Resumos
A doença falciforme é um termo genérico usado para determinar um grupo de alterações genéticas caracterizadas pelo predomínio de hemoglobina (Hb) S. Os principais genótipos que compõem o grupo de doença falciforme são os seguintes: SS, SF [S/beta0 talassemia e S/persistência hereditária de Hb fetal (PHHF)], SFA (S/beta+ talassemia), SC, SD e SH (S/alfa talassemia). O presente trabalho analisa os resultados das avaliações de produtos provenientes da oxidação da Hb S, identificados pela concentração da metemoglobina e de eritrócitos com corpos de Heinz em dois genótipos da doença falciforme (SS e S/beta0 talassemia) e no traço falcêmico (AS), em comparação com o genótipo normal (AA). A análise dos produtos da degradação oxidativa da hemoglobina, evidenciados pelo aumento dos valores das médias referentes à concentração de metemoglobina e do número de eritrócitos com corpos de Heinz, está diretamente relacionada com o aumento da concentração da Hb S. Assim, a degradação oxidativa da hemoglobina decresce entre os genótipos estudados da seguinte forma: SS>SF>AS>AA. É importante destacar que as análises indicaram que a simples presença de Hb S no eritrócito, como é o caso do genótipo AS, é capaz de causar elevação da concentração de metemoglobina em 52,62% das amostras analisadas e de induzir a precipitação de corpos de Heinz em 73,68% dos casos estudados. Explicações referentes aos processos oxidativos e redutores das hemoglobinas estudadas são apresentados no texto. Destaca-se, entre os resultados apresentados, a identificação por meio de eletroforese em agarose alcalina da fração de globina alfa-livre em todas as amostras do genótipo SF provenientes de pessoas com Hb S/beta0 talassemia. É proposto um esquema para explicar a origem da globina alfa-livre, especialmente para o genótipo S/beta0 talassemia, e a importância da sua identificação no diagnóstico laboratorial de Hb S/beta0 talassemia.
Doença falciforme; Degradação oxidativa da Hb S; Metemoglobina; Corpos de Heinz
Sickle cell disease is a generic term used to determine a group of genetic alterations characterized by a dominance of Hb S. The main genotypes which compose the sickle cell disease group are as follows: SS, SF (S/beta0 thalassemia and S/Hereditary Persistence of Fetal Hemoglobin or HPFH), SFA (S/beta+ thalassemia), SC, SD and SH (S/alpha thalassemia). This study analyzes the products resulting from the oxidization of hemoglobin, identified by the methemoglobin concentration and by red blood cells with Heinz bodies, in two sickle cell genotypes (SS and S/beta0 thalassemia) and in the sickle cell trait (AS) compared with the normal genotype (AA). Analysis of the products resulting from hemoglobin oxidative damage, characterized by an increase in the mean levels of methemoglobin and of the number of red blood cells with Heinz bodies, which are directly related to the increase in the Hb S concentration. Thus, oxidative damage of hemoglobin diminishes among the studied genotypes in the following manner: SS>SF>AS>AA. It is important to stress that these results indicate that the simple presence of Hb S in the red blood cell, as in the AS genotype, is capable of increasing the methemoglobin concentration in 52.62% of the assessed samples and inducing the precipitation of Heinz bodies in 73.68% of cases. Elucidation of the oxidative and reductive processes of the studied hemoglobins is presented in the paper. Highlighted among the presented results is the identification, by means of alkaline agarose gel electrophoresis, of the free alpha globin fraction in all SF genotype samples originating from Hb S/beta0 thalassemia individuals. A hypothesis to explain the origin of free alpha globin, especially in the S/beta0 thalassemia genotype is proposed, as is the importance of its identification in the laboratorial diagnosis of S/beta0 thalassemia.
