Abstracts
Aujeszky' s disease (AD) is an infectious disease causing important economic losses to the swine industry worldwide. The disease is caused by an alpha-herpesvirus, Aujeszky' s disease virus (ADV), an enveloped virus with a double stranded linear DNA genome. The ADV genome encodes 11 glycoproteins, which are major targets for the immune system of the host in response to the infection. The glycoprotein E (gE) is a non-essential protein and deletion of the gE gene has been used for the production of marker vaccines. Aiming to develop molecular reagents for the production of a gE specific ELISA test, the gE gene was amplified by PCR, cloned and expressed into a baculovirus expression system. The recombinant DNA vector pFastBac-gE-ADV was used for site-specific transposition into the recombinant baculovirus (bacmid). Colonies with recombinant bacmid-pFastBac-gE-ADV were selected by antibiotic and color selection and the presence of the gE gene was confirmed by PCR. The recombinant bacmid-pFastBac-gE-ADV was cotransfected in insect Trichoplusia ni and the presence of the recombinant DNA and gE protein were detected by PCR, SDS-PAGE and Western blotting, respectively.
Aujeszky; Glycoprotein E; Baculovírus; Recombinant
A doença de Aujeszky (DA) é uma enfermidade infecto-contagiosa que causa grandes perdas econômicas ao produtor e à agroindústria suinícola em todo o mundo. É causada pelo vírus da doença de Aujeszky (VDA), um alfaherpesvírus envelopado com genoma DNA de fita dupla e linear. O genoma do VDA codifica 11 glicoproteínas, as quais são os maiores alvos do sistema imune do hospedeiro em resposta a infecção. A glicoproteína E (gE) é uma proteína não essencial e a deleção do gene da gE é muito utilizada para a produção de vacinas com marcadores. Com o objetivo de desenvolver insumos moleculares para a produção de um teste de ELISA específico para gE do VDA, a seqüência do gene da gE foi amplificada, clonada e expressa no sistema de expressão em baculovírus. O produto da amplificação foi clonado no vetor pGem®-T Easy e subclonado no plasmídeo de expressão pFastBac™1. O DNA recombinante pFastBac-gE-VDA foi usado para a transposição sítio-específica no baculovírus recombinante (bacmid). Após seleção por antibióticos e cor, as colônias com o recombinante bacmid-pFastBac-gE-VDA foram selecionadas e a presença do gene da gE foi confirmada por PCR. O DNA recombinante viral, bacmid-pFastBac-gE-VDA, foi usado para cotransfecção de células de inseto Trichoplusia ni e a presença do recombinante e a proteína gE foi determinada por PCR, por SDS-PAGE e Western blotting, respectivamente.
Aujeszky; Glicoproteína E; Baculovírus; Recombinante
VETERINARY MICROBIOLOGY
Cloning and expression of Aujeszky's disease virus glycoprotein E (gE) in a baculovirus system
Clonagem e expressão da glicoproteina E (gE) do vírus da doença de Aujeszky em sistema de baculovirus
Régia Maria Feltrin DambrosI,II,III; Bergman Moraes RibeiroIV; Raimundo Wagner de S. AguiarIV; Rejane SchaeferII; Paulo Augusto EstevesII; Simone PerecmanisV; Neide Lisiane SimonII; Nayara Cavalcante SilvaIV; Michele ColdebellaII,VI; Janice Reis Ciacci-ZanellaII,** Corresponding Author. Mailing address:Laboratório de Sanidade, Embrapa Suínos e Aves, 89700-000 Concórdia, SC, Brasil. Tel.: (49) 3441 0400 - Fax: (49) 3442 8559. E-mail: janice@cnpsa.embrapa.br
ICompanhia Integrada de Desenvolvimento Agrícola de Santa Catarina, Concórdia, SC, Brasil
IILaboratório de Sanidade, Embrapa Suínos e Aves, Concórdia, SC, Brasil
IIICentro de Ciências Agroveterinárias, Universidade do Estado de Santa Catarina, Lages, SC, Brasil
IVDepartamento de Biologia Celular, Universidade de Brasília, Brasília, DF, Brasil
VDepartamento de Microbiologia Veterinária, Universidade de Brasília, Brasília, DF, Brasil
VIQuímica Industrial de Alimentos, Universidade do Contestado, Concordia, SC, Brasil
ABSTRACT
