
Abstracts
OBJECTIVE: To analyze seizure outcome in individuals with familial mesial temporal lobe epilepsy (FMTLE). METHOD: We followed prospectively 64 individuals with FMTLE and 37 asymptomatic individuals belonging to 28 families. RESULTS: Patients with FMTLE had a mean follow up was 93.4 ± 15.8 months. At baseline they were divided in benign (n = 29), remission (n = 28) and refractory (n = 7). At last follow up visit 41.4% patients with benign FMTLE remained classified as benign, 20.7% became refractory and 37.9% were in remission. In the subgroup of FMTLE in remission 21 75% remained without seizures; 21.4% were classified as benign FMTLE, and one died (3.6%) from cause unrelated to epilepsy. All refractory patients remained refractory. From the asymptomatic group, 10.8% became symptomatic (FMTLE). The mean follow up was 76.0 ± 21.2 months. CONCLUSION: Prospective follow up of more than 7 years in patients with FMTLE revealed that it is unlikely to achieve seizure control in those with refractory seizures. Patients with diagnose of more benign forms of FMTLE for more than one year are likely to either remit or remain under well controlled seizures. The majority of patients who had achieved seizure remission remained seizure-free and none became refractory. Asymptomatic individuals had a greater probability to have seizures compared to the general population in a 6 year period of follow up.
Epilepsy; temporal lobe; familial; seizures
OBJETIVOS: Analisar a evolução de famílias com epilepsia de lobo temporal mesial familiar (ELTMF). METODOLOGIA: Seguimento prospectivo de 64 pacientes com ELTMF e 37 membros assintomáticos pertencente a 28 famílias. RESULTADOS: A média de seguimento dos pacientes com ELTMF foi de 93,4 ± 15,8 meses. Na avaliação inicial os pacientes foram divididos em benignos (n = 29), remissão (n = 28) e refratários (n = 7). Na última visita disponível, 41,4% dos pacientes com ELTMF benigna permaneceram classificados como benignos, 20,7% tornaram-se refratários e 37,9% entraram em remissão. No grupo em remissão, 75% permaneceram livres de crise, 21,4% foram classificados como benignos e um faleceu (3,6%) de causa não relacionada à epilepsia. Todos pacientes refratários permaneceram refratários. Em relação aos assintomáticos 10,8% evoluíram com crises. A média de seguimento dos assintomáticos foi de 76,0 ± 21,2 meses. CONCLUSÃO: O seguimento prospectivo de mais de 7 anos de pacientes com ELTMF revelou que é improvável ocorrer controle de crises no grupo refratário. No grupo benigno é muito provável que estes indivíduos entrem em remissão ou permaneçam com evolução benigna. A maioria dos pacientes do grupo em remissão permaneceu em remissão e nenhum se tornou refratário. Em relação aos assintomáticos a probabilidade de apresentar uma crise no decorrer de aproximadamente 6 anos foi maior que o observado na população geral.
Epilepsia; lobo temporal; familiar; crises
AWARDS WORKS - EXPANDED ABSTRACT
Long term follow up of familial mesial temporal lobe epilepsy* Correspondence should be addressed: Márcia Elisabete Morita Departamento de Neurologia FCM-UNICAMP Cidade Universitária CEP: 13083-970, Campinas, SP, Brasil E-mail: marciamorita@gmail.com
Evolução da epilepsia de lobo temporal mesial familiar
Márcia E. Morita; Lívia Conz; Claudia V. Maurer-Morelli; Eliane Kobayashi; Clarissa L. Yasuda; Luiz E. G. Betting; Fabrício R. S. Pereira; Iscia Lopes-Cendes; Fernando Cendes
Universidade Estadual de Campinas - UNICAMP, Campinas, SP, Brasil
Correspondence should be addressed Correspondence should be addressed: Márcia Elisabete Morita Departamento de Neurologia FCM-UNICAMP Cidade Universitária CEP: 13083-970, Campinas, SP, Brasil E-mail: marciamorita@gmail.com
ABSTRACT
OBJECTIVE: To analyze seizure outcome in individuals with familial mesial temporal lobe epilepsy (FMTLE).
METHOD: We followed prospectively 64 individuals with FMTLE and 37 asymptomatic individuals belonging to 28 families.