Sickle cell disease; Oxidative damage of Hb S; Methemoglobin; Heinz bodies
ARTIGO ORIGINAL ORIGINAL PAPER
Avaliação dos produtos da degradação oxidativa da Hb S nos genótipos SS, SF (S/b0 talassemia) e AS, em comparação com hemoglobinas normais
Evaluation of the products from oxidative damage of Hb S in SS, SF (S/b0 thalassemia) and AS genotypes compared to normal hemoglobins
Paulo Cesar NaoumI; Patrícia Caetano de SouzaII
IProfessor-titular da Universidade Estadual Paulista (UNESP); diretor da Academia de Ciência e Tecnologia de São José do Rio Preto
IIMestra; doutoranda do curso de Pós-Graduação em Genética do Instituto de Biociências, Letras e Ciências Exatas, UNESP de São José do Rio Preto
Endereço para correspondência Endereço para correspondência Paulo Cesar Naoum Academia de Ciência e Tecnologia Rua Bonfá Natale 1.860 CEP 15020-130 São José do Rio Preto-SP Tel.: (17) 233-4490 e-mail: a.c.t@terra.com.br www.ciencianews.com.br
RESUMO
A doença falciforme é um termo genérico usado para determinar um grupo de alterações genéticas caracterizadas pelo predomínio de hemoglobina (Hb) S. Os principais genótipos que compõem o grupo de doença falciforme são os seguintes: SS, SF [S/b0 talassemia e S/persistência hereditária de Hb fetal (PHHF)], SFA (S/b+ talassemia), SC, SD e SH (S/a talassemia). O presente trabalho analisa os resultados das avaliações de produtos provenientes da oxidação da Hb S, identificados pela concentração da metemoglobina e de eritrócitos com corpos de Heinz em dois genótipos da doença falciforme (SS e S/b0 talassemia) e no traço falcêmico (AS), em comparação com o genótipo normal (AA). A análise dos produtos da degradação oxidativa da hemoglobina, evidenciados pelo aumento dos valores das médias referentes à concentração de metemoglobina e do número de eritrócitos com corpos de Heinz, está diretamente relacionada com o aumento da concentração da Hb S. Assim, a degradação oxidativa da hemoglobina decresce entre os genótipos estudados da seguinte forma: SS>SF>AS>AA. É importante destacar que as análises indicaram que a simples presença de Hb S no eritrócito, como é o caso do genótipo AS, é capaz de causar elevação da concentração de metemoglobina em 52,62% das amostras analisadas e de induzir a precipitação de corpos de Heinz em 73,68% dos casos estudados. Explicações referentes aos processos oxidativos e redutores das hemoglobinas estudadas são apresentados no texto. Destaca-se, entre os resultados apresentados, a identificação por meio de eletroforese em agarose alcalina da fração de globina alfa-livre em todas as amostras do genótipo SF provenientes de pessoas com Hb S/b0 talassemia. É proposto um esquema para explicar a origem da globina alfa-livre, especialmente para o genótipo S/b0 talassemia, e a importância da sua identificação no diagnóstico laboratorial de Hb S/b0 talassemia.
unitermos: Doença falciforme, Degradação oxidativa da Hb S, Metemoglobina, Corpos de Heinz
ABSTRACT
Sickle cell disease is a generic term used to determine a group of genetic alterations characterized by a dominance of Hb S. The main genotypes which compose the sickle cell disease group are as follows: SS, SF (S/b0 thalassemia and S/Hereditary Persistence of Fetal Hemoglobin or HPFH), SFA (S/b+ thalassemia), SC, SD and SH (S/a thalassemia). This study analyzes the products resulting from the oxidization of hemoglobin, identified by the methemoglobin concentration and by red blood cells with Heinz bodies, in two sickle cell genotypes (SS and S/b0 thalassemia) and in the sickle cell trait (AS) compared with the normal genotype (AA). Analysis of the products resulting from hemoglobin oxidative damage, characterized by an increase in the mean levels of methemoglobin and of the number of red blood cells with Heinz bodies, which are directly related to the increase in the Hb S concentration. Thus, oxidative damage of hemoglobin diminishes among the studied genotypes in the following manner: SS>SF>AS>AA. It is important to stress that these results indicate that the simple presence of Hb S in the red blood cell, as in the AS genotype, is capable of increasing the methemoglobin concentration in 52.62% of the assessed samples and inducing the precipitation of Heinz bodies in 73.68% of cases. Elucidation of the oxidative and reductive processes of the studied hemoglobins is presented in the paper. Highlighted among the presented results is the identification, by means of alkaline agarose gel electrophoresis, of the free alpha globin fraction in all SF genotype samples originating from Hb S/b0 thalassemia individuals. A hypothesis to explain the origin of free alpha globin, especially in the S/b0 thalassemia genotype is proposed, as is the importance of its identification in the laboratorial diagnosis of S/b0 thalassemia.
