Abstracts
In this study we propose the analysis of genetic diversity of the common three-toed sloth, Bradypus variegatus, in an attempt to understand population structure, identify divergent intraspecific units, and contribute to the knowledge of biodiversity in the neotropical forests. We analyzed a 387 bp segment of the mitochondrial DNA control region in 28 individuals distributed in different localities of both Atlantic and Amazon forests. Our results demonstrated that the genetic diversity of B. variegatus is distributed in six management units, MUs. The observed MUs encompass six phylogenetic lineages and represent respectively north and south regions of Atlantic forest, three regions within the Amazon forest, and a transition region between these two biomes. Considering the fact that these MUs are concordant with phylogroups and endemism areas already described for other vertebrate species, we can say that the study of B. variegatus, a widely distributed and not endangered species, can help to identify areas for conservation biology purposes in neotropical rain forests.
Bradypus; mitochondrial DNA; management units; conservation biology
Neste estudo nós realizamos a análise da diversidade genética da preguiça comum, Bradypus variegatus, a fim de compreender os padrões de estrutura populacional, identificar unidades intraespecíficas divergentes e contribuir para o conhecimento da biodiversidade nas florestas da região neotropical. Nós analisamos um segmento de 387 pb da região controle do DNA mitocondrial de 28 indivíduos distribuídos em diferentes localidades da Floresta Amazônica e da Mata Atlântica. Os resultados obtidos demonstram que a diversidade genética da espécie pode ser representada em seis diferentes unidades de manejo (UM). Tais UMs englobam seis linhagens filogenéticas e estão localizadas em diferentes regiões geográficas sendo elas, as porções norte e sul da Mata Atlântica, três regiões dentro da área de Floresta Amazônica e uma área de transição entre os dois domínios de mata. As diferentes unidades intraespecíficas de B. variegatus são concordantes com grupos filogeográficos e áreas de endemismo já observadas para outras espécies de vertebrados. Levando em consideração o fato de que estas UMs concordam com filogrupos e áreas de endemismo previamente descritos para outras espécies de vertebrados, o estudo da preguiça comum, uma espécie amplamente distribuída e considerada não ameaçada de extinção, pode auxiliar na identificação de áreas destinadas à conservação biológica ao longo das florestas úmidas da região neotropical.
Bradypus; DNA mitocondrial; unidades de manejo; conservação biológica
Identifying management units in non-endangered species: the example of the sloth Bradypus variegatus Schinz, 1825
Identificação de unidades de manejo para espécies não ameaçadas de extinção: o exemplo da preguiça Bradypus variegatus Schinz, 1825
Moraes-Barros, N.I, * * e-mail: namoraes@ib.usp.br ; Miyaki, CY.II; Morgante, JS.I
ILaboratório de Biologia Evolutiva e Conservação de Vertebrados LABEC, Departamento de Genética e Biologia Evolutiva, Instituto de Biociências, Universidade de São Paulo USP, Rua do Matão 277, CEP 05508-090, São Paulo, SP, Brazil
IILaboratório de Genética e Evolução Molecular de Aves, Departamento de Genética e Biologia Evolutiva, Instituto de Biociências, Universidade de São Paulo USP, São Paulo, SP, Brazil
ABSTRACT
In this study we propose the analysis of genetic diversity of the common three-toed sloth, Bradypus variegatus, in an attempt to understand population structure, identify divergent intraspecific units, and contribute to the knowledge of biodiversity in the neotropical forests. We analyzed a 387 bp segment of the mitochondrial DNA control region in 28 individuals distributed in different localities of both Atlantic and Amazon forests. Our results demonstrated that the genetic diversity of B. variegatus is distributed in six management units, MUs. The observed MUs encompass six phylogenetic lineages and represent respectively north and south regions of Atlantic forest, three regions within the Amazon forest, and a transition region between these two biomes. Considering the fact that these MUs are concordant with phylogroups and endemism areas already described for other vertebrate species, we can say that the study of B. variegatus, a widely distributed and not endangered species, can help to identify areas for conservation biology purposes in neotropical rain forests.
