZIO, G.; FEROCI, G. Solubility and solubilization properties of non-steroidal anti-inflammatory drugs. Int. J. Pharm., Amsterdam, v.126, p.95-102, 1995.
- FLOURIÉ, B.; MOLIS, C.; ACHOUR, L.; DUPAS, H.; HATAT, C., RAMBAUD, J.C. Fate of b-cyclodextrin in the human intestine. J. Nutr., Bethesda, v.123, p.676-680, 1993.
- FRIJLINK, H.W.; EISSENS, A.C.; SCHOONEN, A.J.M.; LERK, C.F. The effects of cyclodextrins on drug absorption II: in vivo observations. Int. J. Pharm., Amsterdam, v.64, p.195-205, 1990a.
- FRIJLINK, H.W.; VISSER, J.; HEFTING, N.R.; OOSTING, R.; MEIJER, D.K.F.; LERK, C.F. The pharmacokinetics of b-cyclodextrin and hydroxypropyl-²-cyclodextrin in the rat. Pharm. Res., New York, v.7, n.12, p.1248-1252, 1990b.
- FUJIOKA, K.; KUROSAKI, Y.; SATO, S.; NOGUCHI, T.; NOGUSHI, T.; YAMAHIRA, Y. Biopharmaceutical study of inclusion complexes. I. Pharmaceutical advantages of cyclodextrin complexes of bencyclane fumarate. Chem. Pharm. Bull., Tokyo, v.31, n.7, p.2416-2423, 1983.
- GAL, P.; GILMAN, J.T. Drug disposition in neonates with patent ductus arteriosus. Ann. Pharmacother., Cincinnati, v.27, p.1383-1388, 1993.
- GERLOCZY, A.; HOSHINO, T.; PITHA, J. Safety of oral cyclodextrins: effects of hydroxypropyl cyclodextrins, cyclodextrin sulfates and cationic cyclodextrins on steroid balance in rats. J. Pharm. Sci., New York, v.83, n.2, p.193-196, 1994.
- GROSFELD, J.L.; CHAET, M.; MOLINARI, F.; ENGLE, W.; ENGUM, S.A.; WEST, K.W.; RESCORLA, F.J.; SCHERER, L.R. Increased risk of necrotizing enterocolitis in premature infants with patent ductus arteriosus treated with indomethacin. Ann. Surg., Philadelphia, v.224, n.3, p.350-357, 1996.
- HABIB, M.J.; GHOSH, T.K.; AKOGYERAM, C.O.; AHMADI, B. Effects of cyclodextrines and phospholipids in enhancing dissolution of indomethacin. Drug Dev. Ind. Pharm., New York, v.21, n.15, p.1815-1822, 1995.
- HAMADA, Y., NAMBU, N., NAGAI, T. Interactions of a and b-cyclodextrin with several non-steroidal anti-inflammatory drugs in aqueous solution. Chem. Pharm. Bull., Tokyo, v.23, n.6, p.1205-1211, 1975.
- HAMMERMAN, C. Patent ductus arteriosus: clinical relevance of prostaglandins and prostaglandin inhibitors in PDA pathophysiology and treatment. Clin. Perinatol., Philadelphia, v.22, n.2, p.457-479, 1995.
- HAMMERMAN, C.; KAPLAN, M. Comparative tolerability of pharmacological treatments for patent ductus arteriosus. Drug Saf., Auckland, v.24, n.7, p.537-551, 2001.
- HIRAYAMA, F.; UEKAMA, K. Cyclodextrin-based controlled drug release system. Adv. Drug Deliv. Rev., Amsterdam, v.36, p.125-141, 1999.
- HYMAN, P.E.; CLARKE, D.D.; EVERETT, S.L.; SONNE, B.; STEWART, D.; HARADA, T.; WALSH, J.H.; TAYLOR, I.L. Gastric acid secretory function in preterm infants. J. Pediatr., St. Louis, v.106, n.3, p.467-471, 1985.
- IRIE, T.; FUKUNAGA, K.; PITHA, J. Hydroxypropylcyclodextrins in parenteral use. I: lipid dissolution and effects on lipid transfers in vivo. J. Pharm. Sci., New York, v.81, n.6, p.521-523, 1992a.