key words: Sickle cell disease, Oxidative damage of Hb S, Methemoglobin, Heinz bodies
Introdução
A doença causada pelas células falciformes se caracteriza por um conjunto de sinais e sintomas provocados pela deformação de expressiva quantidade de eritrócitos com hemoglobina S (Hb S) e induzidos pela desoxigenação. Esses eritrócitos com Hb S, quando oxigenados (oxi-Hb S), apresentam-se morfologicamente normais ou discóides no sangue; eventualmente, podem ser observados alguns eritrócitos afoiçados ou falcizados. Entretanto, há situações em que o processo de falcização é muito intenso em função da elevada concentração da Hb S desoxigenada (desoxi-Hb S)(3,14).
A doença falciforme é um termo genérico usado para determinar um grupo de alterações genéticas caracterizadas pelo predomínio da Hb S. A introdução da eletroforese de hemoglobinas como principal método de investigação laboratorial contribuiu para o entendimento clínico, genético e hematológico dos doentes falcêmicos, pois sua aplicação permitiu explicar que a diversidade clínica e hematológica estava relacionada com a concentração da Hb S, bem como sua associação com outras hemoglobinas. Assim, os principais genótipos que compõem o grupo de doença falciforme foram caracterizados e identificados por SS, SF (S/b0 talassemia e S/PHHF), SFA, (S/b+ talassemia), SD, SC, SH (S/a talassemia), entre outros(2, 7, 9).
É evidente que, para o estabelecimento criterioso do genótipo da doença falciforme, é necessário que sejam feitas análises laboratoriais em familiares, especialmente nos pais, incluindo: a qualificação eletroforética em meio alcalino ou cromatográfico por cromatografia líquida de alta pressão (HPLC), com a comprovação eletroforética em gel de agarose ácida da Hb S e suas associações (SS, SD, SF, etc.); a quantificação da Hb S fracionada por eletroforese ou cromatografia; avaliações hematimétricas e morfológicas dos eritrócitos; e a dosagem de Hb fetal por metodologias bioquímica e imunológica(2, 11).
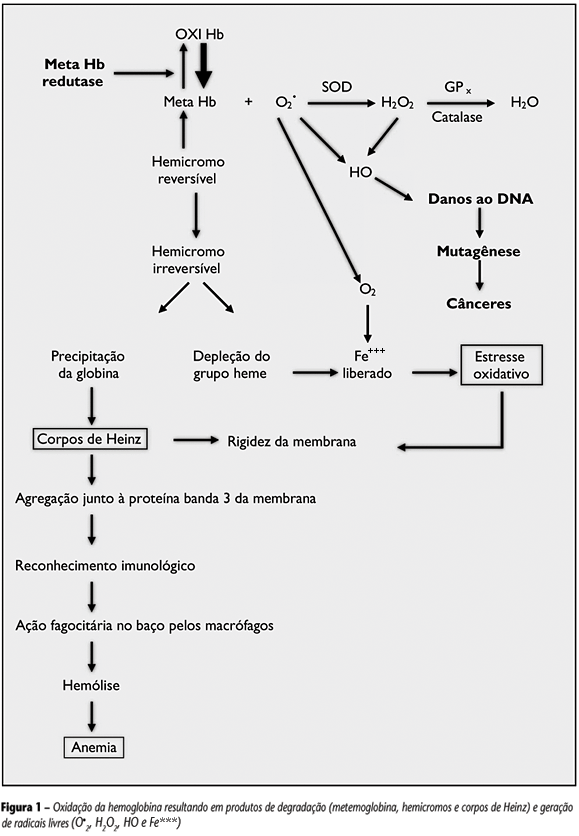
A morbidade e a mortalidade de doentes falcêmicos têm sido relacionadas com várias situações, entre as quais destacam-se: o desconhecimento da doença, a dificuldade de se proceder ao diagnóstico precoce e correto do genótipo e as deficiências do atendimento médico e dos procedimentos terapêuticos, entre outros(10, 14). Entretanto, um dos fatores que predispõe o processo hemolítico das células falciformes induzindo-o às múltiplas conseqüências patológicas da doença falciforme deve-se à degradação oxidativa da Hb S(4, 12, 13). A desoxigenação da Hb S favorece a sua metemoglobinização (meta-Hb S) e a conseqüente elevação desse pigmento dentro do eritrócito. Quando a concentração de meta-Hb S supera a ação da enzima meta-Hb redutase, é desencadeada a degradação da meta-Hb S, com formação de hemicromos ou subprodutos do grupo heme, e a precipitação da globina S, oxidada sob forma de corpos de Heinz (Figura 1)(14, 15). Durante a transformação do eritrócito discóide com Hb S em eritrócito afoiçado, entre os eventos bioquímicos e polimerizantes da célula, ocorre a degradação oxidativa da Hb S, com liberação de espécies ativadas de oxigênio (O2, H2O2, HO) que alteram a distribuição das moléculas de imunoglobulinas G (IgG) na superfície da membrana eritrocitária. As disposições desordenadas