Aujeszky' s disease (AD) is an infectious disease causing important economic losses to the swine industry worldwide. The disease is caused by an alpha-herpesvirus, Aujeszky' s disease virus (ADV), an enveloped virus with a double stranded linear DNA genome. The ADV genome encodes 11 glycoproteins, which are major targets for the immune system of the host in response to the infection. The glycoprotein E (gE) is a non-essential protein and deletion of the gE gene has been used for the production of marker vaccines. Aiming to develop molecular reagents for the production of a gE specific ELISA test, the gE gene was amplified by PCR, cloned and expressed into a baculovirus expression system. The recombinant DNA vector pFastBac-gE-ADV was used for site-specific transposition into the recombinant baculovirus (bacmid). Colonies with recombinant bacmid-pFastBac-gE-ADV were selected by antibiotic and color selection and the presence of the gE gene was confirmed by PCR. The recombinant bacmid-pFastBac-gE-ADV was cotransfected in insect Trichoplusia ni and the presence of the recombinant DNA and gE protein were detected by PCR, SDS-PAGE and Western blotting, respectively.
Key-words: Aujeszky. Glycoprotein E. Baculovírus. Recombinant.
RESUMO
A doença de Aujeszky (DA) é uma enfermidade infecto-contagiosa que causa grandes perdas econômicas ao produtor e à agroindústria suinícola em todo o mundo. É causada pelo vírus da doença de Aujeszky (VDA), um alfaherpesvírus envelopado com genoma DNA de fita dupla e linear. O genoma do VDA codifica 11 glicoproteínas, as quais são os maiores alvos do sistema imune do hospedeiro em resposta a infecção. A glicoproteína E (gE) é uma proteína não essencial e a deleção do gene da gE é muito utilizada para a produção de vacinas com marcadores. Com o objetivo de desenvolver insumos moleculares para a produção de um teste de ELISA específico para gE do VDA, a seqüência do gene da gE foi amplificada, clonada e expressa no sistema de expressão em baculovírus. O produto da amplificação foi clonado no vetor pGem®-T Easy e subclonado no plasmídeo de expressão pFastBac1. O DNA recombinante pFastBac-gE-VDA foi usado para a transposição sítio-específica no baculovírus recombinante (bacmid). Após seleção por antibióticos e cor, as colônias com o recombinante bacmid-pFastBac-gE-VDA foram selecionadas e a presença do gene da gE foi confirmada por PCR. O DNA recombinante viral, bacmid-pFastBac-gE-VDA, foi usado para cotransfecção de células de inseto Trichoplusia ni e a presença do recombinante e a proteína gE foi determinada por PCR, por SDS-PAGE e Western blotting, respectivamente.
Palavras-chave: Aujeszky; Glicoproteína E; Baculovírus; Recombinante.
INTRODUCTION
Aujeszky's disease virus (ADV) is an enveloped virus with a double stranded linear DNA genome, a member of Alphaherpesvirinae. ADV is the causative agent of Aujeszky's disease (AD), which is an economical important disease of swine industry worldwide (1). In addition to severe clinical signs, the infection may curse without clinical signs in swine, the natural reservoir for the virus. In this host, the virus establishes a lifelong latent infection in neuronal ganglia (5,10,14). Control of AD is performed by the use of vaccines and culling of seropositive animals. Many countries succeeded in eradication programs using glycoprotein E (gE) deleted vaccines. Thus, antibodies developed by vaccinated pigs can be differentiated from those produced in response to natural infection by gE-specific enzyme-linked immunosorbent assays (ELISA) differential test.
The ADV genome encodes for 70 different proteins (6), including 11 envelope glycoproteins (9). The gE is a non essential protein for viral replication (5,19-21) and the deletion of the gE gene has been used for the production of marker vaccines (4,13). The immunization strategies are important to control the disease by reducing signs and viral spread, and for differentiation of vaccinated from naturally infected animals. This can be accomplished through an ELISA able to detect anti-gE antibodies and therefore identify naturally infected animals. However, sensitive, fast, available and practical diagnostic tests to detect ADV antibodies are needed in diagnostic laboratories (17). Reagents for diagnosis are frequently imported, raising the costs of animal testing and eradication programs.
