Abstracts
Detailed knowledge of electroencephalographic patterns accompanying epileptic seizures in children is paramount to the correct identification of epileptic seizures and syndromes. In this article, we present a review of ictal patterns of different seizure types in children, illustrating with examples collected in our video-EEG laboratory at Pequeno Príncipe Hospital.
electroencephalography; epilepsy; ictal pattern
O conhecimento detalhado dos padrões eletrencefalográficos que acompanham crises epilépticas em crianças é essencial para a correta identificação de crises e síndromes epilépticas. Neste artigo apresentamos uma revisão dos padrões ictais de diferentes crises em crianças, ilustrando com exemplos coletados na nossa unidade de vídeo-eletrencefalograma no Hospital Pequeno Príncipe.
eletrencefalograma; epilepsia; padrão ictal
REVIEW ARTICLE
Ictal patterns in children: an illustrated review
Padrão ictal em crianças: uma revisão ilustrada
Mônica Jaques Spinosa; Paulo Breno de Noronha Liberalesso; Larissa Mehl; Alfredo Löhr Júnior
Unidade de Neurologia Infantil, Hospital Pequeno Príncipe, Curitiba, Paraná, Brasil
Endereço para correspondência Endereço para correspondência Mônica Jaques Spinosa Rua Johann Sebastian Bach, 71 Vista Alegre CEP 80820-140, Curitiba, PR, Brasil E-mail: < monicaspinosa@yahoo.com>
ABSTRACT
Detailed knowledge of electroencephalographic patterns accompanying epileptic seizures in children is paramount to the correct identification of epileptic seizures and syndromes. In this article, we present a review of ictal patterns of different seizure types in children, illustrating with examples collected in our video-EEG laboratory at Pequeno Príncipe Hospital.
Keywords: electroencephalography; epilepsy; ictal pattern.
RESUMO
O conhecimento detalhado dos padrões eletrencefalográficos que acompanham crises epilépticas em crianças é essencial para a correta identificação de crises e síndromes epilépticas. Neste artigo apresentamos uma revisão dos padrões ictais de diferentes crises em crianças, ilustrando com exemplos coletados na nossa unidade de vídeo-eletrencefalograma no Hospital Pequeno Príncipe.
Unitermos: eletrencefalograma; epilepsia; padrão ictal.
INTRODUCTION
Detailed knowledge of electroencephalographic (EEG) patterns accompanying epileptic seizures in children is paramount to the correct identification of epileptic seizures and syndromes. Furthermore, the observation of an event that is not associated with an ictal alteration of the EEG tracing determines the diagnosis of a non-epileptic event, such as pseudoseizures, behavioral aberrations and parasomnias. In certain seizure types, such as hyperkinetic seizures emerging from the frontal lobe, with bizarre semiology, this differential diagnosis is crucial.1 Theneurophysiologist must be able to recognize an ictal EEG activity among the frequent artifacts produced by profuse movement that is characteristic of this seizure type.
Moreover, different ictal patterns may coexist in a specific seizure type and epileptic syndrome. As such, tonic seizures of Lennox-Gastaut Syndrome may have four different ictal patterns, comprising combinations of fast rhythms and diffuse attenuation of EEG activity.2 A specific seizure type associated with a certain EEG pattern may show subtlevariations of ictal tracing according to epilepsy syndrome. An example is typical absence seizures, which show an ictal pattern of generalized spike-and-wave or polyspike-and-wave discharges of 3Hz or more, with frequency, degree of regularity, duration and number of spikes per dischargevarying slightly depending on the patient's epilepsy syndrome. 3In addition to clinical aspects (seizure semiology and neurological findings), EEG tracing may help differentiate between similar seizure types, such as typical and atypical absences.4
The aim of this article is to describe ictal patterns of different seizure types in children, illustrating with examples collected in our video-EEG laboratory at Pequeno Príncipe Hospital.
METHODS
Current literature concerning the description of ictal patterns in children has been thoroughly reviewed in the Pub Med database. Broad description terms have been used such as "ictal pattern children" and "ictal pattern EEG". Narrow searches were also conducted using keywords referring to specific epileptic syndromes in children. Examples of EEG tracings were selected from video-EEG exams performed at Pequeno Príncipe Hospital in the past 5 years.