da molécula de IgG em determinadas regiões da célula falciforme facilitam as ligações com os receptores Fc das células endoteliais e, dessa forma, induzem a ação fagocitária dos macrófagos no sistema retículo endotelial, causando hemólise e anemia. É provável que os produtos de degradação oxidativa da Hb S se apresentem qualitativa e quantitativamente diferentes entre os principais genótipos da doença falciforme, e esse fato pode estar relacionado principalmente à concentração da Hb S, à interação com outras hemoglobinas (C, fetal, D, etc.) dentro de um mesmo eritrócito e à associação com a deficiência de enzimas eritrocitárias antioxidantes(1, 5, 8).
Os dois genótipos de doença falciforme que apresentam maior gravidade clínica são o SS (anemia falciforme) e SF (interação de Hb S com talassemia beta zero, ou S/b0 talassemia). Ambos apresentam anemia grave com Hb fetal variável entre 2% e 10% no genótipo SS e entre 5% e 20% no genótipo SF. Paradoxalmente, num outro genótipo SF, no qual a elevação da Hb fetal se deve à sua persistência hereditária após o nascimento, a qualidade de vida do doente falcêmico é muito melhor, geralmente com discreto grau de anemia e com índices menores de morbidade e mortalidade(7, 10). A explicação decorre por duas razões: a) no genótipo SF, em função da interação S/b0 talassemia, a distribuição intra-eritrocitária de Hb fetal é heterogênea, ou seja, há eritrócitos apenas com Hb fetal proveniente de clones eritroblásticos com genes da talassemia b0 e eritrócitos somente com Hb S provenientes de clones eritroblásticos com genes da Hb S; b) enquanto que no genótipo SF, em função da associação da Hb S com a persistência hereditária de Hb fetal (PHHF), o mesmo eritrócito contém esses dois tipos de hemoglobinas, pois as sínteses destas provieram dos mesmos clones de eritroblastos com genes da PHHF e da Hb S(7). É importante destacar que a Hb fetal (a2g2) é um tetrâmero molecularmente menos estável que a Hb A (a2bA2), com maior capacidade auto-oxidativa, desencadeando conseqüentemente a formação de subprodutos de hemoglobina caracterizados por metemoglobina e corpos de Heinz(12, 15).
O objetivo do presente trabalho é avaliar a relação entre concentração de Hb S nos genótipos de hemoglobinas SS, SF e AS com produtos da degradação oxidativa da Hb S, em comparação com a Hb A obtida de genótipo AA.
Material e método
Foram analisadas 136 amostras de sangue pertencentes a pessoas com seus genótipos de hemoglobinas previamente qualificados clinica e laboratorialmente em: Hb AA (78 amostras), Hb AS (19 amostras), Hb SS (17 amostras) e Hb SF (16 amostras). As concentrações porcentuais das hemoglobinas S e fetal e da metemoglobina foram respectivamente avaliadas por eletroforese alcalina quantitativa, teste bioquímico da resistência alcalina e teste de absorção espectrofotométrica de pigmentos hemoglobínicos induzidos por cianeto de potássio e ferricianeto de potássio(11). A pesquisa intra-eritrocitária de corpos de Heinz foi realizada por incubação de sangue total com azul de crezil brilhante a 1% na temperatura de 37°C(11), e a investigação de globina alfa-livre em amostras de sangue com genótipo SF pertencentes a pessoas portadoras de Hb S/b0 talassemia foi feita por meio de eletroforese em gel de agarose alcalina com sangue hemolisado por saponina a 1%(11).
