Abstract
Leishmaniasis is a neglected disease endemic in five continents. It is a severe disease that may lead to death, and its early detection is important to avoid severe damage to affected individuals. Molecular methods to detect Leishmania are considered alternatives to overcome the limitations presented by conventional methods. The aim of this study was to develop multiplex PCR systems able to detect small amounts of target DNA of Leishmania infantum and Leishmania braziliensis, and the gene coding for glyceraldehyde-3-phosphate dehydrogenase (G3PD) in mammals, enabling quality evaluation of the sample simultaneously with detection of the specific target. The systems created for G3PD recognition were combined with detection systems for L. infantum and L. braziliensis to compose multiplex PCR systems for visceral (mVL) and cutaneous (mACL) leishmaniasis diagnosis. The multiplex PCR systems developed were assessed in blood samples from five different species of mammal reservoirs involved in the disease cycle in Brazil, and 96 and 52 human samples from patients with suspected visceral leishmaniasis (VL) and cutaneous leishmaniasis (ACL), respectively. Three G3PD detection systems were created (G3PD1, G3PD2 and G3PD3) with different product sizes, G3PD2 was chosen for the formation of multiplex PCR systems. The two multiplex PCR systems (mVL and mACL) were reproducible in all species evaluated. Results of test samples (sensitivity, specificity and efficiency) suggest its use in routine diagnosis, research activities in medicine and veterinary medicine. Additionally, the systems designed to detect the G3PD gene are capable of combining with other targets used for molecular diagnosis of infectious diseases. Concerning leishmaniasis, the multiplex PCR systems can be used in epidemiological studies for the detection of new and classic reservoirs, which may contribute to the reliability of results and development of actions to control the disease.
leishmaniasis; diagnosis; glyceraldehyde-3-phosphate dehydrogenase; quality control
ORIGINAL PAPER
Application of the mammalian glyceraldehyde-3-phosphate dehydrogenase gene for sample quality control in multiplex PCR for diagnosis of leishmaniasis
Gonçalves SC; Régis-da-Silva CG; Brito MEFC; Brandão-Filho SP; Paiva-Cavalcanti M
Department of Immunology Aggeu Magalhães Research Center, Oswaldo Cruz Foundation (Fiocruz), Recife, Pernambuco State, Brazil
Correspondence to Correspondence to: Milena de Paiva-Cavalcanti Departamento de Imunologia, Centro de Pesquisas Aggeu Magalhães Av. Moraes Rego, s/n Cidade Universitária, Recife, PE, 50670-420, Brasil Phone: +55 81 21012679. Fax: +55 81 21012640 Email: mp@cpqam.fiocruz.br
ABSTRACT
Leishmaniasis is a neglected disease endemic in five continents. It is a severe disease that may lead to death, and its early detection is important to avoid severe damage to affected individuals. Molecular methods to detect Leishmania are considered alternatives to overcome the limitations presented by conventional methods. The aim of this study was to develop multiplex PCR systems able to detect small amounts of target DNA of Leishmania infantum and Leishmania braziliensis, and the gene coding for glyceraldehyde-3-phosphate dehydrogenase (G3PD) in mammals, enabling quality evaluation of the sample simultaneously with detection of the specific target. The systems created for G3PD recognition were combined with detection systems for L. infantum and L. braziliensis to compose multiplex PCR systems for visceral (mVL) and cutaneous (mACL) leishmaniasis diagnosis. The multiplex PCR systems developed were assessed in blood samples from five different species of mammal reservoirs involved in the disease cycle in Brazil, and 96 and 52 human samples from patients with suspected visceral leishmaniasis (VL) and cutaneous leishmaniasis (ACL), respectively. Three G3PD detection systems were created (G3PD1, G3PD2 and G3PD3) with different product sizes, G3PD2 was chosen for the formation of multiplex PCR systems. The two multiplex PCR systems (mVL and mACL) were reproducible in all species evaluated. Results of test samples (sensitivity, specificity and efficiency) suggest its use in routine diagnosis, research activities in medicine and veterinary medicine. Additionally, the systems designed to detect the G3PD gene are capable of combining with other targets used for molecular diagnosis of infectious diseases. Concerning leishmaniasis, the multiplex PCR systems can be used in epidemiological studies for the detection of new and classic reservoirs, which may contribute to the reliability of results and development of actions to control the disease.
Key words: leishmaniasis, diagnosis, glyceraldehyde-3-phosphate dehydrogenase, quality control.