ICTAL PATTERNS
Generalized Myoclonic Seizures
Myoclonias are a predominant feature of some severe epilepsy syndromes in childhood. However,these are not always epileptic in origin. Early Myoclonic Epilepsy is a severe epilepsy syndrome in which intractable seizures are accompanied by poor developmental outcome and suppression-burst interictal EEG. Even though myoclonias are the main clinical manifestation, most are not associated with EEG changes, and therefore non-epileptic. 5In Dravet Sydrome (Severe Myoclonic Epilepsy of Infancy), massive myoclonic seizures are associated with bursts of irregular spike-and-wave discharges. Erratic myoclonias, however, have no EEG correlate.6,7
In Doose Syndrome (Myoclonic-Astatic Epilepsy) myoclonic seizures can occur independently or preceding an atonic seizure. A studythat combined video-EEG and electromyography data has shown that drop attacks in this epileptic syndrome can be caused either by massive myoclonic flexor seizures (9/36 seizures, 2/5 patients), myoclonic-atonic seizures (2/36 seizures, 1/5 patients) or, more commonly, atonic seizures (25/36 seizures, 4/5 patients).8 A video-EEG analysis of 30 patients withDoose Syndrome revealed that the ictal EEG is similar in both atonic and myoclonic seizures, being characterized by generalized spike or polyspike-and-wave complexes.9 In a more detailed analysis, the authors described that in atonic seizures the spike-and-wave complex was a positive-negative-deep positive discharge, followed by a large slow wave. There seemed to be a direct correlation between the depth of the second positive component and the intensity of the seizure. In reference to the myoclonic seizures, the duration of the spike-and-wave complex appeared to be shorter than that related to atonic seizures.
Juvenile Myoclonic Epilepsy (JME) is a generalized idiopathic epileptic syndrome that may begin in late childhood and adolescence. As the name implies, JME's main seizure type is myoclonic jerks. These occur frequently upon awakening and may be precipitated by sleep deprivation, emotional stress and alcohol consumption. Ictal EEG shows a fast spike-and-wave and polyspike-and-wave pattern of 4 to 6Hz. Ictal epileptic discharges are generalized, bilaterally symmetric and have a higher amplitude at fronto-central regions.10 The number of spikes per polyspike-and-wave complex may be as high as 20 and is directly correlated with the intensity of seizures.11
In Benign Myoclonic Epilepsy of Infancy, generalized myoclonic seizures such as in JME are accompanied by bursts of generalized spike-and-wave or polyspike-and-wave discharges.7
Epileptic Spasms
Epileptic spasms of West Syndrome are represented in the EEG by a slow wave of medium to high amplitude, positive at central EEG channels. Fusco & Vigevano12 reported a review of 955 spasms and concluded that this pattern was observed in all spasms and that the start of the muscle contraction correlated with the start of the slow wave. Other additional EEG patterns described were a superimposed spindle-like fast activity of 14 to 16Hz of medium amplitude and a decremental activity. The spindle-like activity was observed by the authors in 69% of the cryptogenic cases and 35% of the symptomatic cases, and was clinically associated to a motionless stare. This activity occurred either isolated, generally at the beginning or the end of a cluster (40% of the cases studied), or immediately before the slow wave. In the latter, a stare was closely followed by a spasm. The decremental activity, consisting of a diffuse flattening of the EEG tracing, corresponded to a post ictal pattern and followed the slow wave in 69% of the cryptogenic cases and 70% of the symptomatic cases. Fusco & Vigevano also observed that during a cluster of spasms, the persistence of the hypsarrhythmic background was significantly associated with cryptogenic West Syndrome (80% in the cryptogenic group versus 32% in the symptomatic group). Figure 1 shows the EEG of a 10 month-old patient with West Syndrome. The epileptic spasm is associated with a high amplitude slow wave, followed by diffuse attenuation.