Os valores obtidos das avaliações quantitativas foram submetidos à análise estatística descritiva e agrupados em dois conjuntos: a) de acordo com o genótipo; b) de acordo com a concentração de Hb S. As análises incluíram as porcentagens de Hb fetal e metemoglobina e número de eritrócitos com corpos de Heinz, para os quais determinou-se para cada variável a média, mediana, valor mínimo, valor máximo, desvio padrão (DP) e erro padrão (EP), além da distribuição porcentual. Para comparação entre duas médias foi utilizado o teste t para observações independentes, usando o nível de significância de a = 5% (p b 0,05).
Resultados
As amostras analisadas tiveram seus genótipos definidos previamente com bases clínica e laboratorial. Mesmo assim, todas foram submetidas a eletroforeses qualitativa e quantitativa de hemoglobinas, em meios alcalino e ácido, com o objetivo de confirmar os genótipos, bem como o de quantificar a concentração de Hb S nos genótipos AS, SS e SF. Todas as 19 amostras com genótipo AS apresentavam concentrações de Hb S abaixo de 50%, com valores mínimo e máximo de 23,4% e 45,5%, respectivamente. As 17 amostras com genótipo SS apresentaram concentrações de Hb S acima de 72,3%, enquanto que entre as 16 amostras do genótipo SF, as concentrações de Hb S variaram entre 65,2% e 88,8% (Tabelas 1, 2 e 3).
Para todas as amostras de sangue analisadas, referentes aos genótipos AA, AS, SS e SF, avaliaram-se as concentrações porcentuais de Hb fetal e metemoglobina, além da quantificação média de eritrócitos com precipitados de corpos de Heinz em relação a mil eritrócitos. Os valores obtidos e que resultaram em média, mediana, mínimo, máximo, desvio padrão (DP) e erro padrão (EP) estão apresentados na Tabela 4. A análise descritiva desses valores para a Hb fetal mostrou-se moderadamente elevada no genótipo SS ( : 4,94%) e acentuadamente elevada no genótipo SF (: 14,06%). Por outro lado, os valores de metemoglobina apresentaram-se moderadamente elevados nos genótipos SS (: 3,97%) e SF (: 3,3%). A presença de corpos de Heinz mostrou-se discretamente elevada no genótipo AS (: 2,42/1000), moderadamente elevada no genótipo SF (: 9,43/1000) e acentuadamente elevada no genótipo SS (: 28/1000). Os valores comparativos entre duas médias para se obter o grau de significância estatística entre os genótipos estudados (AA, AS, SS e SF), com avaliações específicas para Hb fetal, metemoglobina e corpos de Heinz, estão expostos na Tabela 5. Por meio da análise desses resultados é possível constatar que a diferença dos valores da concentração média de Hb fetal entre os genótipos somente não teve significância estatística no confronto de Hb AA com Hb AS (p = 0,068). Com relação aos valores da concentração média de metemoglobina, apenas os confrontos entre os genótipos AS e SF (p = 0,077) e SS e SF (p = 0,549) não tiveram significância estatística. Por outro lado, a presença de corpos de Heinz entre os quatro genótipos foi estatisticamente diferente.
: 4,94%) e acentuadamente elevada no genótipo SF (: 14,06%). Por outro lado, os valores de metemoglobina apresentaram-se moderadamente elevados nos genótipos SS (: 3,97%) e SF (: 3,3%). A presença de corpos de Heinz mostrou-se discretamente elevada no genótipo AS (: 2,42/1000), moderadamente elevada no genótipo SF (: 9,43/1000) e acentuadamente elevada no genótipo SS (: 28/1000). Os valores comparativos entre duas médias para se obter o grau de significância estatística entre os genótipos estudados (AA, AS, SS e SF), com avaliações específicas para Hb fetal, metemoglobina e corpos de Heinz, estão expostos na Tabela 5. Por meio da análise desses resultados é possível constatar que a diferença dos valores da concentração média de Hb fetal entre os genótipos somente não teve significância estatística no confronto de Hb AA com Hb AS (p = 0,068). Com relação aos valores da concentração média de metemoglobina, apenas os confrontos entre os genótipos AS e SF (p = 0,077) e SS e SF (p = 0,549) não tiveram significância estatística. Por outro lado, a presença de corpos de Heinz entre os quatro genótipos foi estatisticamente diferente.