Keywords:Bradypus, mitochondrial DNA, management units, conservation biology.
RESUMO
Neste estudo nós realizamos a análise da diversidade genética da preguiça comum, Bradypus variegatus, a fim de compreender os padrões de estrutura populacional, identificar unidades intraespecíficas divergentes e contribuir para o conhecimento da biodiversidade nas florestas da região neotropical. Nós analisamos um segmento de 387 pb da região controle do DNA mitocondrial de 28 indivíduos distribuídos em diferentes localidades da Floresta Amazônica e da Mata Atlântica. Os resultados obtidos demonstram que a diversidade genética da espécie pode ser representada em seis diferentes unidades de manejo (UM). Tais UMs englobam seis linhagens filogenéticas e estão localizadas em diferentes regiões geográficas sendo elas, as porções norte e sul da Mata Atlântica, três regiões dentro da área de Floresta Amazônica e uma área de transição entre os dois domínios de mata. As diferentes unidades intraespecíficas de B. variegatus são concordantes com grupos filogeográficos e áreas de endemismo já observadas para outras espécies de vertebrados. Levando em consideração o fato de que estas UMs concordam com filogrupos e áreas de endemismo previamente descritos para outras espécies de vertebrados, o estudo da preguiça comum, uma espécie amplamente distribuída e considerada não ameaçada de extinção, pode auxiliar na identificação de áreas destinadas à conservação biológica ao longo das florestas úmidas da região neotropical.
Palavras-chave:Bradypus, DNA mitocondrial, unidades de manejo, conservação biológica.
1. Introduction
Conservation Biology is concerned with the study of biodiversity and the factors that threaten it (Meffe and Carrol, 1997). Some of the goals of conservation programs encompass describing and prioritizing areas for protection and defining specific biodiversity components as target units to conservation efforts (Margules and Pressey, 2000; Myers et al., 2000; Araújo 2002; Moritz, 2002). Therefore, it is important to know the components of biological diversity as well as to identify the threats.
For conservation genetics the main concern is how, within this process, to identify the major elements of intraspecific diversity and the processes that generate these elements (Frankham et al., 2002). The first known approach to delineate intraspecific units for conservation and management is the identification of subspecies. Although this practice continues until the present, it has been shown to be deficient in some aspects, such as inadequate or confused criteria and incorrect identification across taxa (Moritz, 2002). This apparent deficiency led to several studies that, along the last two decades, have been discussing the identification and characterization of such intraspecific units. The concept of evolutionary significant unit (ESU) is the main subject of these discussions.
The term ESU was first used by Ryder (1986) to guide prioritization of intraspecific units for captive management and was defined as one or a set of conspecific populations with a distinct and long-term evolutionary history, mostly separated from other such units. This term was then adopted as a more general definition for distinct population segments: groups with some level of reproductive isolation and adaptive distinctiveness (Waples, 1991). According to Moritz (1994a), an ESU can be identified as a group of populations reciprocally monophyletic for mitochondrial DNA haplotypes and also differing significantly for allele frequencies at nuclear loci. The author suggests a second category or a subdivision, the management unit (MU). The MU is characterized by a distinct population that should be managed to ensure the viability of the larger ESU. The criterion to recognize a MU is that it represents conspecific populations that are demographically autonomous and have a significant divergence of allele frequencies at nuclear or mitochondrial loci, regardless of the phylogenetic distinctiveness of the alleles (Avise, 1995; Moritz, 1999).