Menezes & Rho13 retrospectively reviewed video-EEG data of 26 patients of 4 to 17 years of age (mean 7. 75 years) with epileptic spasms persisting beyond the second year of life. The average age at onset of spasms was 6. 8 months and the average age at the most recent video-EEG documentation of spasms was 75 months. The majority of patients (17/26) had other associated seizure types, such as tonic seizures (13 patients), partial seizures (11), myoclonic seizures (11), generalized tonic-clonic seizures (6), atypical absences (2) and atonic seizures (1). Interictal EEG analysis revealed hypsarrhythmic pattern in only 4 patients (15.4%), ranging from 44 to 89 months of age. As observed in spasms associated with West Syndrome, the most common ictal pattern reported in spasms persisting beyond infancy was a slow wave transient (73%). Other ictal patterns observed were: slow wake superimposed with fast activity (4 patients); multiphasic slow wave (6 patients); sharp wave or blunted sharp or slow wave (4 patients). Decremental activity, frequently superimposed by a rhythmic beta or alpha activity, followed a slow wave in all spasms of 19 patients, a few spasms of 2 patients and never occurred in 2 patients. Voltage attenuation was not associated with a slow wave in 5 patients. During longer (greater than 3 seconds) decremental periods, a tonic muscle contraction was reported in 8 cases, which was described by the authors as a spasm-tonic seizure. This finding was interpreted as a transitional seizure type in the evolution of a spasm to a tonic seizure, which may be incomplete in these patients due to lack of brain maturation. Ictal EEG asymmetry was seen in 4 patients. The authors concluded that the electroclinical presentation of epileptic spasms persisting beyond infancy resembles that of West Syndrome.
Generalized Clonic Seizures
Korff & Nordli Jr14 described ictal semiology and EEG pattern of a series of 101 seizures of 69 infants. Generalized clonic seizures were recorded in 6 patients. Ictal EEG consisted of repetitive generalized sharp waves or spikes with an occipital predominance in one patient and frontal predominance in two. Seizure termination was abrupt in 33%, with sudden replacement of the ictal pattern by either the usual background activity, or generalized slowing or generalized attenuation. More commonly (66%), seizure end was poorly defined, with alternating epochs of ictal rhythm and usual background activity.
Generalized Tonic-Clonic Seizures
In infants, classic generalized tonic-clonic seizures are rarely observed. However, in a series of 101 seizures in this age group, one seizure described as generalized symmetric myoclonic jerks followed by generalized tonic posturing and erratic myoclonia and oroalimentary automatisms was reported in a 9 month old patient with Dravet Syndrome. Ictal EEG consisted of a sequence of generalized sharp waves, diffuse attenuation, bicentral theta-alpha rhythm with subsequent spread, generalized spikes and abrupt termination.6Figure 2 shows the EEG during a generalized tonic-clonicseizure of a one year-old boy with epileptic encephalopathy, probably related to a metabolic disorder. Ictal tracing is characterized by diffuse attenuation during thetonic phase, followed by rhythmic slow waves with intermixed spikes during theclonic phase.