A Tabela 6 destaca quatro bandas de valores porcentuais para as concentrações de Hb fetal e metemoglobina, onde a banda correspondente a 0%-2% é considerada normal para ambas avaliações. Observa-se que para o genótipo AA a Hb fetal esteve normal em 98,72% das amostras analisadas, enquanto que para o genótipo AS a normalidade caiu para 73,68%. Por outro lado, nenhuma das amostras com genótipos SS e SF mostrou valor normal de Hb fetal. Com relação à metemoglobina, quase 90% das amostras com genótipos AA resultaram normais, enquanto que nos genótipos AS, SS e SF apenas 47,38%, 41,18% e 50%, respectivamente, apresentaram-se dentro da banda da normalidade. Por fim, a avaliação da presença de corpos de Heinz mostrou que no genótipo AA 98,72% das amostras analisadas estavam normais; no genótipo AS, apenas 26,32% apresentaram valores normais; e nos genótipos SS e SF, nenhuma amostra mostrou-se normal.
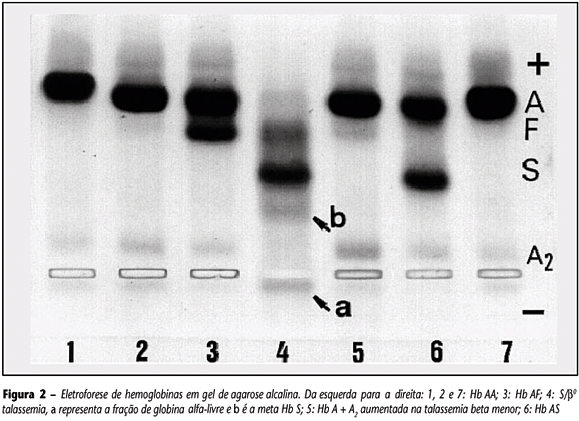
Nas análises eletroforéticas em agarose alcalina de todas as 16 amostras com genótipo SF foi identificada a presença de globinas alfa-livres com diferentes concentrações, que variaram entre 2% e 10% (Figura 1), fato não observado em nenhuma das amostras de outros genótipos (AA, AS e SS).
Discussão
Os resultados obtidos nos estudos das amostras de sangue extraído de portadores de hemoglobinas normais (AA) e com três diferentes genótipos de Hb S (AS, SS e SF) mostraram que a presença de Hb S desencadeia processos oxidativos com diferentes graus de intensidade entre seus genótipos. Os produtos resultantes da degradação oxidativa da hemoglobina, caracterizados pelo aumento dos valores das médias referentes à concentração de metemoglobina e do número de eritrócitos com corpos de Heinz, estão diretamente relacionados com o aumento da concentração da Hb S. Assim, a degradação oxidativa da hemoglobina decresce entre os genótipos estudados da seguinte forma: SS > SF > AS > AA. Apesar disso, os resultados individuais dos genótipos AS, SS e SF, expostos nas Tabelas 1, 2 e 3, indicaram que nem sempre a elevação da concentração de Hb S está relacionada com o aumento dos produtos de degradação oxidativa.
Outra constatação importante se deveu ao fato da elevação da concentração média de Hb fetal estar relacionada diretamente com a diminuição dos produtos de degradação oxidativa (Tabela 4). Os resultados expostos na Tabela 6 evidenciaram que a simples presença de Hb S no eritrócito, como é o caso do genótipo AS, é capaz de causar elevação da concentração de metemoglobina em 52,62% das amostras analisadas e induzir a precipitação de corpos de Heinz em 73,68%. Entretanto, é necessário destacar que elevações de metemoglobina acima de 5% ocorreram em 35,29% das amostras com genótipo SS, em 12,5% do genótipo SF, e não se constatou nenhum caso entre as amostras do genótipo AS. Essa mesma tabela revela que 100% das amostras dos genótipos SS e SF apresentaram corpos de Heinz elevados, enquanto que no genótipo AS a freqüência foi de 73,68%.