AD is present in Brazil since 1912 (2) and in swine herds in Santa Catarina State (SC) since 1983 (16). AD was a significant problem for SC, an important swine producer and pork exporter. Until 2000, there were no official control program for AD in SC and several swine herds were registered as infected or vaccine users. The vaccine used in Brazil since 1995 is an inactivated gE deleted vaccine. Therefore, antibodies from vaccinated pigs can be detected by a gE-specific ELISA test. In 2001, an eradication program funded by a joined effort of industry, swine producers and government has eradicated AD gradually from swine herds in the state, and no AD case has been reported since July 2004 (11).
The objective of this work was to implement technology to produce reagents to improve diagnostic tools for more sensitive, specific and fast tests to be used in AD's eradication efforts. For this, coding sequence for gE of ADV was amplified by PCR, cloned and the gE recombinant protein was expressed in a baculovirus expression system. Recombinant baculoviruses are widely used as vectors to express heterologous genes in cultured insect cells and insect larvae (15). Due to its restrict host range, the baculovirus can be used in free areas of determined agent, with no risks for the region or country, since no infectious agent will be manipulated.
MATERIALS AND METHODS
Virus, cells and DNA extraction
The ADV strain used in this work was obtained from the collection of viruses of the Virology Laboratory of Embrapa Swine and Poultry Research Center in Concórdia (SC) Brazil, identified as protocol numbers 0261/83 (Embrapa). Viruses were cultivated in swine kidney cells (SK-6). Confluent SK-6 monolayers were inoculated with ADV at a multiplicity of infection of 0.1 1. After 18 hours, the culture supernatant was harvested and centrifuged on a sucrose cushion 25% at 100,000 x g for 3 hours at 4ºC. The viral pellet was re-suspended in TNE (Tris-Cl 10 mM, pH 7,4; NaCl 150 mM; EDTA 1 mM, pH 8.0) and treated with 1% sodium dodecyl sulfate (SDS) and 20 µg/µL proteinase K for 1 h at 37ºC (19). After digestion, the viral DNA was extracted with phenol, precipitated with cold ethanol, re-suspended in TE pH 8.0 and maintained at 4ºC (18). Viral DNA was subjected to electrophoresis and the gels were stained with ethidium bromide (5 ug/mL, Sigma®) and photographed under UV light. A 1 Kb DNA Ladder (Invitrogen) was included as a marker and to estimate the DNA concentration.
Polimerase Chain Reaction (PCR)
In order to amplify the complete coding sequence of gE, two primers containing EcoRI and BamHI sites were designed. The forward primer contains a restriction site of EcoRI (5'- cacaccggggttgaattccatgc-3') and the reverse primer contains a restriction site of BamHI (5'-gaccggatcccccggtatttaagc-3'). Both primers were synthesized at IDT® Integrated DNA Technologies, Inc (Dialab Diagnostics, SA, USA).
Reaction mixes for PCR amplification were prepared in ultrapure Millipore water containing 0.8 mM dNTP mix, 0.1 µg/µL of each primer, 4 mM of MgCl2, 5% (V/V) glycerol, 2.5 U/50 µL of Taq DNA polymerase (Invitrogen) and Taq DNA polymerase buffer (Invitrogen). The amplifications were performed with an initial cycle of 4 minutes at 95°C and annealing (56ºC, 90 seconds), then the Taq DNA polimerase was added and the reactions were incubated at 72ºC for 2 minutes. Afterwards, the reactions were submitted to 5 cycles of denaturation (95ºC, 1 minute); annealing (58ºC, 90 seconds) and extension (70ºC, 2 minutes), followed by 30 cycles of denaturation (95ºC, 1 minute); annealing (60ºC, 90 seconds) and extension (72ºC, 2 minutes). An additional incubation of 70ºC for 10 min was performed at the end of the reaction. PCR products were submitted to gel electrophoresis 0.8% agarose gel in TAE (Tris-base 40 mM; acetate 40 mM; EDTA 1 mM, pH 8.0) using a molecular weight marker of 1 kb DNA Ladder (Invitrogen). PCR reactions containing the amplified gE gene were purified with Wizardâ SV Gel kit and PCR Clean-Up System (Promega, USA), according to manufacturer's recommendations.