Generalized tonic-clonic seizure ictal EEG is described as generalized fast rhythmic spikes, accentuated at frontal leads, during the tonic phase. As the seizure progresses to the clonic phase, the fast rhythm is fragmented by rhythmic slow waves. Polyspikes accompany the clonic jerk, while the slow wave is related to the relaxation period.15
Generalized Tonic Seizures
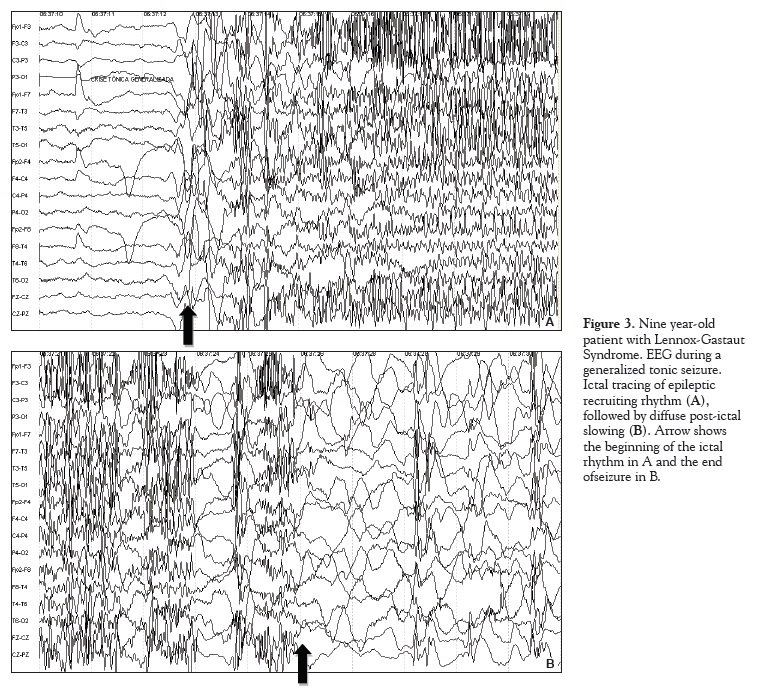
Tonic seizures are the most characteristic seizures of Lennox-Gastaut Syndrome and are represented in the EEG by a generalized desynchronization and/or rhythmic fast activity. Four ictal patterns comprising a variation of these two findings have been described: attenuation of EEG activity; fast rhythmic activity of 15 to 25Hz, initially of low amplitude with progressive increase to 50-100µV; initial attenuation followed by the fast rhythm previously described; fast rhythm of 10 to 15 Hz with high amplitude from the seizure start. 2 Gastaut has described the "epileptic recruiting rhythm" as a fast rhythmof 10Hz with progressive increase in amplitude characteristic of generalized tonic seizures.16Figure 3 shows the epileptic recruiting rhythm during a generalized tonic seizure of a nine year-old patient with Lennox-Gastaut Syndrome.
Generalized Atonic Seizures
Atonic seizures are observed in Doose Syndrome and Lennox-Gastaut Syndrome, causing falls. It is interesting to note that in Doose Syndrome most seizures that provoke falls are atonic, according to video-polygraphic studies, while in Lennox-Gastaut Syndrome most drop-attacks are caused by tonic seizures. As previously mentioned, the ictal correlate of an atonic seizure is a generalized spike or polyspike-and-wave complex.2,9
Generalized atonic seizures in infants were accompanied by an EEG ictal pattern consisting of generalized sharp waves or spikes in 80% of patients (4/5), with occipital predominance in one and frontal in two. In the remaining infant ictal rhythm consisted of generalized slow waves.6
Absence Seizures
Typical absences are seizures mainly characterized by impairment of consciousness of sudden start and termination, with a generalized spike-and-wave or polyspike-and-wave discharge of 3 Hz or more. Discharges may vary according to epilepsy syndrome. The International League Against Epilepsy (ILAE) has recognized four epilepsy syndromes which include this seizure type: Childhood Absence Epilepsy (CAE), Juvenile Absence Epilepsy (JAE), JME and Epilepsy with Generalized Grand Mal on Awakening (EGMA).3
In CAE, ictal EEG consists of 3-4Hz generalized spike-and-wave discharges, commonly with a frontal predominance, lasting 4 to 20 seconds, with the initial 1-2 seconds of fast discharges, followed by a gradual slowing of 0. 5 to 1Hz.3 Koutromanidis & Smith state that the typical absence seizure may be divided in three phases: the initial phase, comprising the first second, showing a greater frequency and irregularity; the second phase (next 3 seconds) of regular and stable discharges of 3Hz; followed by the terminal phase of gradual slowing.4Figure 4 shows a typical absence seizure in athree year-old patient with Early Onset CAE, with ictal rhythm consisting of generalized spike-and-wave complexes of 2,5Hz.