Diante dos valores obtidos, é possível admitir que a presença da Hb S, independente do genótipo, ao tornar-se desoxigenada (desoxi-Hb S) expõe o ferro à oxidação e conseqüente metemoglobinização, cuja intensidade é aproximadamente cinco vezes maior que na hemoglobina normal (AA). Se considerarmos que a elevação do número de eritrócitos com corpos de Heinz representa o produto final da degradação oxidativa da hemoglobina, a Hb S é pelo menos 57 vezes mais susceptível a sua formação que a Hb A ao compararmos a relação entre as freqüências médias de corpos de Heinz alterados nos genótipos AA (1,28%) e AS (73,68%).
Desses fatos apresentados, conclui-se que a intensidade da degradação oxidativa da Hb S está relacionada com a concentração dessa hemoglobina, porém com comportamentos individualizados e nem sempre relacionados. Assim, é possível considerar que o processo oxidativo da Hb S depende de processos biológicos que ocorrem com especificidades individuais. Entre esses processos destacam-se a capacidade antioxidativa do eritrócito, o funcionamento das bombas de Na+, K+ e Ca++, a oxigenação celular e a elevação da Hb fetal, principalmente(1, 5, 6).
Nossos resultados podem ser embasados em evidências de que, na anemia falciforme, os mecanismos redutores dos eritrócitos contra as lesões oxidativas estão alterados pela diminuição das atividades da glutationa peroxidase (GPx) e da catalase, pelo aumento da concentração de superóxido dismutase (SOD) e pela redução dos níveis de vitamina E no plasma e na membrana eritrocitária(1, 5). Alterações similares foram também relatadas na talassemia beta maior e na deficiência de G-6PD, cujos eritrócitos sofrem processos oxidativos por desequilíbrio entre globinas alfa e beta e pela deficiência enzimática, respectivamente (4, 5).
No caso das doenças falciformes, em especial aquelas com os genótipos SS e SF (S/b0 talassemia), o comportamento individual referente às alterações morfofuncionais dos eritrócitos falcêmicos e às suas conseqüências clínicas se devem em grande parte aos eritrócitos falciformes irreversíveis (EFI). Há doentes falcêmicos cujos EFI têm baixa proporção (< 5%), enquanto que outros têm EFI entre 5% e 25%(5). Esses últimos contêm concentrações elevadas de produtos secundários provenientes das lesões oxidativas gerados pela constante oxidação da desoxi-Hb S, enquanto que doentes com EFI abaixo de 5% mantêm suas atividades redutoras em equilíbrio(4). Por essas razões, é possível explicar os valores obtidos nas análises individuais dos genótipos AS, SS e SF (Tabelas 1, 2 e 3), em contraste com os valores das médias e dos produtos de degradação oxidativa (metemoglobina e corpos de Heinz) apresentados nas Tabelas 4, 5 e 6.
A identificação de globina alfa-livre nas 16 amostras de sangue com o genótipo SF provenientes de doentes falcêmicos com interação entre Hb S e talassemia beta (b0) é indicativa do desequilíbrio entre globinas alfa e beta. A Figura 2 mostrou a eletroforese de hemoglobinas efetuada em gel de agarose alcalina, com destaque para uma das amostras com o genótipo SF, em que é possível visualizar a globina alfa-livre bem como a meta-Hb S, além das outras frações: S, A2 e fetal.
As globinas alfa-livres constituem um fato que é demonstrado pela primeira vez por meio de eletroforese. Para obter a globina alfa em amostras com genótipo SF é necessário que a hemólise dos eritrócitos seja feita por meio do uso de saponina a 1% (v:w) e que a qualidade do suporte eletroforético no presente caso, a agarose alcalina seja de boa qualidade. Para explicar a origem da globina alfa-livre na Hb S/talassemia bo, propomos a representação esquemática da Figura 3, por meio da qual a completa ausência de síntese da globina beta (b0), proveniente da herança do gene beta defeituoso de um dos pais, impede que a globina alfa normalmente sintetizada estabeleça a combinação a2b2. Assim, somente serão formados os tetrâmeros a2b2S (Hb S), a2g2 (Hb fetal) e a2d2 (Hb A2). A falta de síntese de globina beta normal (b0) induz a precipitação da globina alfa em forma de dímeros (a2). Estes são conhecidos por alfa-livre, cuja fração pode ser identificada neste trabalho por meio da eletroforese alcalina em sangue total hemolisado com saponina a 1% (Figura 2, a).