INTRODUCTION
Leishmaniasis is a vector-borne disease that is caused by obligate intramacrophage protozoa (family Trypanosomatidae, order Kinetoplastida). It is a neglected disease, endemic in rural areas of five continents and is the third most important vector-borne disease in the world (1). The annual incidence is about 1.5 to 2 million cases worldwide, with up to 350 million people at risk of infection (2). There are an estimated 500,000 new cases of visceral leishmaniasis (VL) and more than 50,000 deaths from the disease each year (3). Such death rate is surpassed, among parasitic diseases, only by malaria (4).
There are at least 20 species of Leishmania, which may cause different clinical diseases. The prevalence of Leishmania spp. infection varies according to geographical distribution (5). VL is caused by L. donovani complex that consists of Leishmania (Leishmania) infantum syn. Leishmania (Leishmania) chagasi and Leishmania (Leishmania) donovani (6). American cutaneous leishmaniasis (ACL) is caused by species of Leishmania braziliensis and Leishmania mexicana complexes (7, 8). In Brazil, L. infantum and Leishmania (Viannia) braziliensis are the most common species isolated from VL and ACL carriers, respectively (7).
Early diagnosis and treatment are important as individual and community control measures. Untreated patients can act as reservoirs and contribute to the anthroponotic transmission (8). The clinical signs and symptoms are not pathognomonic for VL or ACL, and may be confused with other similar conditions such as malaria, tropical splenomegaly, schistosomiasis or cirrhosis with portal hypertension, African trypanosomiasis, milliary tuberculosis, brucellosis, typhoid fever, bacterial endocarditis, histoplasmosis, malnutrition, lymphoma, leukemia and numerous primary and secondary skin diseases (9).
The conventional routine diagnosis of leishmaniasis requires microscopic demonstration of Leishmania amastigotes in aspirates from spleen tissue, scraped skin smears, or in bone marrow. Blood culture is also commonly used (10). However, these conventional methods of diagnosis do have some limitations (11). Microscopic examination has low sensitivity and requires invasive samples (bone marrow, spleen, liver), whereas culture is susceptible to contamination and is time-consuming. Serological methods may be useful, but have problems with cross-reactions and differentiation between current and past infections, furthermore they are not accurate in immunosuppressed patients (12-16).
In the past ten years, the molecular biology has become increasingly relevant to the diagnosis and control of infectious diseases. Information on DNA sequences has been extensively exploited for the development of polymerase chain reaction (PCR)-based assays for various applications, including understanding of parasites and diagnosis of the diseases (10).
With application of PCR and its variations (nested, multiplex, real-time), molecular biology has contributed to the detection of parasite DNA with high sensitivity and specificity (17-19). However, for accurate diagnosis, with a minimum margin of error, it is necessary the use of internal controls, such as the housekeeping genes. The positivity for these genes demonstrates the quality of the sample under test and guarantees the issuance of a negative result.
Variations of PCR have been developed and used to identify Leishmania spp. such as the quantitative polymerase chain reaction (qPCR), nested PCR, and multiplex PCR (20-28). The possibility of combining multiple targets in the same assay led to expansion of applications of the multiplex PCR technique on leishmaniasis diagnosis, since the identification of complexes and species of the parasite to evaluation of sample integrity and PCR performance on sandfly pools (23-28).
Several targets have been used to detect Leishmania including kinetoplast DNA (kDNA) with high conserved multiple copies (29-31). Recently the gene for glucose-6-phosphate dehydrogenase (G6PD), single copy, has been used to identify the subgenus Viannia being able to distinguish L. braziliensis from other species (32).
Glyceraldehyde-3-phosphate dehydrogenase (G3PD) is an enzyme with a crucial role in glucose metabolism, with nuclear, cytoplasmic and membrane functions in cells of organisms which use glycolysis to produce energy. The presence of this gene in all mammalian cells ensures its detection in samples whose conditions are suitable for diagnostic tests, making it a potential tool for sample quality control in realtime PCR, as well as the genes of albumin, β-actin and β-globin (20, 33-35). However, in these trials, sample quality assessment is done in separate reactions, generating more costs and increasing the time for determining the diagnosis.
The limitations in diagnosis methods and the lack of access of the population to diagnostic tools indicate the need to develop technologies with quality assurance of results, readily accessible to affected population, and that have a favorable cost-benefit. In this context, this paper aims at developing multiplex PCR systems to limit the possibility of errors in the definition of the final diagnosis, in human or animal patients, to promote the simultaneous detection of specific target (L. infantum or L. braziliensis) and quality control of the samples (G3PD gene from mammals) in the same reaction, which will reduce costs with reagents and time for the release of diagnosis. This tool will be useful for the diagnosis of leishmaniasis as well as for epidemiological studies.