In JAE ictal frequency of discharges is higher, ranging from 3. 5 to 4Hz. Both spike and polyspike components may occur, and seizures may last longer.3,10
The differentiation between typical and atypical absences is both clinical and electrographical. Atypical absence seizures may have a gradual onset and the degree of consciousness impairment is often difficult to access, with possible detainment of some level of awareness. Atypical absences occur in cognitively impaired children with symptomatic or cryptogenic epilepsy syndromes that include frequent seizures of various types such as tonic, atonic and myoclonic. Examples of such syndromes are Myoclonic-Astatic Epilepsy and Lennox-Gastaut Syndrome. The ictal EEG in atypical absences consists of irregular generalized spike-and-wave discharges of a lesser frequency, below 2. 5Hz.4Figure 5 shows the EEG of an eleven year-old patient with atypical absence epileptic status, characterized by generalized and irregular slow (2Hz) spike-and-wave complexes.
Focal Seizures
In Korff's and Norli's series of 69 infants,6 focal seizures had various semiologies: tonic-clonic with secondary generalization (3); clonic (11); tonic (11); focal tonic-clonic (3); behavioral arrest (1); behavioral arrest with version (14) and hypermotor (1). Although EEG showed an ictal pattern restricted to a specific brain region, clinical manifestation was sometimes bilateral and symmetrical: one patient out of three with tonic-clonic seizure with secondary generalization and two patients out of eleven with focal tonic seizure had symmetrical tonic posturing; some patients with focal clonic seizure had bilateral asymmetric myoclonic jerks (number of patients not specified in the article). Ictal EEG in focal seizures consisted of spikes (66%) or rhythmic theta-alpha activity (33%) in tonic-clonic seizures with secondary generalization; rhythmic spikes (73%), low voltage fast activity (9%), attenuation (9%) or rhythmic delta (9%) in clonic seizures; low voltage fast activity (36%), rhythmic theta-alpha activity with or without delta waves or attenuation (27%), spikes (27%) or attenuation (9%) in tonic seizures; rhythmic beta activity (66%) or rhythmic theta-alpha activity (33%) in focal tonic-clonic seizures; unilateral posterior spikes in the sole behavioral arrest seizure observed; low voltage fast activity (29%), rhythmic theta-alpha activity (29%), rhythmic delta activity (21%), rhythmic beta activity (7%), irregular slowing (7%) or spikes (7%) in behavior arrest with version seizures; hemispheric rhythmic delta activity in the single hypermotor seizure reported.
In Malignant Migrating Partial Seizures in Infancy, described by Coppola in 1995, cryptogenic refractory migrating focal motor seizures are accompanied by severe developmental delay.17 The ictal EEG correlate is a focal rhythmic pattern that shifts from one brain region to the next, involving both cerebral hemispheres independently. Ictal rhythm may be in the alpha, theta or delta range, the last one having been reported with intermixed spikes.17-20
Ictal video-EEG recordings of 30 patients with Benign Childhood Epilepsy with Centro-Temporal Spikes (BCECTS) were reported in a study that collected data of 10 epilepsy centers in Italy.21 Two patients had electrographical seizures only, 3 had electroclinical and electrographic seizures and the remaining 25 had electroclinical seizures only. Ictal onset occurred at the right hemisphere in 13, left hemisphere in 16 and both hemispheres in one. Four different ictal patterns were observed: low voltage fast rhythmic spikes with a gradual increase in amplitude and decrease in frequency (observed in 14/30 patients); spikes mixed with sharp waves of increasing frequency and amplitude (6/30 patients); monomorphic theta activity progressively forming a discharge of increasing amplitude and decreasing frequency (7/30 patients); initial focal attenuation of EEG activity, followed by one of the 3 previous patterns (5/30 patients). In 21 patients the initial ictal pattern modified during the seizure, switching to another pattern among those described. Secondary generalization, which occurred in 6 patients, was observed in all 4 different patterns.