Construction of recombinant plasmid expressing gE of ADV
Escherichia coli DH5a (Invitrogen) e DH10Bac (Invitrogen) competent cells were used as hosts in all DNA cloning and transposition procedures as Sambrook et al. (18) and manufacturer's recommendations. The plasmids used in this work were the pGem®-T Easy Vector Systems (Promega, USA), pFastBac1 and bacmid of Bac-to-Bac® Baculovirus Expression Systems kit (Invitrogen).
The purified gE fragment was cloned into the pGem®-T Easy (Promega, USA) according to manufacturer's recommendations. The recombinant DNA pGem-gE.ADV was digested with restriction enzyme EcoRI (New England Biolabs), the digested fragment (1772 pb) was excised from the 1% agarose LMP (Low Melting Point; Invitrogen) and purified with the kit Wizard® SV Gel and PCR (Promega, USA).
The plasmid expression pFastBac1 was linearized after digestion with EcoRI, dephosphorylated and ligated with the purified gE fragment. The products of ligation were introduced in E. coli DH5a competent cells. Following selection, DNA of the selected colonies was extracted and analyzed by restriction digestion with EcoRI and BamHI for the presence and correct orientation of the insert.
Competent E. coli DH10Bac cells were transformed with recombinant DNA pFastBac-gE.ADV and pFastBac-Gus (control of transposition) as recommended by the manufacturer's. Recombinant bacmid (white colonies) containing the pFastBac-gE.ADV were selected, the DNA was isolated and the quality of the recombinant DNA of bacmid.pFastBac-gE.ADV was analyzed in 0.5% agarose gel and also tested by PCR using the primers and conditions described above.
Transfection of insect cells with recombinant DNA of bacmid.pFastBac-gE.ADV
Recombinant DNA of bacmid.pFastBac-gE.ADV was diluted in medium TC-100 without serum, mixed with lipofectin (CellFECTIN®; Invitrogen) and incubated 30 minutes at room temperature. Recombinant DNA pFastBac-Gus (transposition control) and bacmid-pFastBac empty vector were used as controls. Insect cells Trichoplusia ni (BTI-Tn5B1-4) (3) were transfected with this recombinant DNA and lipofectin mix. After 3 hours, the mix was removed, medium was added and the cells were incubated at 27ºC. At the fifth day, supernatant and co-transfected cells were ressuspended and centrifuged. The supernatant containing recombinant viruses were recovered and submitted to DNA extraction. The cell pellet was washed twice with PBS (pH 7.2) and frozen at -70ºC. Uninfected BTI-Tn5B1-4 cells were used as negative control.
Analysis of gE transfection and expression by PCR and SDS-PAGE
A sample of recombinant DNA gE.ADV was tested by PCR using primers and conditions described above to detect the presence of gE sequence. Protein extracts from BTI-Tn5B1-4 cells infected with the recombinant virus were prepared in denaturing buffer and analyzed in an 12% sodium dodecyl sulphate-polyacrilamide gel electrophoresis (SDS-PAGE) as described by Laemmli (7). The separated proteins on the gel were transferred to a nitrocellulose membrane (Millipore) for Western blotting. The reaction was blocked with PBS-T20 0.05%. The primary antibodies used were porcine antiserum anti-gE.ADV (IDEXX Laboratories, USA), porcine SPF antiserum (collected from Embrapa's SPF herd and ADV ELISA tested negative) and porcine polyclonal antibody ADV positive (ELISA and serum-neutralization; Cedisa). The secondary antibody used an was IgG anti-pig peroxidase (HRP) (KPL Kirkegaard & Perry Laboratories, Guildford, UK) diluted 1:7.000 in PBS-BSA 1% and incubated for 1 h. The membrane was washed and incubated with DAB (cromatogen 3 - 3' diaminobenzidine; Sigma®) for 12 min.