Several other definitions of ESU were described and extensively discussed. However, a consensus definition seems to be as difficult and controversial as the species concept issue (Moritz, 1994a,b, 1999; Moritz et al., 1995; Taylor and Dizon, 1996; Ledge et al., 1996; Pennock and Dimick, 1997; Waples, 1998). Crandall et al. (2000) argue that intraspecific subdivisions can only be considered ESUs when they represent adaptive differentiation. The authors proposed a broader categorization of population distinctiveness based on concepts of ecological and genetic exchangeability. The authors also state that neutral markers are not enough to identify these units because they may not reflect adaptive genetic variation and substantial adaptive divergence can occur despite gene flow. Fraser and Bernatchez (2001) review the different ESU concepts and state that, depending on the taxa, the evolutionary forces that are evaluated, and the temporal scale that is considered, some ESU criteria will work better than others. Furthermore, the authors propose the "adaptive evolutionary conservation" concept as an integrative framework for imputing conservation units. In a similar argumentation, Moritz (2002) agrees with an integrative approach by incorporating knowledge of evolutionary and ecological processes, phylogeography, species, and genetic diversity data to geographic tools and concepts to prioritize conservation areas. Besides, the author argues that conservation strategies for species may be improved, and should be more flexible, by considering the genetic diversity in two dimensions, one concerned with adaptive variation and the other with neutral divergence caused by isolation. Adaptive diversity can be replaced, depending on the maintenance of viable populations in functional landscapes, whereas the genetic diversity due to historical isolation represents unique geographical lineages that once lost cannot be replaced (Moritz, 1999). Therefore, the use of neutral molecular markers to recognize ESUs is justified.
No matter which definition is adopted to identify an ESU, an integrative approach and the knowledge of all biodiversity components are needed to target a unit for conservation purposes, whether for a species, a subspecies, a biogeographic partition, or evolutionary lineages.
Despite this discussion and all works concerned with conservation units of species threatened with extinction, few studies about management units have been carried out in non-endangered species. The study of intraspecific diversity can reveal important information regarding the identification of areas and biogeographic regions to conservation planning. According to Araújo (2002), the study of a large number of different species can help target appropriate areas for conservation purposes by identifying the amount and distribution of species diversity. This becomes more evident for those species distributed in biodiversity hotspots and areas of endemism.
The tropical rain forests of South America encompass two of the most diverse forested areas. The Atlantic forest is the most fragmented and disturbed environment and it is also a biodiversity hotspot with 567 endemic vertebrate species (Myers et al., 2002). The Amazon forest is considered the largest and most diverse of the tropical forest wilderness areas, compounded by an archipelago of distinct areas of endemism separated by the major rivers (Da Silva et al., 2005). Bradypus variegatus, the common three-toed sloth, is one among the several mammal species that can be found in both forests. This species is not considered threatened with extinction. Nevertheless, it is an arboreal species and its survival depends on the forests preservation. B. variegatus is one of the few extant representatives of the original megadiverse xenarthran split, a mammal exclusive to the neotropics and one of the most emblematic species of the Brazilian fauna. A recent study on Atlantic forest B. variegatus revealed that sloths distributed on northern Atlantic forest are genetically divergent from those from the southern region and represent two distinct populations, each one considered a MU. Each MU is also compounded by a distinct mitochondrial DNA lineage and the divergence among them was attributed to isolation by distance (Moraes-Barros et al., 2006). This was the first phylogeographic study on this species and it shows the necessity of a better understanding of the common three-toed sloth diversity.
In this study we investigate the genetic diversity within B. variegatus in a wide geographical area, covering two different biomes, the Atlantic and the Amazon forests. Our goal is, based on mitochondrial DNA sequences, to analyze the neutral intraspecific genetic divergence. We intend to identify demographically independent populations, divergent phylogenetic lineages, and investigate whether these populations and lineages reveal the existence of intraspecific units for the common sloth, which can help to delineate conservation units for both Atlantic and Amazon forests.
2. Materials and Methods
2.1. Sampling and DNA isolation
We analyzed 28 B. variegatus specimens from different geographical regions of both the Amazon and Atlantic forests. Some of these individuals were sampled in nature (DNA sample donations from Universidade Federal do Pará, Brazil BVAR specimens), while most samples were from museum collections (MPEG - Museu Paraense Emílio Goeldi, Belém, PA, Brazil; MZUSP - Museu de Zoologia da Universidade de São Paulo, São Paulo, SP, Brazil; MVZ - Museum of Vertebrate Zoology, Berkeley, CA, USA) (Table 1). For comparative purposes, B. variegatus DNA sequences from two regions of Atlantic forest were also included in the analysis. These sequences were previously described in Moraes-Barros et al. (2006).