MATERIALS AND METHODS
Blood Samples
Blood was collected from Homo sapiens, Canis familiaris, Oryctolagus cuniculus, Necromys lasiurus and Rattus rattus to assess the reproducibility of the multiplex PCR systems in samples from different mammals. Informed consent was obtained from all studied subjects, from the database of Leishmaniasis Reference Service (LRS), Fiocruz, PE, Brazil. In addition, the present study was approved by the Research Ethics Committee (CEP-FIOCRUZ/PE, 42/2010) and by the Ethics Committee in Animal Use (CEUA- FIOCRUZ/RJ, LW-41/10).
DNA Extraction
The extraction of DNA from blood samples was performed with Illustra® blood genomic Prep Mini Spin kit (GE Healthcare, USA) according to the manufacturer's recommendations.
Primers Design
Based on the results of Castilho et al. (33), MEGA software (4.0) (available online at www.megasoftware.net) was employed for alignment of G3PD sequences from different mammals involved in the transmission cycle of leishmaniasis, using a NCBI BLAST database (http://www.ncbi.nlm.nih.gov). From this alignment, two primers were designed capable of binding to DNA to the following mammalian species: Homo sapiens, Sus scrofa, Mus musculus, Rattus norvegicus, Sigmodon hispidus, Equus caballus, Felis catus, Bos taurus, Macaca fascicularis, Macaca auratus, Oryctolagus aries, Canis lupus and Canis familiaris.
G3PD Detection Systems
To develop G3PD detection systems, the primers presented by Castilho et al. (33), GAPD-F and GAPD-R, were combined with primers designed in this study, looking for a better differentiation among bands for detection of Leishmania spp. and composition of multiplex PCR systems. For a preliminary assessment of G3PD gene systems, an experiment was conducted in which the reaction was composed of sample without DNA (negative control), dog blood samples that tested positive and negative for VL, and genomic DNA of L. chagasi (MHOM/ BR/1974/PP75) as a positive control under the conditions and system described by Lachaud et al. (36).
Multiplex PCR System Development
Two multiplex PCR systems were developed and tested under the conditions of the Linf 1B and B1/B2 for simultaneous detection of L. infantum or L. braziliensis and G3PD gene, respectively (20, 29).
Optimization of Multiplex PCR Systems
Reproducibility evaluation in different mammalian species
Blood samples from Homo sapiens, Canis familiaris, Oryctolagus cuniculus, Rattus rattus and Necromys lasiurus were used.
The analysis of positive results was performed by adding genomic DNA of L. infantum or L. braziliensis in samples of each animal species used in the study, based on the optimal range of detection of Linf 1B and B1/B2 (20, 29). When it was necessary, modifications in cycling conditions (annealing temperatures and extension) were made, as well as alterations in quantity and concentration of reagents.
Detection range determination
A dilution curve of factor 10 (10 ng to 0.1 fg) of parasite DNA, tested in both multiplex PCR reactions, to check possible loss of sensitivity of multiplex PCR reaction compared to conventional systems was performed.
Documentation of Results
The analysis, interpretation and registry of test results were performed using 1.5% agarose gel electrophoresis, stained with ethidium bromide (10 mg/mL), and a molecular weight marker 100 bp ladder DNA (GibcoBRL Life Technologies, USA).
Testing of Patient Samples
After optimization, the multiplex PCR for visceral leishmaniasis (mVL) and the multiplex PCR for American cutaneous leishmaniasis (mACL) systems were evaluated in 96 blood and bone marrow samples from human and dog patients, and 52 biopsy samples from human patients. These samples had been previously tested positive for leishmaniasis by conventional PCR, at LRS, Fiocruz/PE, Brazil.
Sensitivity, specificity and efficiency of mVL and mACL were calculated according to Ferreira and Ávila (37).
RESULTS
G3PD Detection Systems
The designed primers (Figure 1) were combined with GAPD-F and GAPD-R and three detection systems were generated: G3PD1, G3PD2 and G3PD3 (Table 1) (33). The result of preliminary experiment showed an excellent performance of the three systems designed for G3PD gene detection (Figure 2).
Based on the products generated for simultaneous detection of L. infantum, L. braziliensis and G3PD gene, the G3PD2 system was chosen for optimization of the multiplex PCR for L. infantum detection, and the multiplex PCR for L. braziliensis detection (Table 2).