In Early Onset Benign Childhood Epilepsy with Occipital Spikes, seizures are characterized by autonomical and behavioral symptoms associated with eyes deviation and ictal vomiting. Seizures are infrequent but may be prolonged, with progressive impairment of consciousness, sometimes evolving to hemi or generalized tonic-clonic seizures. Ictal EEG shows unilateral rhythmic slow activity, usually superimposed by fast rhythms and spikes. In Idiopathic Childhood Epilepsy of Gastaut seizures are brief, diurnal and frequent, consisting generally of elementary visual hallucinations and/or ictal blindness. EEG shows a marked decrease in occipital interictal paroxysms preceding an ictal pattern of lower amplitude fast rhythms, fast spikes, or both.22
The ILAE classification of seizures of 198123 is frequently difficult to apply to infants and nonverbal children due to the limitation of assessment of the degree of consciousness impairment during the event. Seizure types such as complex partial and absence are, consequently, poorly defined in this substantial group of pediatric patients. Therefore, the definition of hypomotor seizure24 as an epileptic ictal event in which the main clinical characteristic is a sudden decrease in behavioral activity is a useful tool whenever the level of consciousness cannot be determined. Both focal and generalized seizures fall into the hypomotor classification. Källén et al.25 described video-EEG recordings of 110 hypomotor seizures in 34 patients. Among these, 79% were younger than 2 years old, 68% had multifocal epilepsy and 29% had generalized epilepsy. Clinical signs, observed preceding behavioral arrest in 5 patients with generalized epilepsy, were myoclonic jerks or epileptic spasms. Following the hypomotor phase, version of the head and eyes, clonic unilateral arm movement, bilateral clonic blinking occurred in patients with focal or lateralized ictal onset. Epileptic spasms followed hypomotor seizures in patients with generalized epilepsy. Complex motor behavior was observed evolving from hypomotor seizures with focal, lateralized or generalized onset. During the behavioral arrest phase, subtle motor signs such as oral automatisms, deviation of eye or head, irregular eye blinking and stereotyped limb movements were observed in 75% of the events. Seizures that were either lateralized or regional at ictal onset were significantly longer in duration than generalized ones. Among those seizures with regional ictal patterns (50/110), 14% were frontal, 42% were temporal (temporal, frontotemporal or parietotemporal), 42% were parietal (parietal or parietooccipital) and 2% were occipital. Generalized seizures were accompanied at the EEG by electrodecremental activity in 16/39 seizures; diffuse rhythmic slowing in 11/39; slow spike-and-wave complexes in 6/39; paroxysmal fast activity in 4/39 and 3 per second spike-and-wave complexes in 2/39. Figure 6 shows the EEG tracing of a hypomotor seizure of a premature 11 month old patient, characterized by ictal diffuse rhythmic slowing. The authors noted that the fact that generalized hypomotor seizures were predominantly associated with ictal patterns other than spike-and-wave complexes, could indicate a different mechanism from absence seizures. They also suggested that maturational effects, which may be still lacking in infants, could play a role in the generation of synchronized rhythmic spike-and-wave complexes.
Hypermotor seizures, as defined by Lüders et al.24 are partial seizures consisting mainly of prominent complex movements involving proximal muscles. These seizures are usually observed in frontal lobe epilepsies but may also arise from the temporal lobe. Weinstock et al.1 described video-EEG findings of 5 children with hypermotor seizures. Ictal patterns consisted of rhythmic theta activity in 2, one involving the midline central region and one over the frontotemporal areas. One patient had ictal bifrontal rhythmic spike-and-wave pattern. In two patients the ictal EEG was obscured by diffuse electromyographic artifact. Postictally, delta activity was observed diffusely in one patient and solely over the left hemisphere in the other.
Battaglia et al.26 described 174 seizures of 18 patients under 12 years of age with lesional epilepsy of the frontal lobe in order to depict the semiology of frontal onset seizures in children. Ictal EEG onset was localized at frontal electrodes, frequently involving ipsilateral central and vertex areas. Secondary spreading was either temporal or diffuse. Only one patient had seizures with secondary parietal spreading of the ictal rhythm. Three patterns of ictal EEG activity were observed: high amplitude sharp transient followed by low-amplitude fast activity; rhythmic fast activity or decremental activity followed by slow waves with superimposed spikes; localized theta activity and slow waves. Seizure semiology varied widely. The authors used Lüder's classification of seizure semiology in children24 and observed complex motor seizures in 61.1% of patients (comprising hypermotor seizures in 27.8% and mixed hypermotor, asymmetric epileptic spasm, gelastic, automotor, tonic and versive components in 33.3%) and simple motor seizures in 61.1% (comprising focal or asymmetric tonic in 11.1%, clonic in 11.1%, asymmetric epileptic spasms in 22.2% and versive seizures in 16.6%).