DNA was extracted from tissue samples of museum specimens (study skin) by using the protocol described in Moraes-Barros and Morgante (in press). A standard Proteinase K and Phenol protocol (Sambrook et al., 1989) was used to obtain DNA from ethanol preserved tissues.
2.2. Data collection
For each individual, a segment of 387 base pairs (bp) of the mitochondrial DNA (mtDNA) control region was analyzed. According to the integrity of DNA samples, two different Polymerase Chain Reaction (PCR) protocols were used to amplify the mtDNA segment. For DNA samples obtained from fresh tissues, primers and PCR conditions followed the protocol described in Moraes-Barros et al. (2006). DNA samples obtained from museum study skin usually present high levels of fragmented molecules. PCR performed upon these DNA samples was conducted by using different combinations of internal primers previously described in Moraes-Barros and Morgante (in press). These primer combinations resulted in overlapping and adjacent segments that together compound the original 400 pb fragment. All PCR products were purified using Shrimp Alkaline Phosphatase and Exonuclease I. Sequencing reactions were conducted using the BigDye Kit (Applied Biosystems, Inc.) with 100-200 ng of PCR product in 10 µl reactions. Sequencing reactions were precipitated using isopropanol and run on ABI 377 automated sequencer (Applied Biosystems, Inc.). In attempt to ensure the quality of the sequences and avoid analyzing unspecific amplifications upon fragmented DNA, all PCR and sequencing reactions were performed at least twice. We only used samples that resulted in sequences with no individual ambiguities. Sequence data were edited and aligned using Sequence Navigator 1.0.1 (Applied Biosystems, Inc.). DNA sequences were submitted to GenBank under accession numbers EU124524 to EU124550.
2.3 Data analysis
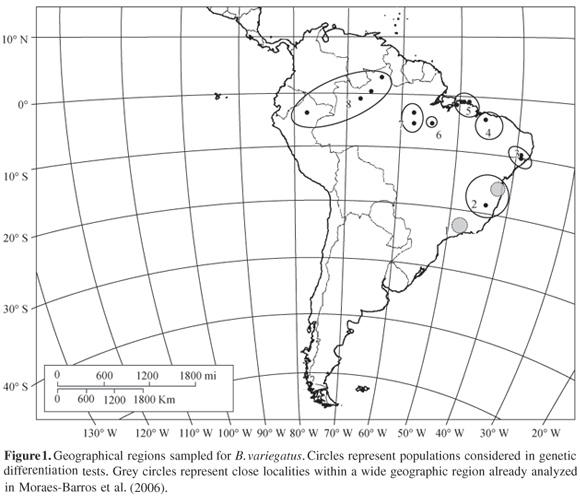
To test whether each different sampled locality of B. variegatus constituted a demographically autonomous population, the genetic divergence among them was tested. Analyses of molecular variance (AMOVA) were performed to determine the amount of variation due to population structure, considering each sampled locality as a population. Interpopulational component of genetic variation (Fst - Wright, 1965) was used as a measure of population subdivision. Fst values were calculated considering haplotype frequencies and nucleotide differences among DNA sequences, the jst index (Excoffier et al., 1992). For some localities, only one individual was sampled, therefore they were grouped together in a wide geographical region and each region was considered a population (Figure 1). These analyses were conducted on the software Arlequin 2.0 (Schneider et al., 2000).
Mitochondrial genealogies were also estimated to investigate the phylogenetic relationship among B. variegatus lineages and to compare them to previous described clades from Atlantic forest. Phylogenetic trees were inferred by using PAUP* version 4b10 (Swoford, 2002). Models of molecular evolution were estimated in Modeltest 3.5 (Posada and Crandall, 1998). A DNA sequence from B. torquatus (GenBank accession number AY762814) was used as the out group. The observed lineages were then compared to their sampled localities.
3. Results
In the population analyses, we first tested each sampled region as a population. AMOVA results showed that 89% (0.896) of the B. variegatus genetic variation was due to differences among populations. jst values estimated among the eight populations ranged from 0.138 to 0.966 and only two values were considered statistically non significant. According to these jst values, population 5 was not significantly differentiated from population 1 (jst = 0.134, P = 0.991 ± 0.003). The same result was observed when comparing populations 3 and 4 (jst = 0.203, P = 0.171 ± 0.03) (Table 2a).