mVL Optimization
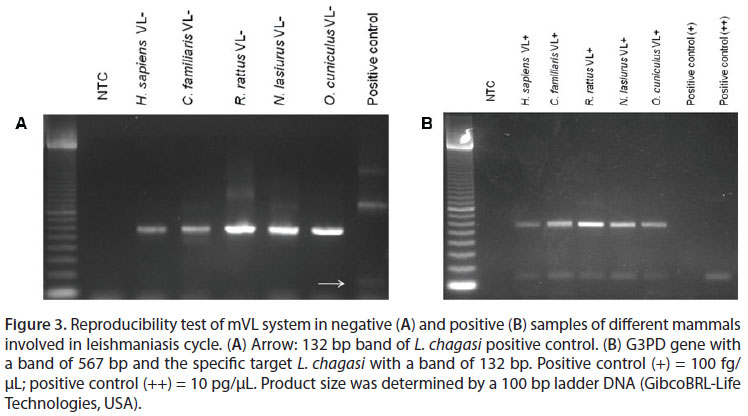
The analysis of experiments under conditions of Linf 1B showed no reproducible results (20). In order to generate reproducibility, the reactions were modified using the conditions described by Lachaud et al. (36) with primers of Linf 1B. After the modification, excellent reproducibility was obtained among different mammalian species (Figure 3).
Detection range
To avoid loss of sensitivity for the detection of a specific target (L. infantum), the concentration of primers to detect the G3PD gene was reduced, from 5 pmol/µL to 1 pmol/µL, in order to equilibrate the competition among reagents and encourage the Linf 1B system.
The optimal conditions of functioning of mVL, in a final volume of 25 µL, were: Taq polymerase buffer 0.5 µM Tris, 1.25 µM KCl, pH 8.4; 0.3 µM MgCl2; 5 nM dNTP; 5 µM of each primer Linf 1B; 1 µM G1F; 1 µM G2R; 0,25U Taq polymerase and 2 µL DNA template. In these conditions, the mVL showed a detection limit of 0.1 fg.
Testing of Patient Samples
A total of 32 (33%) samples did not pass quality control and were rejected for validation tests. The evaluation of mVL in patient samples showed: 25% sensitivity, 97% specificity and 79% efficiency.
The low sensitivity obtained in contrast with the excellent detection limit created the hypothesis of sample degradation due to long storage time (samples stored since 2005). To check the sensitivity of mVL in blood samples and rule out possible flaws in this diagnostic system developed, a dilution curve similar to that aforementioned was performed, by diluting DNA from L. infantum in freshly collected human blood. The analysis of this experiment showed that the mVL system does not have failures in the detection limit (detection of 0.1 fg) and tested positive all blood samples recently collected.
mACL Optimization
Reproducibility in different mammals
The mACL worked well under the following reaction conditions: Taq polymerase buffer 0.5 µM Tris, 1.25 µM KCl, pH 8.4; 0.3 µM MgCl2; 5 nM dNTP; 25 µM of B1/B2 primers mixture; 5 µM G1F; 5 µM G2R; 0.25 U Taq polymerase and 2 µL DNA template. It showed reproducibility among different mammalian species, and both negative and positive samples (Figure 4).
Detection range
A temperature gradient was applied to verify the conditions under which mACL has a better detection limit. The optimal cycling conditions in a final volume of 25 µL were: initial denaturation: 94ºC for five minutes; denaturation: 94ºC for 30 seconds; annealing: 63ºC for one minute; extension: 72ºC for one minute; final extension: 72ºC for five minutes. In these conditions, the mACL showed a detection limit of 10 pg.
Testing of Patients Samples
The evaluation of the mACL system in biopsy samples showed sensitivity and specificity of 100% and 87.5% respectively, and 92% efficiency.
DISCUSSION
The main deficiencies in applying PCR assays include false-positive results (from background DNA contamination) and potential falsenegative results, besides the detection limits of the assay. Clinical samples often contain substances that may partially or completely inhibit the amplification reaction by the DNA polymerase (38, 39). Appropriate care and correct conditioning of the sample may prevent such problems, but the integrity of target DNA must be monitored. Methods that ensure the best sample processing include incorporation of internal amplification controls into the PCR assay to monitor the presence of purified sample DNA and potential PCR inhibitors (40).
The need to include internal controls has been observed in various types of clinical samples, especially in materials in which purification of nucleic acids is difficult due to abundance of organic matter such as feces and food (4146). The inclusion in PCR assays of genes that constitute cells such as β-actin, β-globin, albumin and G3PD has been useful to evaluate the quality of samples in qPCR and PCR experiments (20, 34, 47, 48). Due to the recognized need for evaluating the integrity of DNA, controls are usually used in separate reactions, generating more costs and consuming more time to establish the diagnosis (49).
In the present study, three detection systems were created to allow distinction among the various targets used in the molecular diagnosis of leishmaniasis. In addition, the developed control systems have a wide applicability in molecular diagnostics, and can be optimized in a similar way with PCR-based diagnosis of bacteria, virus, and other protozoa, which did not evaluate the possibility of false-negative results in their tests (50-53). The goal of the current study was to evaluate the quality of samples associated with detection of a target in the same reaction, in order to provide a safe outcome for patient diagnosis and also to apply it in epidemiological studies. The evaluation of the reproducibility of mVL and mACL systems showed that they can be used in research of new reservoirs, contributing to the expansion of leishmaniasis eco-epidemiology knowledge and adequacy of control measures for each source of the disease.