- IRIE, T.; FUKUNAGA, K.; GARWOOD, M.K.; CARPENTER, T.O.; PITHA, J.; PITHA, J. Hydroxypropylcyclodextrins in parenteral use. II: effects on transport and disposition of lipids in rabbits and humans. J. Pharm. Sci., New York, v.81, n.6, p.524-528, 1992b.
- IRIE, T.; UEKAMA, K. Pharmaceutical applications of cyclodextrins III: toxicological issues and safety evaluation. J. Pharm. Sci., New York, v.86, n.2, p.147-162, 1997.
- JONES, S.P.; GRANT, D.J.W.; HADGRAFT, J.; PARR, G.D. Cyclodextrins in the pharmaceutical sciences. Part I: preparation, structure and properties of cyclodextrins and cyclodextrin inclusion compounds. Acta Pharm. Technol., Stuttgart, v.30, n.3, p.213-223, 1984.
- KADIMA, L.N.; LESNE, M. Le métabolisme des médicaments chez le nouveau-né et le nourrisson. J. Pharm. Belg., Bruxelles, v.35, n.4, p.301-310, 1980.
- KAUKONEN, A.M.; LENNERNÄS, H.; MANNERMAA, J.P. Water-soluble b-cyclodextrins in paediatric oral solutions of spironolactone: preclinical evaluation of spironolactone bioavailability from solutions of b-cyclodextrin derivatives in rats. J. Pharm. Pharmacol., London, v.50, p.611-619, 1998.
- KRASNA, I.H.; LEE, R.T. Allopurinol protects the bowel from necrosis caused by indomethacin and temporary intestinal ischemia in mice. J. Pediatr. Surg., New York, v.28, n.9, p.1175-1177, 1993.
- KRASOWSKA, H. Kinetics of indomethacin hydrolysis. Acta Pharm. Jugosl., Zagreb, v.24, p.193-201, 1974.
- KUROZUMI, M.; NAMBU, N.; NAGAI, T. Inclusion compounds of non-steroidal anti-inflammatory and other slightly water soluble drugs with a and b-cyclodextrins in powdered form. Chem. Pharm. Bull., Tokyo, v.23, n.12, p.3062-3068, 1975.
- LEFF, R.D.; ROBERTS, R.J. Problems in drug therapy for pediatric patients. Am. J. Hosp. Pharm., Bethesda, v.44, p.865-870, Apr, 1987.
- LIN, S.Z. Isolation and solid-state characteristics of a new crystal form of indomethacin. J. Pharm. Sci., New York, v.81, n.6, p.572-576, 1992.
- LIN, S.Z.; WOUESSIDJEWE, D.; POELMAN, M-C., DUCHÊNE, D. Indomethacin and cyclodextrin complexes. Int. J. Pharm., Amsterdam, v.69, p.211-219, 1991.
- LIVERSIDGE, G.G.; DENT, J.; EICKHOFF, W.M. Influence of indomethacin amphoteric gel on gastric ulcerogenicity and absorption of indomethacin in rats. Pharm. Res., New York, v.6, n.1, p.44-48, 1989.
- LOFTSSON, T.; BREWSTER, M.E. Pharmaceutical applications of cyclodextrins. 1. Drug solubilization and stabilization. J. Pharm. Sci., New York, v.85, n.10, p.1017-1025, 1996.
- LOFTSSON, T.; BREWSTER, M.E. Cyclodextrins as pharmaceutical excipients. Pharm. Tech. Eur., Eugene, v.5, p.26-34, 1997.
- LOFTSSON, T.; OLAFSSON, J.H. Cyclodextrins: new drug delivery systems in dermatology. Int. J. Dermatol., Philadelphia, v.37, n.4, p.241-246, 1998.
- LUGEA, A.; SALAS, A.; GUARNER, F.; MALAGELADA, J-R. Influence of dietary fat on duodenal resistance to acid. Gut., London, v.34, p.1303-1309, 1993.