Based on these results and considering the geographic region of each population, we rearranged populations, considering populations 3 and 4 a panmitic population. Region 5 contains a single haplotype, the same present in region 1, and both were considered the same genetic population despite the geographical distance between them. The jst values resultant after comparing the six populations (1+5, 2, 3+4, 6, 7, 8) were all statistically significant (Table 2b). Also, most of the B. variegatus genetic diversity was still due to variation among populations (0.879).
The inferred phylogenetic trees revealed six mtDNA lineages within B. variegatus (Figure 2, lineages I to VI). Four of these lineages are exclusive to specific geographical regions. Lineage I can be subdivided in two clades, described here as Ia and Ib. Clade Ib is exclusive to the region 2 whereas clade Ia is represented in three distinct regions (1, 3, and 5). A similar result was observed in lineage V that is distributed in regions 3 and 4 (Figure 2).
4. Discussion
Our analysis on population divergence showed that Bradypus variegatus genetic diversity is distributed in six geographic regions significantly divergent from each other in mtDNA haplotypes (Table 2b). Therefore, these groups can be considered demographically autonomous and genetically divergent populations and we can infer that each population is a MU, according to the definition of Moritz (1994a).
Within the Atlantic forest, we identified two divergent populations, distributed respectively in northern and southern regions of the forest. These MUs were already observed in a previous study (Moraes-Barros et al., 2006) and here we confirmed that they are genetic divergent from other B. variegatus populations, with the exception of Atlantic forest southern region (region 1) which is not significantly differentiated from region 5, in the northeast edge of the Brazilian Amazon forest (Figure 1). Values of jst indicate that we can not discard the panmitic hypothesis (Table 2), and the inference of phylogenetic relationships among mtDNA lineages indicates that both regions share the same lineage, clade Ia (Figure 2). The poor sampling in region 5 and the presence of the same highly frequent haplotype in two geographically distant localities may be responsible for this genetic similarity. It is possible that a more detailed sampling reveals more haplotypes in region 5. However, according to our data, we can not consider these two regions as genetically divergent populations. We can only infer the presence of an ancestral and high frequent haplotype in both regions.
The phylogenetic division of Atlantic forest in northern and southern units was already reported for other mammals species like marsupials, rodents, and bats (Ditchfield, 2000; Costa, 2003), different species of vertebrates (Muller, 1973; Lynch, 1979; Vanzolini, 1988; Bates et al., 1998; Pellegrino et al., 2005), and also invertebrates (Pinto-da-Rocha and Da Silva, 2005). The pattern observed in the common sloth was also described in the crab-eating fox, Cerdocyon thous (Tchaicka et al., 2007). Besides the north and south division of Atlantic forest, Tchaicka et al. (2007) also demonstrated a similarity between the southern region of Atlantic forest and the eastern region of Amazon forest. This similarity was also observed in the common sloth and is represented by the panmitic population composed by individuals distributed in region 1 and region 5 (Figure 1).
Another MU is represented by population 3+4. The geographic region enclosed by this MU encompasses two localities that belong to different biomes, locality 3 and locality 4. Locality 3 is in the Atlantic forest and locality 4 represents a transition region between the Amazon and the Atlantic forests, located at the Maranhão State, considered part of the Cerrado biome. According to our results, sloths sampled at these two regions can be considered as a single panmitic population (Table 2). This population is composed by mtDNA lineages V, VI, and Ia (Figure 2). Lineages V and VI are exclusive to this population while lineage Ia is also present in the southern region of Atlantic forest and in the eastern edge of Amazon forest, evidencing a relationship between these two biomes (Figure 2). We consider that the genetic composition of population 3+4 might indicate a connection between the Atlantic and Amazon forests, through the northeast region of Brazil. We also consider that the region of Cerrado, in central Brazil, between the region 5 and region 1, could represent another connection between the two neotropical forested biomes.