The detection limit presented by mVL was higher than that recorded by Paiva-Cavalcanti et al. (20), with no reduction of efficiency in multiplex PCR compared to real-time PCR. Disch et al. (54) suggested a duplex PCR for L. donovani complex with 4.7 fg of detection limit, lower than the limit reached by mVL. Harris et al. (26) proposed a multiplex PCR with 1 fg of detection limit for characterization of new world Leishmania complexes. The proposal presented in the present study has better detection ability and is species-specific.
The evaluation of patient samples revealed a significant loss of reliability due to poor quality samples or their degradation (33% of the samples tested negative in internal control). Despite of low sensitivity obtained in mVL, the experiment conducted on recently collected blood, maintained an excellent detection limit and showed that losses due to degradation occur in samples stored for long periods (such as samples from LRS, stored since 2005), and the DNA of etiological agent is the most affected, probably because there is less of its DNA material than of the host. Despite the low sensitivity obtained in mVL, the system showed specificity and detection limits in accordance with the objective.
The mACL showed better sensitivity than that reported by Bruijn and Barker (29), for the system B1/B2 alone. Marcussi et al. (55) presented the LBF1/LBR1 primers for identification of L. braziliensis with a limit of 50 ng of DNA in the reaction, a detection limit lower than mACL.
The tested samples for mACL showed excellent results that suggest its use in routine diagnoses. Gomes et al. (56), evaluating PCR for detection of L. braziliensis in biopsy samples, obtained 96% sensitivity. Despite the fact that the SL RNA gene was the target, the results of Gomes et al. (56) are similar to mACL.
The detection limits provided by the two multiplex showed that this molecular diagnostic tool can be used without loss of efficiency, besides promoting reduction of reagent costs and time for the institution of therapy. These data are extremely important to regions where the disease is endemic, since the developed techniques may promote improvements in quality of life of affected persons by reducing the risk of serious injuries due to ACL.
During the validation of this method, the importance of having sample quality control was proved by the percentage of degraded samples detected by mVL. Some methodologies employed in recent studies do not perform the evaluation of sample quality, and in this case, there is no guarantee of the accuracy of negative results. The use of sample control in the same reaction without sacrificing sensitivity (mACL) or detection limits (mVL) of the original system is an advantage for the economy of reagents and time compared to reactions conducted separately.
Given to the limitations presented by conventional methods, the development of a tool that is able to promote an accurate diagnosis is essential. In this context, this study presents two multiplex PCR that allows assessment of the quality of the sample simultaneously with the detection of L. infantum or L. braziliensis, therefore reducing the number of false negatives and assuring the quality of results. The methods can be applied both for diagnosis in humans and animals, and their use is particularly indicated in routine diagnosis and research.
CONCLUSION
The development of mVL and mACL systems may be implemented in the diagnosis in LRS, Brazil. Additionally, the systems designed to detect the G3PD gene are able to combine with other agents for molecular diagnosis. For leishmaniasis, the multiplex systems might be used in epidemiological studies for detection of new and classic reservoirs, contributing to the reliability of results and development of actions to control the disease.
ACKNOWLEDGEMENTS
The authors would like to thank Luciana A. Figueredo for her substantial comments on this paper.
REFERENCES
1. Herwaldt BL. 1999;354(9185):1191-9.
2. Murray HW, Berman NG. Advances in 2005;366(9496):1561-77.
3. Desjeux P. The increase in risk factors for leishmaniasis worldwide. Trans R Soc Trop Med Hyg. 2004;95(3):239-43.
4. WHO. World Health Organization. Health topics: leishmaniasis [Internet]. Geneva: Special Programme for Research & Training in Tropical Diseases (TDR). [cited 2010 Jan 26]. Available from: http://apps.who.int/tdr/svc/diseases/leishmaniasis.
5. Lainson R, Shaw JJ. Evolution, classification and geographical distribution. In: Peters W, Killick-Kendrick R, editors. The leishmaniasis in biology and medicine. London: Academic Press; 1987. p. 1-120.
6. Dantas-Torres F. Canine leishmaniosis in South America. Parasit Vectors. 2009;2(Suppl 1):S1.
7. Singh S. New developments in diagnosis of leishmaniasis. Indian J Med Res. 2006;123(1):311-30.
8. Chappuis F, Sundar S, Hailu A, Ghalib H, Rijal S, Peeling RW et al. Visceral leishmaniasis: what are the needs for diagnosis, treatment and control? Nat Rev Microbiol. 2007;5(11):873-82.