RESULTS: Patients with FMTLE had a mean follow up was 93.4 ± 15.8 months. At baseline they were divided in benign (n = 29), remission (n = 28) and refractory (n = 7). At last follow up visit 41.4% patients with benign FMTLE remained classified as benign, 20.7% became refractory and 37.9% were in remission. In the subgroup of FMTLE in remission 21 75% remained without seizures; 21.4% were classified as benign FMTLE, and one died (3.6%) from cause unrelated to epilepsy. All refractory patients remained refractory. From the asymptomatic group, 10.8% became symptomatic (FMTLE). The mean follow up was 76.0 ± 21.2 months.
CONCLUSION: Prospective follow up of more than 7 years in patients with FMTLE revealed that it is unlikely to achieve seizure control in those with refractory seizures. Patients with diagnose of more benign forms of FMTLE for more than one year are likely to either remit or remain under well controlled seizures. The majority of patients who had achieved seizure remission remained seizure-free and none became refractory. Asymptomatic individuals had a greater probability to have seizures compared to the general population in a 6 year period of follow up.
Key words: Epilepsy, temporal lobe, familial, seizures.
RESUMO
OBJETIVOS: Analisar a evolução de famílias com epilepsia de lobo temporal mesial familiar (ELTMF).
METODOLOGIA: Seguimento prospectivo de 64 pacientes com ELTMF e 37 membros assintomáticos pertencente a 28 famílias.
RESULTADOS: A média de seguimento dos pacientes com ELTMF foi de 93,4 ± 15,8 meses. Na avaliação inicial os pacientes foram divididos em benignos (n = 29), remissão (n = 28) e refratários (n = 7). Na última visita disponível, 41,4% dos pacientes com ELTMF benigna permaneceram classificados como benignos, 20,7% tornaram-se refratários e 37,9% entraram em remissão. No grupo em remissão, 75% permaneceram livres de crise, 21,4% foram classificados como benignos e um faleceu (3,6%) de causa não relacionada à epilepsia. Todos pacientes refratários permaneceram refratários. Em relação aos assintomáticos 10,8% evoluíram com crises. A média de seguimento dos assintomáticos foi de 76,0 ± 21,2 meses.
CONCLUSÃO: O seguimento prospectivo de mais de 7 anos de pacientes com ELTMF revelou que é improvável ocorrer controle de crises no grupo refratário. No grupo benigno é muito provável que estes indivíduos entrem em remissão ou permaneçam com evolução benigna. A maioria dos pacientes do grupo em remissão permaneceu em remissão e nenhum se tornou refratário. Em relação aos assintomáticos a probabilidade de apresentar uma crise no decorrer de aproximadamente 6 anos foi maior que o observado na população geral.
Unitermos: Epilepsia, lobo temporal, familiar, crises.
INTRODUCTION
Familial mesial temporal lobe epilepsy (FMTLE) has been described as a well characterized syndrome, with different degrees of seizure severity, with the majority of patients having good seizure control. However, there has been no report of long term follow up of FMTLE.
The best definition of FMTLE is based on the familial recurrence of mesial temporal lobe epilepsy (MTLE) in the absence of any suggestion of other partial (including lateral TLE symptoms) or generalized epilepsy syndromes in other affected family members.1,3,4,5 The finding of at least two MTLE patients in one family is suggestive of FMTLE. The observation of an autosomal dominant inheritance pattern with incomplete penetrance implies the presence of asymptomatic carriers of the genetic abnormalities, who can transmit the disease to their offspring. Therefore, we should consider inclusion of families not only with affected first-degree relatives, but also those with affected second and third degree relatives. This criterion has not been employed in some reported series, leading to exclusion of many possible FMTLE kindreds.6
The purpose of this study was to analyze seizure outcome in individuals with FMTLE.
METHODS
We followed prospectively 64 individuals with clinical diagnose of FMTLE from 28 families and 37 asymptomatic patients (first or second degree relatives from families with FMTLE). Seizures and epilepsy syndromes were classified according to the ILAE. Patients who fulfilled the clinical criteria for FMTLE were divided into three groups, according to seizure outcome: Remission: seizure free for at least two years; Benign: patients, with less than six complex partial seizures (CPS) per year and no more than two secondary generalized tonic-clonic seizures per year; and Refractory: more than six CPS per year despite adequate anti-epileptic drugs (AEDs). Patients who did not fulfilled criteria for MTLE or who underwent epilepsy surgery (n = 16) during this follow up were excluded from this study.