RESULTS AND DISCUSSION
Genomic DNA viral samples, 0261/83 (Embrapa), were digested with BamHI restriction enzyme and presented the genotype II pattern (19). The digestion revealed the presence of fragment BamHI 7, which corresponds to a part of US region (unique short) of the ADV genome fragment 7, in which the gE gene is located (9,10).
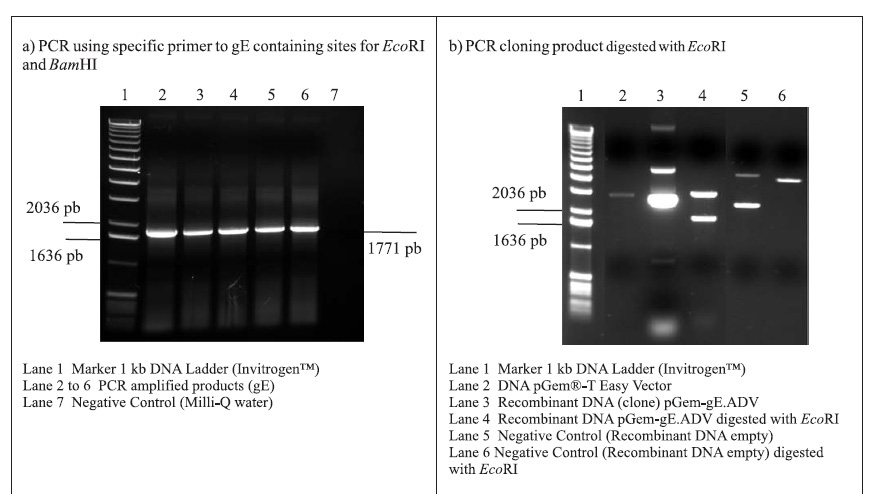
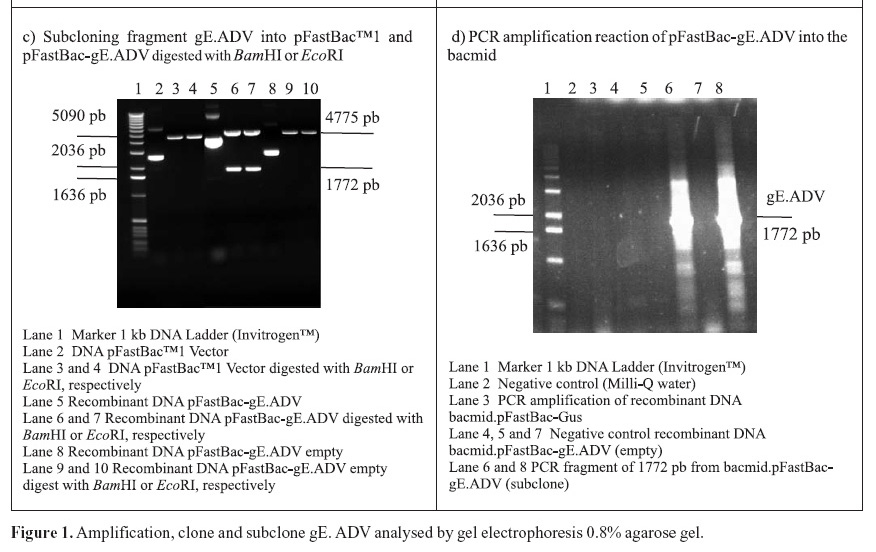
PCR amplifications resulted in a fragment of 1771 pb, corresponding to the gE coding sequence (1734 pb) plus 37 oligonucleotides of the primers (Fig. 1A). Following the amplifications, the 1771 pb product of complete gE gene was purified and cloned into the pGem-T Easy Vector (3015 pb), originating the plasmid pGem-gE.ADV of 4786 pb (Fig. 1B). In order to subclone the gE sequence into the donor expression vector pFastBac1, plasmid DNA of the pGem-gE.ADV was digested with EcoRI and the gE.ADV insert of approximately 1772 pb (Fig. 1B) was ligated into the pFastBac1 at the EcoRI restriction site. The product of this ligation was the plasmid pFastBac-gE.ADV of approximately 6547 pb, which presented the insert in the correct orientation, as demonstrated by BamHI digestion (Fig. 1C).