Complex Partial Seizures
Mesial Temporal Lobe Epilepsy with Hippocampal Sclerosis is the most common refractory epilepsy syndrome in adulthood. Recent studies have demonstrated that seizures frequently start in childhood and adolescence. The importance of a correct diagnosis and treatment lies in the fact that early surgical intervention, whenever refractoriness to antiepileptic drugs is shown, improves cognitive and social prognosis.27 Cersósimo et al.28 described 42 children with this epileptic syndrome. The age of seizure onset varied from 1 to 6 years, with mean of 8 years. Overall seizure semiology did not differ from the classical description of complex partial seizures due to mesial temporal sclerosis in adults (epigastic, emotional, déjà vu and olfactory-gustatory auras; complex partial seizures with consciousness impairment, motor arrest, oroalimentary and gestural automatisms, sometimes accompanied by motor signs such as head deviation, unilateral dystonic posturing and unilateral clonic limb movements). When the authors separated the patients in 3 groups according to age, it was possible to determine that in those whose seizures started before 2 years of age motor seizures predominated, with clonic version of the head and unilateral clonic seizures of the upper limb, associated with behavioral arrest and oroalimentary automatisms. In the age group of patients with seizure start between 2 and 10 years old, less motor manifestations were observed and automatisms were more complex. In the group with latest epilepsy start (between 10 and 16 years of age), seizures were characterized by epigastric aura followed by a complex partial seizure with gestural, oroalimentary and verbal automatisms and dystonic posturing. Ictal EEG also differed between groups, being restricted to the temporal lobe in the older group, while in the younger group ictal pattern was apparently generalized in 2 patients. In 30 patients (71.5%) ictal EEG revealed the distinctive rhythmic theta activity with progressive increase in amplitude. In the remaining 28.5%, ictal start was related to a focal flattening of the EEG tracing at the temporal region, followed by fast activity.
FINAL REMARKS
Ictal patterns in children vary widely according to seizure type and epilepsy syndrome. Additionally, brain maturational processes, affected by age and, whenever present, by a primary neurological disorder, seem to be involved in the determination of seizure semiology and EEG ictal activity. A comprehensive knowledge of the interaction of these factors and potential EEG presentations is essential to the pediatric electroencephalographist.
Received Oct. 05, 2011; accepted Nov. 05, 2011.
- 1. Weinstock A, Giglio P, Kerr SL, Duffner PK, Cohen ME. Hyperkinetic seizures in children. J Child Neurol 2003; 18(8):517-24.
- 2. Markland ON. Lennox-Gastaut syndrome (childhood epileptic encephalopathy). J Clin Neurophisiol 2003; 20(6):426-41.
- 3. Duron RM, Medina MT, Martínez-Juárez IE, Bailey JN, Perez-Gosiengfiao KT, Alonso ME, et al. Seizures of idiopathic generalizes epilepsies. Epilepsia 2005; 46(Suppl 9):34-47.
- 4. Koutroumanidis M, Smith S. Use and abuse of EEG in the diagnosis of idiopathic generalized epilepsies. Epilepsia 2005; 46(Suppl 9):96-107.
- 5. Lombroso CT. Early myoclonic encephalopathy, early infantile epileptic encephalopathy, and benign and severe infantile myoclonic epilepsies: a critical review and personal contributions. J Clin Neurophysiol 1990; 7(3):380-408.
- 6. Korff CM, Nordli Jr DR. Epilepsy Syndromes in Infancy. Pediatr Neurol 2006; 34:253-63.
- 7. Dulac O, Plouin P, Shermon A. Contributors to the Royamount Workshop. Epilepsy Res 1998; 30:91-106.
- 8. Oguni H, Fukuyama Y, Imaizumi Y, Uchara T. Video-EEG analysis of drop seizures in myoclonic astatic epilepsy of early childhood (Doose syndrome). Epilepsia 1992; 33(5):805-13.