According to Costa (2003), patterns of connection between the Atlantic and Amazon forests were also observed in marsupials and rodents. One of these "connection bridges" pointed to the gallery forests in central Brazil, connecting the south portion of Atlantic forest and the northern region of Brazil. Another connection is through the northeastern region of Brazil. These connection patterns already described for other mammals are congruent with the similarity observed between regions 1 and 5 of B. variegatus distribution and also with the region represented by population 3+4 (Figure 1).
Within the Amazon forest, we can delimit three MUs. Despite the significant distinctiveness of mtDNA sequences among these MUs, it is also possible to observe a correspondence between the geographical regions and the inferred phylogeny (Figure 2). Each one of these three MUs is compounded exclusively by a single lineage: population 6, in the northeast region of Amazon, right margin of the Xingu River lineage III; population 7, near the upper Tapajós River lineage II; population 8, surrounding the northwestern Amazon lineage IV. Two of the Amazon mtDNA lineages show high phylogenetic support. However, the basal relationship between them is unsolved which prevent us from inferring whether there is reciprocal monophyly. Nevertheless, the geographical distribution of exclusive lineages within populations 6 and 7 is coincident with Amazonian endemism areas already described for terrestrial vertebrates: the Xingu and Tapajós areas (Da Silva et al., 2005). A review of the genus Bradypus pointed to the need of a detailed study of geographic variation of sloths and also claimed that some populations of Amazon and Central America should deserve the subspecies status, specially a population localized by the upper Tapajós River (Anderson and Handley, 2001). This region, along with the endemism area of Tapajós, is concordant with one of our exclusive populations, localized in region 7 (Figure 1).
According to our results, we can observe five main geographic regions that represent the neutral genetic diversity of B. variegatus. The first region encloses the Atlantic forest and the eastern edge of Amazon forest. Within this region, we found a divergent population representing the north component of Atlantic forest. The third region encompasses a transition area between these two forested areas. We also detected a west component and two central regions within Amazon forest. In the Amazon forest, each population is composed by an exclusive mtDNA lineage.
It is important to mention that this study was conducted with few localities and sloths sampled through Atlantic forest, Cerrado, and Amazon forest. A detailed sampling can reveal more divergent intraspecific units. Also, the mtDNA phylogenetic analysis did not reveal a solved relationship among mtDNA lineages, preventing us from further phylogeographic inferences. Moreover, the pattern evidenced by the mtDNA shows only the matrilineal evolutionary history and it is not sufficient to discard possible bias due to differential migration between males and females or introgression. However, since there are no studies describing sex-biased dispersal for sloths and considering that they are not volant mammals, the observed marked population structure is as expected for this mammal.
Therefore, if these genetic divergent populations constitute ESUs or how populations from Amazon and Atlantic forest are related, are questions that are yet to be addressed through the use of additional and unlinked molecular markers, a more detailed locality sampling, and the implementation of ecological data.
Nevertheless, considering the geographical distribution of the six observed populations and the significant mtDNA divergence among them, we have enough evidence that permit us to consider each one as a management unit, MU. These intraespecific units are congruent with described endemism areas of Amazon forest and phylogroups on Atlantic forest evidencing the importance of such studies as a tool to understand the biogeographic patterns on neotropical region and helping to delimit geographic areas as conservation units for both the Atlantic and Amazon forests.
Acknowledgments The authors would like to thank all researchers and institutions that contributed to this work by tissue donations and permission to access the museum collections: MPEG, MZUSP, MVZ, and Universidade Federal do Pará. We also wish to thank Diogo Meyer, Juliana Machado Ferreira, and the two anonymous reviewers for comments and suggestions, and Karen O. for help on the English review. This paper is the result of a project supported by FAPESP (Grants and fellowships: 99/01310-4, 00/14629-8, 00/13213-2, 03/03212-7, 05/55438-4).
Received April 30, 2007
Accepted August 21, 2007
Distributed December 1, 2007
- ANDERSON, RP. and HANDLEY, CO. JR., 2001. A new species of three-toed sloth (Mammalia: Xenarthra) from Panamá, with a review of the genus Bradypus. Proc. Biol. Soc. Wash, vol. 114, no. 1, p. 1-33.