A identificação de cadeias alfa-livre em sangue de pessoa com o genótipo SF pode ser considerada como importante informação para o diagnóstico laboratorial entre Hb S e talassemia b0, ou S/b0 talassemia. Finalmente, a presença da meta Hb S (Figura 2, b) na amostra do paciente com S/b0 talassemia pode ser identificada em casos em que a concentração da metemoglobina for b 5% (Tabela 3, caso 115, que corresponde à amostra 4 da Figura 2).
Agradecimentos
Os autores agradecem às biólogas Janaína Radispiel e Magaly da Silva Moraes a colaboração técnica durante a realização dos exames laboratoriais e ao médico Flávio Augusto Naoum a análise estatística.
Primeira submissão em 30/10/03
Última submissão em 29/01/04
Aceito para publicação em 12/04/04
Publicado em 20/08/04
- 1. ANDERSEN, H. R.; NIELSEN, J. B.; NIELSEN, F.; GRANDJEAN, P. Antioxidative enzyme activities in human erythrocytes. Clin Chem, v. 43, p. 562-8, 1997.
- 2. BAIN, B. J. Hemoglobinopathy diagnosis London: Blackwell Science Ltd, 2001.
- 3. BUNN, H. F. Pathogenesis and treatment of sickle cell disease. N Engl J Med, v. 337, p. 762-9, 1997.
- 4. DAS, D. K.; ERSMAN, W. B. Oxygen radicals. Sistemic events and disease processes Basel: Karger, 1990. 196 p.
- 5. DAS, S. K.; NAIR, R. C. Superoxide dismutase, glutathione peroxidase, catalase and lipid peroxidation of normal and sickled erythrocytes. Br J Haematol, v. 44, p. 87-92, 1980.
- 6. HUISMAN, T. H. J. The structure and function of normal and abnormal haemoglobins. Clinical Haematology, v. 6: 1-30, 1993.
- 7. MARCUS, S. J. et al. Quantitative analysis of erythrocytes containing fetal hemoglobin (F cells) in children with sickle cell disease. Am J Hematol, v. 54, p. 40-6, 1997.
- 8. MEDEIROS, M. G. G.; BECHARA, E.; NAOUM, P. C.; MOURÃO, C. A. Oxygen toxicity of hemoglobinemia in subjects from a highly polluted town. Arch Env Health, v. 38, p. 11-6, 1983.
- 9. NAGEL, R. L. Haemoglobinopathies due to structural mutations. In: PROVAN, D.; GRIBBEN, J. Molecular haematology Oxford: Blackwell Science Ltd, 2000. p. 121-33.
- 10. NAOUM, P. C.; NAOUM, F. A. Doença das células falciformes São Paulo: Sarvier, 2004. prelo.
- 11. NAOUM, P. C. Hemoglobinopatias e talassemias São Paulo: Sarvier, 1997.
- 12. NAOUM, P. C. Radicais livres em eritrócitos falcêmicos e talassêmicos. Bol Soc Bras Hematol Hemot, v. 18, p. 75-81, 1996.
- 13. SOUZA, P. C. Avaliação dos produtos de degradação oxidativa da Hb S em eritrócitos de doentes falcêmicos 1999. Dissertação de mestrado. UNESP de São José do Rio Preto.
- 14. STEINBERG, M. H. Pathophysiology of sickle cell disease. Clinical Haematology, v. 11, p. 163-84, 1998.
- 15. WINTERBOURN, C. Oxidative denaturation in congenital hemolityc anemias: the unstable hemoglobins. Semin Hematol, v. 27, p. 41-50, 1990.
Datas de Publicação
-
Publicação nesta coleção
18 Ago 2004 -
Data do Fascículo
Ago 2004
Histórico
-
Aceito
20 Ago 2004 -
Revisado
12 Abr 2004 -
Recebido
30 Out 2003