- 9. Oguni H, Fukuyama Y, Tanaka T, Hayashi K, Funatsuka M, Sakauchi M, et al. Myoclonic-astatic epilepsy of early childhood clinical and EEG analysis of myoclonic-astatic seizures, and discussions on the nosology of the syndrome. Brain Dev 2001; 23:757-64.
- 10. Dreifuss FE. Juvenile myoclonic epilepsy: characteristics of a primary generalized epilepsy. Epilepsia 1989; 30(Suppl. 4):S1-7.
- 11. Janz D. Epilepsy with impulsive petit mal (juvenile myoclonic epilepsy). Acta Neurol Scand 1985; 72:449-59.
- 12. Fusco L, Vigevano F. Ictal clinical electroencephalographic findings of spasms in West Syndrome. Epilepsia 1993; 34(4):671-8.
- 13. Menezes MAS, Rho JM. Clinical and electrographic features of epileptic spasms persisting beyond the second year of life. Epilepsia 2002; 43(6):623-30.
- 14. Korff CM, Nordli Jr DR. The clinical-electrographic expression of infantile seizures. Epilepsy Res 2006; 70:S116-31.
- 15. Niedermeyer E, da Silva FL. Electroencephalography: basic principles, clinical applications and related fields. Baltimore MD. Lippincott, Williams & Wilkins; 2005.
- 16. Gastaut H, Broughton R. Epileptic Seizures. Springfield IL: Charles C Thomas; 1972.
- 17. Coppola G, Poulin P, Chiron C, Robain O, Dulac O. Migrating partial seizures in infancy: a malignant disorder with developmental arrest. Epilepsia 1995; 36:1017-24.
- 18. Veneselli E, Perrone MV, Di Rocco M, Gaggero R, Biancheri R. Malignant migrating partial seizures in infancy. Epilepsy Res 2001; 46:27-32.
- 19. Ben-Zeev B, Shalev RS. Malignant migrating partial seizures in infancy. Pediatr Neurol 2004; 31:287-90.
- 20. Hmaimess G, Kadhim H, Nassogne M-C, Bonnier C, van Rijckevorsel K. Levetiracetam in a neonate with malignant migrating partial seizures. Pediatr Neurol 2006; 34:55-9.
- 21. Capovilla G, Beccaria F, Bianchi A, Canevini MP, Giordano L, Gobbi G, et al. Ictal EEG patterns in epilepsy with centro-temporal spikes. Brain Dev 2011; 33:301-9.
- 22. Panayiotopoulos CP, Michael M, Sanders S, Valeta T, Koutroumanidis M. Benign childhood focal epilepsies: assessment of established and newly recognized syndromes. Brain 2008; 131(9):2264-86.
- 23. Commission on Classification and Terminology of the International League Against Epilepsy: Proposal for revised clinical and electrographic classification of epileptic seizures. Epilepsia 1981; 22: 489-501.
- 24. Lüders H, Acharya J, Baumgartner C, Benbadis S, Beasel A, Burgess R, et al. Semiological seizure classification. Epilepsia 1998; 39(9): 1006-13.
- 25. Källén K, Wyllie E, Lüders HO, Lachhwani D, Kotagal P. Hypomotor seizures in infants and children. Epilepsia 2002; 43(8):882-8.
- 26. Battaglia D, Lettori D, Contaldo I, Veredice C, Sacco A, Vasco J, et al. Seizure semiology of lesional frontal lobe epilepsies in children. Neuropediatrics 2007; 38:287-91.
- 27. Wheless JW, Kim HL: Adolescent seizures and epilepsy syndromes. Epilepsia 2002; 43(Suppl 3):S33-52.
- 28. Cersósimo R, Flesler S, Bartuluchi M, Soprano AM, Pomata H, Caraballo R: Mesial temporal lobe epilepsy with hippocampal sclerosis: study of 42 children. Seizure 2011; 20(2):131-7.
Publication Dates
-
Publication in this collection
13 July 2012 -
Date of issue
2011
History
-
Received
05 Oct 2011 -
Accepted
05 Nov 2011