RESULTS
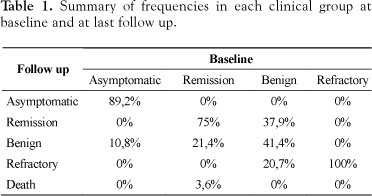
Patients with FMTLE had a mean follow up of 93.4 ±15.8 months (mean ± SD; ranging from 45 to 121.9 months). The mean baseline duration of epilepsy was 28.7 ±15.3 years ranging from 1 to 62 years. At baseline they were divided in benign (n = 29), remission (n = 28) and refractory (n = 7). Patients who have been submitted to epilepsy surgery (n=16) during this follow up were not included in this study. At last follow up visit 12 (41.4%) patients with benign FMTLE remained classified as benign, 6 (20.7%) became refractory, 11 (37.9%) were in remission. In the subgroup of FMTLE in remission 21 (75%) remained without seizures; 6 (21.4%) were classified as benign FMTLE, and one died (3.6%) from cause unrelated to epilepsy. All refractory patients remained refractory. From the asymptomatic group, 4 (10.8%) became symptomatic (FMTLE). The mean follow up was 76.0 ± 21.2 months (mean ± SD; ranging from 34.7 to 94.73 months).

DISCUSSION
This work in progress suggests that patients with FMTLE presenting with refractory seizures are unlikely to remit in a short term follow up. Patients with more benign forms of FMTLE for more than one year are likely to either remit or remain under well controlled seizures. The majority of patients who had achieved seizure remission remained seizure-free and none became refractory. Asymptomatic individuals had a greater probability to have seizures compared to the general population in a 6 year period of follow up.2 Whether patients with benign FMTLE are more likely to remit than patients with sporadic MTLE secondary to hippocampal sclerosis remains to be investigated. Further analyses and comparison with a cohort of patients with sporadic MTLE are necessary to confirm these preliminary observations.
ACKNOWLEDGMENTS
Supported by a Grant from FAPESP - Fundação de Amparo à Pesquisa do Estado de São Paulo.
Received June 16, 2008; accepted July 18, 2008.
* Trabalho premiado com o Prêmio Jovem Pesquisador (Estudos Clínicos) durante o XXXII Congresso Brasileiro de Eplepsia 2008.
- 1. Andrade-Valença LP, Valença MM, Velasco TR, Carlotti CG Jr, Assirati JA, Galvis-Alonso OY, Neder L, Cendes F, Leite JP. Mesial temporal lobe epilepsy: Clinical and neuropathologic findings of familial and sporadic forms. Epilepsia 2008; 49(6):1046-54.
- 2. Hauser WA. The natural history of temporal lobe epilepsy. In: Lüders HO ed. Epilepsy Surgery. New York: Raven Press 1992: 133-141.
- 3. Kobayashi E, D'Agostino MD, Lopes-Cendes I, Berkovic SF, Li ML, Andermann E, Andermann F, Cendes F. Hippocampal atrophy and T2-weighted signal changes in familial mesial temporal lobe epilepsy. Neurology 2003; 60(3):405-9.
- 4. Kobayashi E, Li LM, Lopes-Cendes I, Cendes F. Magnetic resonance imaging evidence of hippocampal sclerosis in asymptomatic, first-degree relatives of patients with familial mesial temporal lobe epilepsy. Arch Neurol 2002; 59(12):1891-4.
- 5. Kobayashi E, Santos NF, Torres FR, Secolin R, Sardinha LA, Lopez-Cendes I, Cendes F. Magnetic resonance imaging abnormalities in familial temporal lobe epilepsy with auditory auras. Arch Neurol 2003 Nov; 60(11): 1546-51. Erratum in: Arch Neurol 2004; 61(2):199.
- 6. Striano P, Gambardella A, Coppola A, Di Bonaventura C, Bovo G, Diani E, Boaretto F, Egeo G, Ciampa C, Labate A, Testoni S, Passarelli D, Manna I, Sferro C, Aguglia U, Caranci F, Giallonardo AT, Striano S, Nobile C, Michelucci R. Familial mesial temporal lobe epilepsy (FMTLE): a clinical and genetic study of 15 Italian families. J Neurol 2008; 255(1):16-23.
Publication Dates
-
Publication in this collection
05 Jan 2009 -
Date of issue
Sept 2008
History
-
Accepted
18 July 2008 -
Received
16 June 2008