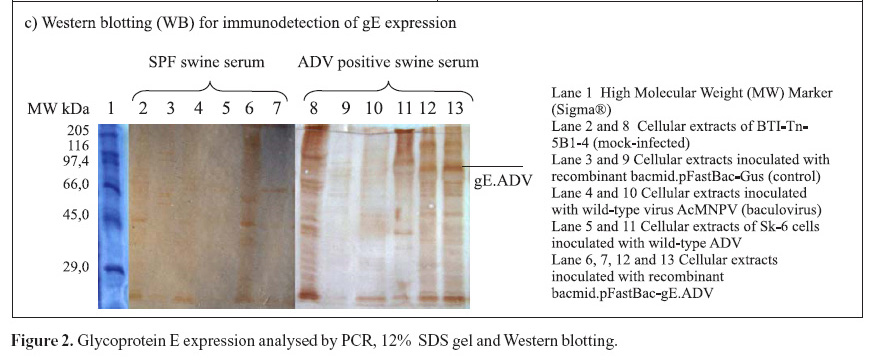
The recombinant pFastBac-gE.ADV containing the gE gene cloned into pFastBac1 under the polyedhrin promoter (8) was transformed in host cells E. coli DH10Bac by transposition into the bacmid. Minipreps of DNA extracted from recombinant colonies (white), a blue colony (empty) and a transposition control colony (pFastBac-Gus) were analysed by gel eletrophoresis (data not shown) and PCR (Fig. 1D). Recombinant DNA from colonies of bacmid.pFastBac-Gus, empty vector bacmid-pFastBac and Milli-Q water were also used as negative controls in this PCR (Fig. 1D). PCR reactions indicated amplification of gE.ADV sequence on samples of bacmid.pFastBac-gE.ADV. Extracted recombinant DNA of bacmid.pFastBac-gE.ADV was used to co-transfect insect cells which were harvested 5 days pos-infection. DNA was analyzed by PCR (Fig. 2A) and protein extracts by denaturing 12% SDS-PAGE gel stained with Coomassie blue (Fig. 2B) (11).
Furthermore, mock-infected insect cells BTI-Tn5B1-4, the recombinant bacmid.pFastBac-Gus (control) and the virus AcMNPV (wild-type with polyhedrin gene) protein extracts were also analyzed as shown in Fig. 2C. In addition to those, a cellular extract of SK-6 cells infected with wild type ADV was also analyzed and compared to recombinant gE.ADV expressed protein (Fig. 2C). The recombinant-gE baculovirus was inoculated in cultured cells and expressed the recombinant gE, detected by Western blotting. The recombinant gE.ADV expressed a protein of molecular weight between 82.2 and 115.5 kDa (Fig. 2C).
The recombinant-gE baculovirus will be used for antigen and monoclonal antibody production. Thus, gE.ADV will aid in the development of a more sensitive, specific and safer diagnostic test for the ADV.
Submitted: January 18, 2007; Returned to authors for corrections: April 20, 2007; Approved: July 16, 2007
- 1. Bascuñana, C.R.; Bjornerot, L.; Ballagi-Pordany, A.; Robertsson, J.A.; Belak, S. (1997). Detection of psudorabies virus genomic sequences in apparently uninfected 'single reactor' pigs. Vet. Microbiol., 55: 37-47.
- 2. Carine, A.; Maciel, J. (1912). La pseudo-rage ou paralisie bulbaire infectieuse au Brésil. Bull. Soc. Pathol. Exot., 5: 576-578.
- 3. Granados, R.R.; Guoxun, L.; Derksen, C.G.; Mckenna, K.A.A. (1994). A new insect cell line from Trichoplusia ni (BTI-Tn-5B1-4) susceptible to Trichoplusia ni single enveloped nuclear polyhedrosis virus. J. Invertebr. Pathol, 64(3): 260-266.
- 4. Kit, S.; Kit, M. (1991). Genetically engineered herpesvirus vaccines. Prog. Med. Virol, 38: 128-166.