- MAHMUD, T.; SOMASUNDARAM, S.; SIGTHORSSON, G.; SIMPSON, R.J.; RAFI, S.; FOSTER, R.; JACOB, M.; PACY, J.; BJARNASON, I.; SCOTT, D.L.; TAVARES, I.A.; WRIGGLESWORTH, J.M.; ROSETH, A. Enantiomers of flurbiprofen can distinguish key pathophysiological steps of NSAID enteropathy in the rat. Gut., London, v.43, p.775-82, 1998.
- MALVIYA, M.; OHLSSON, A.; SHAH, S. Surgical versus medical treatment with cyclooxygenase inhibitors for symptomatic patent ductus arteriosus in preterm infants. COCHRANE NEONATAL GROUP. In The Cochrane Database of Systematic Reviews. v.3, 2003. Acessível em: http://gateway1.ovid.com/ovidweb.cgi . Consultado em: 21.01.2004.
- MASON, S. Some aspects of gastric function in the newborn. Arch. Dis. Child., London, v.37, p.387-391, 1962.
- MENGUY, R.; DESBAILLETS, L. Role of inhibition of gastric mucous secretion in the phenomenon of gastric mucosal injury by indomethacin. Am. J. Dig. Dis., Philadelphia, v.12, n.9, p.862-866, 1967.
- MIYAJI, T.; INOUE, Y., ACARTÜRK, F.; IMAI, T.; OTAGIRI, M.; UEKAMA, K. Improvement of oral bioavailability of fenbufen by cyclodextrin complexations. Acta Pharm. Nord., Stockholm, v.4, n.1, p.17-22, 1992.
- MIYAJIMA, K.; YOKOI, M.; KOMATSU, H.; NAKAGAKI, M. Interaction of b-cyclodextrin with bile salts in aqueous solutions. Chem. Pharm. Bull., Tokyo, v.34, n.3, p.1395-1398, 1986.
- MIYAZAKI, S.; YAMAYRA, T.; NADAI, T. Effect of bile flow on indomethacin absorption in rats. Acta Pharm. Suec., Stockholm, v.18, p.135-138, 1981.
- MOSHER, G.; THOMPSON, D.O. Complexation and Cyclodextrins. In SWARBRICK, J., BOYLAN, J.C. Encyclopedia of Pharmaceutical Technology. 2nd ed. v.1. New York; Basel: Marcel Dekker, Inc., p.531-558, 2002.
- NAKANISHI, K.; MASADA, M.; NADAI, T.; MIYAJIMA, K. Effect of the interaction of drugs-²-cyclodextrin complex with bile salts on the drug absorption from rat small intestinal lumen. Chem. Pharm. Bull., Tokyo, v.37, n.1, p.211-214, 1989.
- NAKANISHI, K.; NADA, T.; MASADA, M.; MIYAJIMA, K. Effect of cyclodextrins on biological membrane. I. Mechanism of enhancement on the intestinal absorption of non-absorbable drug by cyclodextrins. Chem. Pharm. Bull., Tokyo, v.40, n.5, p.1252-1256, 1992.
- O'BRIEN, M.; McCAULEY, J.; COHEN, E. Indomethacin. In FLOREY, K. (Ed.) Analytical Profiles of Drug Substances. San Diego: Academic Press Inc., v.13, 1984. p.211-238.
- PARENT, L.S. NSAID: induced gastropathy. Drug Topics, New York, v.135, n.3, p.63-71, 1991.
- PELLEGRIN, P.; LESNE, M. La posologie des médicaments chez le nouveau-né, le nourrisson et l'enfant et leurs contre-indications. J. Pharm. Belg., Bruxelles, v.35, n.4, p.289-300, 1980.
- PITHA, J.; PITHA J. Amorphous water-soluble derivatives of cyclodextrins: nontoxic dissolution enhancing excipients. J. Pharm. Sci., New York, v.74, n.9, p.987-990, 1985.
- PITHA, J.; MILECKI, J.; FALES, H.; PANNELL, L.; UEKAMA, K. Hydroxypropryl-b-cyclodextrin: preparation and characterization; effects on solubility of drugs. Int. J. Pharm., Amsterdam, v.29, p.73-82, 1986.