9. Singh S, Dey A, Sivakumar R. Applications of molecular methods for Leishmania control. Expert Rev Mol Diagn. 2005;5(1):251-65.
10. Schallig HD, Oskam L. Molecular biological applications in the diagnosis and control of leishmaniasis and parasite identification. Trop Med Int Health. 2002;7(8):641-51.
11. Camargo JB, Langoni H, Troncarelli MZ, Machado JG, Lucheis SB, Padovani CR. Performance of IFAT, ELISA, direct parasitological examination and PCR on lymph node aspirates for canine visceral leishmaniasis diagnosis. J Venom Anim Toxins incl Trop Dis. 2010;16(3):414-20.
12. Piarroux R, Gambarelli F, Dumon H, Fontes M, Dunan S, Mary C, et al. Comparison of PCR with direct examination of bone marrow aspiration, myeloculture, and serology for diagnosis of visceral leishmaniasis in inmunocompromised patients. J Clin Microbiol. 1994;32(1):746-9.
13. Wilson SM. DNA-based methods in the detection of Leishmania parasites: fields applications and practicalities. Ann Trop Med Parasitol.1995;89(Suppl 1):95-100.
14. Ozensoy S, Ozbel Y, Turgay N, Alkan MZ, Gul K, Gilman-Sachs A, et al. Serodiagnosis and epidemiology of visceral leishmaniasis in Turkey. Am J Trop Med Hyg. 1998;59(3):363-9.
15. Hu XS, Yang WT, Lu HG, Yan HP, Cheng JP, Ma Y, et al. Sequencing a specific kinetoplast DNA fragment of Leishmania donovani for polymerase chain reaction amplification in diagnosis of leishmaniasis in bone marrow and blood samples. J Parasitol. 2000;86(4):822-6.
16. Ikonomopoulos J, Kokotas S, Gazouli M, Zavras A, Stoitsiou M, Gorgoulis VG. Molecular diagnosis of leishmaniosis in dogs. Comparative application of traditional diagnostic methods and the propose assay on clinical samples. Vet Parasitol. 2003;113(2):99-113.
17. Paiva-Cavalcanti M, Régis-da-Silva CG, Gomes YM. Comparison of real-time PCR and conventional PCR for detection of Leishmania (Leishmania) infantum infection: a mini-review. J Venom Anim Toxins incl Trop Dis. 2010;16(4):537-42.
18. Santamaría E, Ponce N, Puerta C, Ferro C. Validación de la PCR en la detección de parásitos de Leishmania (Viaña) spp. en Lutzomyia (Diptera:Psychodidae) como herramienta en la definición de especies vectores. Biomedica. 2005;25(1):271-9.
19. Schwartz E, Hatz C, Blum J. New world cutaneous leishmaniasis in travellers. Lancet Infect Dis. 2006;6(6):342-9.
20. Paiva-Cavalcanti M, Felinto de Brito ME, de Souza WV, de Miranda Gomes Y, Abath FC. The development of a real-time PCR assay for the quantification of Leishmania infantum DNA in canine blood. Vet J. 2009;182(2):356-8.
21. Fisa R, Riera C, Gállego M, Manubens J, Portús M. Nested PCR for diagnosis of canine leishmaniasis in peripheral blood, lymph node and bone marrow aspirates. Vet Parasitol. 2001;99(2):105-11.
22. Akhavan AA, Mirhendi H, Khamesipour A, Alimohammadia MH, Rassi Y, Bates P, et al. Leishmania species: detection and identification by nested PCR assay from skin samples of rodent reservoirs. Exp Parasitol. 2010;126(4):552-6.
23. de Pita-Pereira D, Cardoso MA, Alves CR, Brazil RP, Britto C. Detection of natural infection in Lutzomyia cruzi and Lutzomyia forattinii (Diptera: Psychodidae: Phlebotominae) by Leishmania infantum chagasi in an endemic area of visceral leishmaniasis in Brazil using a PCR multiplex assay. Acta Trop. 2008;107(1):66-9.
24. Rodríguez-González I, Marín C, Logoni SS, Mateo H, Alunda JM, Minaya G, et al. Identification of New World Leishmania species from Peru by biochemical techniques and multiplex PCR assay. FEMS Microbiol Lett. 2007;267(1):9-16.
25. Jorquera A, González R, Marchán-Marcano E, Oviedo M, Matos M. Multiplex-PCR for detection of natural Leishmania infection in Lutzomyia spp. captured in an endemic region for cutaneous leishmaniasis in state of Sucre, Venezuela. Mem Inst Oswaldo Cruz. 2005;100(1):45-8.