- 5. Kluge, J.P.; Beran, G.W.; Hill, H.T.; Platt, K.B. Pseudorabies (Aujeszky's disease). (1999). In: Straw, B.E.; D'allaire, S.; Mengeling, W.L.; Taylor, D.J. (eds). Diseases of swine 8th ed. Iowa State University Press, Ames, Iowa, p.233-246. 1 CD-ROM.
- 6. Klupp, B.G.; Hengartner, C.J.; Mettenleiter, T.C.; Enquist, L.W. (2004). Complete, annotated sequence of the pseudorabies virus genome. J. Virol, 78: 224-440.
- 7. Laemmli, U.K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature (London). 227: 680-685.
- 8. Luckow, V.L.; Lee, S.C.; Barry, G.F.; Olins, P.O. (1993). Eficiente generation of infectious recombinant baculoviruses by site-specific transposon-mediated insertion of foreign genes into a baculovirus genome propagated in Escherichia coli. J. Virol, 67(8): 4566-4579.
- 9. Mettenleiter, T.C.; Lukacs, N.; Rziha, H.J. (1985). Pseudorabies virus avirulent strains fail to express a major glycoprotein. J. Virol, 56: 307-311.
- 10. Mettenleiter, T.C. (2000). Aujeszky's disease (pseudorabies) virus: the virus and molecular pathogenesisstate of the art. Vet. Rec, 31: 99-115.
- 11. Morés, N.; Amaral, A.L.; Ventura, L.; Zanella, J.R.C.; Silva, V.S. (2005). Programa de erradicação da doença de Aujeszky no Estado de Santa Catarina Concórdia: Embrapa Suínos e Aves, 8p. Circular técnica, 44.
- 12. O'Reilly, D.; Miller, L.K.; Luckow, V.A. (1992). Baculovirus expression vectors: a laboratory manual. Freeman and Company, New York, 347p.
- 13. Pensaert, M.; Gielkens, A.L.; Lomniczi, B.; Kimman, T.G.; Vannier, P.; Eliot, M. (1992). Round table on control of Aujeszky' s disease and vaccine development based on molecular biology. Vet. Microbiol, 33(1-4): 53-67.
- 14. Pomeranz, L.E.; Reynolds, A.E.; Hengartner, C. (2005). Molecular biology of pseudorabies virus: Impact on neurovirology and veterinary medicine. Microbiol. Mol. Biol. Rev, 69(3): 462-500.
- 15. Ribeiro, B.M.; Souza, M.L.; Kitajima, E.W. (2004). Anticarsia gemmatalis and baculovirus infection XV National Meeting of Virology, São Pedro, SP, p.40.
- 16. Rowe, C.A.; Romero, C.H. (1986). Isolamento e identificação do vírus da doença de Aujeszky de surtos em suínos no Estado de Santa Catarina. Pesq. Vet. Bras, Rio de Janeiro, 6: 99-103.
- 17. Toma, B.; Moutou, B.; Fortier, B. (1979). Recherche dês anticorps anti-virus de la maladie dÁujeszky par la technique Elisa. Rec.Med. Vet, 155: 455-463.
- 18. Sambrook, J.; Fritsch, E.F.; Maniatis, T. (1989). Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor Laboratory Press, New York.
- 19. Schaefer, R.; Zanella, J.R.C.; Pan, K.A.; Dambros, R.M.F.; Schiochet, M.F.; Coldebella, M. (2005). Characterization of Aujeszky' s disease virus isolated in south Brazil in the last twenty years based on restriction enzyme analysis XVI National Meeteng of Virology, Salvador, p.10.
- 20. Spear, P.G. (1993). Entry of alphaherpesviruses into cells. Seminars in Virology, 4: 167-180.
- 21. Spear, P.G.; Longnecker, R. (2003). Herpesvirus entry: an update. J. Virol, 77(19): 10179-10185.
- 22. Zuckermann, F.A.; Mettenleiter, T.C.; Schreurs, C.; Sugg, N.; Ben-Porat, T. (1988). Complex between glycoproteins gI and gp63 of pseudorabies virus; its effect on virus replication. J. Virol, 62: 4622-4626.
Publication Dates
-
Publication in this collection
17 Oct 2007 -
Date of issue
Sept 2007
History
-
Accepted
16 July 2007 -
Received
18 Jan 2007 -
Reviewed
20 Apr 2007