- RAINSFORD, K.D. NSAID gastropathy: novel physicochemical approaches for reducing gastric mucosal injury by drug complexation with cyclodextrins. Drug Invest., Auckland, v.2, suppl.4, p.3-10, 1990.
- RAJEWESKI, R.A.; STELLA, V.J. Pharmaceutical applications of cyclodextrins. 2. In vivo drug delivery. J. Pharm. Sci., New York, v.85, n.11, p.1142-1169, 1996.
- REDENTI, E.; SZENTE, L.; SZEJTLI, J. Drug/Cyclodextrin/hydroxyl acid multicomponent systems. Properties and Pharmaceutical applications. J. Pharm. Sci., New York, v.89, n.1, p.1-8, 2000.
- REDENTI, E.; SZENTE, L.; SZEJTLI, J. Cyclodextrin complexes of salts of acidic drugs: thermodynamic properties, structural features, and pharmaceutical applications. J. Pharm. Sci., New York, v.90, n.8, p.979-986, 2001.
- RENFRO, W.H.; BURR, J.M.; RAWLINGS, D.J. Criteria for use of indomethacin injection in neonates. Clin. Pharm., Bethesda, v.12, p.232-234, 1993.
- RICHARDSON, D. A woman with an extremely premature newborn. J. Am. Med. Ass., Chicago, v.286, n.12, p.1498-1505, 2001.
- ROMAGNOLI, C.; ZECCA, E.; PAPACCI, P.; CAROLIS, M.P.; GIANNINI, R.; GALLINI, F.; TORTOROLO, G. Furosemide does not prevent indomethacin-induced renal side effects in preterm infants. Clin. Pharmacol. Ther., St. Louis, v.62, n.2, p.181-186, 1997.
- SALTÃO, R.; VEIGA, F. Ciclodextrinas em novos sistemas terapêuticos. Rev. Bras. Cien. Farm., São Paulo, v.37, n.1, p.1-17, 2001.
- SHARGEL, L.; YU, A. B. C. Biopharmaceutics. In: SWARBRICK, J., BOYLAN, J. C. Enciclopedia of Pharmaceutical Technology, 2nd. ed. v.1. New York; Basel: Marcel Dekker, Inc. 2002. p.156-176.
- SCHMIDT, B.; DAVIS, P.; MODDEMANN, D.; OHLSSON, A.; ROBERTS, R.S.; SAIGAL, S.; SOLIMANO, A.; VINCER, M.; WRIGHT, L.L. Long-term effects of indomethacin prophylaxis in extremely-low-birth-weight infants. New Engl. J. Med., Boston, v.344, n.26, p.1966-1972, 2001.
- SHAMAT, M.A. The role of the gastrointestinal microflora in the metabolism of drugs. Int. J. Pharm., Amsterdam, v.97, p.1-13, 1993.
- SOMASUNDARAM, S.; RAFI, S.; HAYLLAR, J.; SIGTHORSSON, G.; JACOB, M.; PRICE, A.B.; MACPHERSON, A.; MAHMOD, T.; SCOTT, D.; WRIGGLESWORTH, J.M.; BJARNASON, I. Mitochondrial damage: a possible mechanism of the "topical" phase of NSAID induced injury to the rat intestine. Gut, London, v.41, n.3, p.344-353, 1997.
- STELLA, V.J.; RAO, V.M.; ZANNOU, E.A.; ZIA, V. Mechanisms of drug release from cyclodextrin complexes. Adv. Drug Deliv. Rev., Amsterdam, v.36, p.3-16, 1999.
- STELLA, V.J.; RAJEWSKI, R.A. Cyclodextrins: their future in drug formulation and delivery. Pharm. Res., New York, v.14, n.5, p.556-567, 1997.
- STEWART, C.F.; HAMPTON, E.M. Therapy review: effect of maturation on drug disposition in pediatric patients. Clin. Pharm., Bethesda, v.6, p.548-564, 1987.
- STRATTAN, C.E. 2-Hydroxypropyl-b-cyclodextrin, part II: safety and manufacturing issues. Pharm. Tech., Eugene, v.2, p.52-58, 1992.