26. Harris E, Kropp G, Belli A, Rodriguez B, Agabian N. Single-step multiplex PCR assay for characterization of new world Leishmania complexes. J Clin Microbiol. 1998;36(7):1989-95.
27. de Pita-Pereira D, Souza GD, Pereira T de A, Zwetsch A, Britto C, Rangel EF. Lutzomyia (Pintomyia) fischeri (Diptera: Psychodidae: Phlebotominae), a probable vector of American cutaneous leishmaniasis: detection of natural infection by Leishmania (Viannia) DNA in specimens from the municipality of Porto Alegre (RS), Brazil, using multiplex PCR assay. Acta Trop. 2011;120(3):273-5.
28. Oliveira DM, Reinhold-Castro KR, Bernal MV, Legriffon CM, Lonardoni MV, Teodoro U, et al. Natural infection of Nyssomyia neivai by Leishmania (Viannia) spp. in the state of Paraná, southern Brazil, detected by multiplex polymerase chain reaction. Vector Borne Zoonotic Dis. 2011;11(2):137-43.
29. Bruijn MH, Barker DC. Diagnosis of New World leishmaniasis: specific detection of species of the Leishmania braziliensis complex by amplification of kinetoplast DNA. Acta Trop. 1992;52(1):45-58.
30. Lopez M, Inga R, Cangalaya M, Echevarria J, Llanos-Cuentas A, Orrego C et al. Diagnosis of leishmania using the polymerase chain reaction: a simplified procedure for field work. Am J Trop Med Hyg. 1993;49(3):348-56.
31. Uliana SR, Nelson K, Beverley SM, Camargo EP, Floeter-Winter LM. Discrimination amongst Leishmania by polymerase chain reaction and hybridization with small subunit ribosomal DNA derived oligonucleotides. J Eukaryot Microbiol. 1994;41(4):324-30.
32. Castilho TM, Shaw JJ, Floeter-Winter LM. New PCR assay using glucose-6-phosphate dehydrogenase for identification of Leishmania species. J Clin Microbiol. 2003;41(2):540-6.
33. Castilho TM, Camargo LMA, McMahon-Pratt D, Shaw JJ, Floeter-Winter LM. A real-time polymerase chain reaction assay for the identification and quantification of American Leishmania species on the basis of glucose-6-phosphate dehydrogenase. Am J Trop Med Hyg. 2008;78(1):122-32.
34. Gilsbach R, Kouta M, Bönisch H, Brüss M. Comparison of in vitro and in vivo reference genes for internal standardization of real-time PCR data. Biotechniques. 2006;40(2):173-7.
35. Solano-Gallego L, Rodriguez-Cortes A, Trotta M, Zampieron C, Razia L, Furlanello T, et al. Detection of Leishmania infantum DNA by fret-based realtime PCR in urine from dogs with natural clinical leishmaniasis. Vet Parasitol. 2007;147(3-4):315-9.
36. Lachaud L, Chabbert E, Dubessay P, Dereure J, Lamothe J, Dedet JP, et al. Value of two PCR methods for the diagnosis of canine visceral leishmaniasis and the detection of asymptomatic carriers. Parasitology. 2002;125(Pt 3):197-207.
37. Ferreira AW, Ávila SLM. Sorologia: importância e parâmetros. In: Ávila SLM, Ferreira AW, editors. Diagnósticos de laboratório das principais doenças infecciosas e auto-imunes. 2nd ed. Rio de Janeiro: Guanabara Koogan Press; 2001. 3-8 p.
38. Jackson DP, Haden JD, Quirke P. Extraction of nucleic acid from fresh and archival material. In: McPherson MJ, Quirke P, Taylor GR, editors. PCR: a pratical approach. New York: Oxford University Press; 1991. 29-50 p.
39. Degrave W, Fernandes O, Campbell D, Bozza M, Lopes U. Use of molecular probes and PCR for detection and typing of Leishmania - a mini-review. Mem Inst Oswaldo Cruz. 1994;89(3):463-9.
40. Yang S, Rothman RE. PCR-based diagnostics for infectious diseases: uses, limitations, and future applications in acute-care settings. Lancet Infect Dis. 2004;4(6):337-48.
41. Thoreson AC, Borre M, Andersen LP, Jorgensen F, Kiilerich S, Scheibel J, et al. Helicobacter pylori detection in human biopsies: a competitive PCR assay with internal control reveals false results. FEMS Immunol Med Microbiol. 1999;24(2):201-8.
42. LeBlanc-Maridor M, Garénaux A, Beaudeau F, Chidaine B, Seegers H, Denis M, et al. Quantification of Campylobacter spp. in pig feces by direct realtime PCR with an internal control of extraction and amplification. J Microbiol Methods. 2011;85(1):53-61.