- ARAÚJO, MB., 2002. Biodiversity hotspots and zones of ecological transition. Conserv. Biol, vol. 16, no. 6, p. 1662-1663.
- AVISE, JC., 1995. Mitochondrial DNA polymorphism and connection between genetics and demography of relevance to conservation. Conserv. Biol, vol. 9, no. 3, p. 686-690.
- BATES, JM., HACKETT, SJ. and CRACRAFT, J., 1998. Area-relationships in the Neotropical lowlands: a hypotheses based on raw distribution of passerine birds. J. Biogeogr, vol. 25, no. 4, p. 783-93.
- COSTA, LP., 2003. The historical bridge between the Amazon and the Atlantic forest of Brazil: a study of molecular phylogeography with small mammals. J. Biogeogr, vol. 30, no. 1, p. 71-86.
- CRANDALL, KA., BINIDA-EMONDS, ORP., MACE, GM. and WAYNE, RK., 2000. Considering evolutionary processes in conservation biology. TREE, vol. 15, no. 7, p. 290-295.
- DA SILVA, JMC., RYLANDS, AB. and FONSECA, GAB., 2005. The fate of the Amazonian areas of endemism. Conserv. Biol, vol. 19, no. 3, p. 689-694.
- DITCHFIELD, AD., 2000. The comparative phylogeography of Neotropical mammals: patterns of intraspecific mitochondrial DNA variation among bats contrasted to non-volant small mammals. Mol. Ecol, vol. 9, no. 2, p. 1307-1318.
- EXCOFFIER, L., SMOUSE, P. and QUATTRO, J., 1992. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics, vol. 131, no. 2, p. 479-491.
- FRANKHAN, R., BALLOU, JD. and BRISCOE, DA., 2002. Introduction to conservation genetics Cambridge, UK, Cambridge University Press, 617p.
- FRASER, DJ. and BERNATCHEZ, L., 2001. Adaptive evolutionary conservation: towards a unified concept for defining conservation units. Mol. Ecol, vol. 10, no. 12, p. 2741-2752.
- LEDGE, JT., ROUSH, R., DESALLE, R., VOGLER, AP. and MAY, B., 1996. Genetic criteria for establishing Evolutionary Significant Units in Cryans buckmouth. Conserv. Biol, vol. 10, no. 1, p. 85-98.
- LYNCH, JD., 1979. The amphibians of the lowland tropical forests. IN DUELLMAN, WE. (ed.). The South American herpetofauna: its origin, evolution, and dispersal Monographs of the Museum of Natural History, Lawrence, University of Kansas, p. 189-215.
- MARGULES, CR. and PRESSEY, RL., 2000. Systematic conservation planning. Nature, vol. 405, no. 6783, p. 243-253.
- MEFFE, GK. and CARROL, CR., 1997. Principles of conservation biology Sunderland, Massachusetts, Sinauer Associates Inc., 729p.
- MORAES-BARROS, N., SILVA, JAB., MIYAKI, CY. and MORGANTE, JS., 2006. Comparative phylogeography of the Atlantic forest endemic sloth (Bradypus torquatus) and the widespread three-toed sloth (Bradypus variegatus). Genetica, vol. 126, no. 1-2, p. 189-198.
- MORAES-BARROS, N. and MORGANTE, JS. A simple protocol for DNA extraction and sequence analysis of museum study skin specimens. Genet. Mol. Biol (in press).
- MORITZ, C., 1994a. Defining "Evolutionarily Significant Units" for conservation. TREE, vol. 9, no. 10, p. 373-375.
- -, 1994b. Applications of mitochondrial DNA analysis on conservation: a critical review. Mol. Ecol, vol. 3, no. 4, p. 401-411.
- -, 1999. Conservation units and translocations: strategies for conserving evolutionary processes. Hereditas, vol. 130, no. 3, p. 217-228.
- -, 2002. Strategies to protect biological diversity and the evolutionary processes that sustain it. Syst. Biol, vol. 51, no. 2, p. 238-254.