- SZEJTLI, J. Cyclodextrin inclusion complexes. In: SZEJTLI, J. Ed. Cyclodextrin Technology. London: Kluwer Academic Publishers, 1988, p. 79-185.
- SZEJTLI, J. Cyclodextrins: properties and applications. Drug Invest., Auckland, v.2, suppl.4, p.11-21, 1990.
- SZEJTLI, J. Cyclodextrins in drug formulations: part I. Pharm. Tech. Int., Eugene, v.2, p.15-22, 1991a.
- SZEJTLI, J. Cyclodextrins in drug formulations: part II. Pharm. Tech. Int., Eugene, v.3, p.16-24, 1991b.
- TAN, X.; LINDENBAUM, S. Studies on complexation between b-cyclodextrin and bile salts. Int. J. Pharm., Amsterdam, v.74, p.127-135, 1991.
- THOMPSON, D.O. Cyclodextrins - enabling excipients: their present and future use in pharmaceuticals. Crit. Rev. Ther. Drug Carrier Syst., New York, v.14, n.1, p.1-104, 1997.
- TöTTERMAN, A.N.; SCHIPPER, N.G.M.; THOMPSON, D.O.; MANNERMAA, J-P. Intestinal safety of water-soluble b-cyclodextrins in paediatric oral solutions of spironolactone: effects on humanintestinal epithelial caco-2 cells. J. Pharm. Pharmacol., London, v.49, p.43-48, 1997.
- TRESZL, A.; SZABÓ, M.; DUNAI, G.; NOBILIS, A.; KOCSIS, I.; MACHAY, T.; TULASSAY, T.; VÁSÁRHELYI, B. Angiotensin II type 1 receptor A1166C polymorphism and prophylactic indomethacin treatment induced ductus arteriosus closure in very low birth weight neonates. Pediatr. Res., Baltimore, v.54, n.5, p.753-755, 2003.
- VEIGA, F.; TEIXEIR-DIAS, J.J.C.; KEDZIEREWICZ, F.; SOUSA, A.; MAINCENT, P. Inclusion complexation of tolbutamide wiyh b-cyclodextrin and hydroxypropyl-b-cyclodextrin. Int. J. Pharm., Amsterdam, v.129, n.63-71, 1996.
- VERT, P.; BIANCHETTI, G.; MARCHAL, F.; MONIN, P.; MORSELLI, P.L. Effectiveness and pharmacokinetics of indomethacin in premature newborns with patent ductus arteriosus. Eur. J. Clin. Pharmacol., Berlin, v.18, p.83-88, 1980.
- WATKINS, J.B.; INGALL, D.; SZCZEPANIK, P.; KLEIN, P.D.; LESTER R. Bile-salt metabolism in the newborn: measurement of pool size and synthesis by stable isotope technique. N. Eng. J. Med., Boston, v.288, n.9, p.431-433, 1973.
- WIEST, D.B.; PINSON, J.B.; GAL, P.S.; BRUNDAGE, R.C.; SCHALL, S.; RANSOM, J.L.; WEAVER, R.L.; PUROHIT, D.; BROWN, Y. Population pharmacokinetics of intravenous indomethacin in neonates with symptomatic patent ductus arteriosus. Clin. Pharmacol. Therap., St. Louis, v.49, n.5, p.550-557, 1991.
- YEH, T.F.; CARR, I. Pharmacological closure of patent ductus arteriosus. In: YEH, T.F. Neonatal Therapeutics. St. Louis: Mosby Year Book, 1991. p.123-138.
- YOSHIDA, A.; ARIMA, H.; UEKAMA, K.; PITHA, J. Pharmaceutical evaluation of hydroxyalkyl ethers of ²-cyclodextrins. Int. J. Pharm., Amsterdam, v.46, p.217-222, 1988.
- YOSHIOKA, H.; ISEKI, K.; FUJITA, K. Development and differences of intestinal flora in the neonatal period in breast-fed and bottle-fed infants. Pediatrics, Evaston, v.72, n.3, p.317-321, 1983.
Datas de Publicação
-
Publicação nesta coleção
13 Mar 2006 -
Data do Fascículo
Set 2005
Histórico
-
Aceito
22 Ago 2005 -
Recebido
02 Jun 2004