- MORITZ, C., LAVERY, S. and SLADE, R., 1995. Using allele frequency and phylogeny to define units for conservation and management. In NIELSEN, JL. (ed.). Evolution and the aquatic ecosystems: defining unique units in population conservation. Maryland, American Fisheries Society, Bethesda, p. 249-262.
- MULLER, P., 1973. Dispersal centres of terrestrial vertebrates in the Neotropical realm Biogeographica, vol. 2. The Hague: Junk, University of Saarland, 244p.
- MYERS, N., MITTERMEIER, RA., MITTERMEIER, CG., FONSECA, GAB. and KENT, J., 2000. Biodiversity hotspots for conservation priorities. Nature, vol. 403, no. 6772, p. 853-858.
- PELLEGRINO, KCM., RODRIGUES, MT., WAITE, AN., MORANDO, M., YASSUDA, Y. Y. and SITES, JW., 2005. Phylogeography and species limits in the Gymnodactylus darwinii complex (Gekkonidae, Squamata): genetic structure coincides with river systems in the Brazilian Atlantic Forest. Biol. J. Linn. Soc, vol. 85, no. 1, p. 13-26.
- PENNOCK, DS. and DIMMICK, WW., 1997. Critique of the Evolutionary Significant Unit as a definition for "distinct populations segments" under the U.S. Endangered Species Act. Conserv. Biol, vol. 11, no. 3, p. 611-619.
- PINTO-DA-ROCHA, R. and DA SILVA, MB., 2005. Faunistic similarity and historic biogeography of the harvestmen of southern and southeastern Atlantic rainforest of Brazil. J. Arach, vol. 33, no. 2, p. 290-99.
- POSADA, D. and CRANDALL, KA., 1998. Modeltest: testing the model of DNA substitution. Bioinformatics, vol. 14, no. 9, p. 817-818.
- RYDER, OA., 1986. Species conservation and systematics: The dilemma of subspecies. TREE, vol. 1, no. 1, p. 9-10.
- SAMBROOK, J., FRITSCH, EF. and MANIATIS, T., 1989. Molecular cloning: a laboratory manual vol. 3. New York, NY, Cold Spring Harbor Laboratory Press.
- SCHNEIDER, S., ROESSLI, D. and EXCOFFIER, L., 2000. Arlequin Ver. 2000: A software for population genetics data analysis Switzerland, Genetics and Biometry Laboratory, Univeristy of Geneva.
- SWOFORD, DL., 2002. PAUP*. Phylogenetic analysis using parsimony (*and other methods) Version 4. Sunderland, Massachussets, Sinauer Associates.
- TAYLOR, BL. and DIZON, AE., 1996. The need to estimate power to link genetics and demography for conservation. Conserv. Biol, vol. 10, no. 2, p. 661-664.
- TCHAICKA, L., EIZIRIK, E., DE OLIVEIRA, TG., CÂNDIDO, JF. JR. and FREITAS, TRO., 2007. Phylogeography and population history of the crab-eating fox (Cerdocyon thous). Mol. Ecol, vol. 16, no. 4, p. 819-838.
- VANZOLINI, PE., 1988. Distributional patterns of South American lizards. In VANZOLINI, PE. and HAYER, WR. (eds.). Proceedings of a workshop on Neotropical distribution patterns Rio de Janeiro, Academia Brasileira de Ciências, p. 317-343.
- WAPLES, RS., 1991. Pacific salmon, Oncorhynchus spp., and the definition of "species" under the Endangered Species Act. Mar. Fish. Rev, vol. 53, no. 3, p. 11-22.
- -, 1998. Evolutionary Significant Units, distinct population segments, and the Endangered Species Act: reply to Pennock and Dimmick. Conserv. Biol, vol. 12, no. 3, p. 718-721.
- WRIGHT, S., 1965. The interpretation of population structure by F-statistics with special regard to systems of mating. Evolution, vol. 19, no. 3, p. 395-420.
Publication Dates
-
Publication in this collection
11 Feb 2008 -
Date of issue
Dec 2007
History
-
Accepted
21 Aug 2007 -
Received
30 Apr 2007