43. Bezold G, Volkenandt M, Gottlöber P, Peter RU. Detection of herpes simplex virus and varicellazoster virus in clinical swabs: frequent inhibition of PCR as determined by internal controls. Mol Diagn. 2000;5(4):279-84.
44. Lund M, Madsen M. Strategies for the inclusion of an internal amplification control in conventional and real time PCR detection of Campylobacter spp. in chicken fecal samples. Mol Cell Probes. 2006;20(2):92-9.
45. Murphy NM, McLauchlin J, Ohai C, Grant KA. Construction and evaluation of a microbiological positive process internal control for PCRbased examination of food samples for Listeria monocytogenes and Salmonella enterica. Int J Food Microbiol. 2007;120(1-2):110-9.
46. Cubero J, van der Wolf J, van Beckhoven J, López MM. An internal control for the diagnosis of crown gall by PCR. J Microbiol Methods. 2002;51(3):387-92.
47. Mary C, Faraut F, Lascombe L, Dumon H. Quantification of Leishmania infantum DNA by a realtime PCR assay with high sensitivity. J Clin Microbiol. 2004;42(11):5249-55.
48. Kompalic-Cristo A, Frotta C, Suárez-Mutis M, Fernandes O, Britto C. Evaluation of a real-time PCR assay based on the repetitive B1 gene for the detection of Toxoplasma gondii in human peripheral blood. Parasitol Res. 2007;101(3):619-25.
49. Müller N, Zimmermann V, Forster U, Bienz M, Gottstein B, Welle M. PCR-based detection of canine Leishmania infections in formalinfixed and paraffin-embedded skin biopsies: elaboration of a protocol for quality assessment of the diagnostic amplification reaction. Vet Parasitol. 2003;114(3):223-9.
50. Palaniappan RU, Chang YF, Chang CF, Pan MJ, Yang CW, Harpending P, et al. Evaluation of ligbased conventional and real time PCR for the detection of pathogenic leptospires. Mol Cell Probes. 2005;19(2):111-7.
51. Ferrari HF, Luvizotto MC, Rahal P, Cardoso TC. Detection of bovine Herpesvirus type 5 in formalinfixed, paraffin-embedded bovine brain by PCR: a useful adjunct to conventional tissue-based diagnostic test of bovine encephalitis. J Virol Methods. 2007;146(12):335-40.
52. Miyamoto CT, Gomes ML, Marangon AV, de Araújo SM, Bahia MT, Martins-Filho OA, et al. Usefulness of the polymerase chain reaction for monitoring cure of mice infected with different Trypanosoma cruzi clonal genotypes following treatment with benznidazole. Exp Parasitol. 2008;120(1):45-9.
53. Montoya A, Miró G, Mateo M, Ramírez C, Fuentes I. Detection of Toxoplasma gondii in cats by comparing bioassay in mice and polymerase chain reaction (PCR). Vet Parasitol. 2009;160(1-2):159-62.
54. Disch J, Caligiorne RB, Maciel F, Oliveira MC, Orsini M, Dias-Neto E, et al. Single-step duplex kDNA-PCR for detection of Leishmania donovani complex in human peripheral blood samples. Diagn Microbiol Infect Dis. 2006;56(4):395-400.
55. Marcussi VM, Marcussi LM, Barbosa-Tessmann IP, Lonardoni MV, Silveira TG. Leishmania (Viannia) braziliensis: new primers for identification using polymerase chain reaction. Exp Parasitol. 2008;120(4):300-5.
56. Gomes AH, Armelin IM, Menon SZ, Pereira-Chioccola VL. Leishmania (V.) braziliensis: detection by PCR in biopsies from patients with cutaneous leishmaniasis. Exp Parasitol. 2008;119(3):319-24.
Received: November 17, 2011.
Accepted: February 24, 2012.
Abstract published online: March 6, 2012.
Full paper published online: May 31, 2012.
CONFLICTS OF INTEREST: The authors declare no conflicts of interest.
FINANCIAL SOURCE: The National Council for Scientific and Technological Development (CNPq), The State of Pernambuco Research Foudation (FACEPE) and CPqAM-FIOCRUZ/PE provided the financial grants
ETHICS COMMITTEE APPROVAL: The present study was approved by the Research Ethics Committee of Oswaldo Cruz Foundation, Recife, PE, Brazil (CEP-FIOCRUZ/PE, 42/2010) and by the Ethics Committee in Animal Use of Oswaldo Cruz Foundation, Recife, PE, Brazil (CEUA-FIOCRUZ/RJ, LW-41/10).
- 3. Desjeux P. The increase in risk factors for leishmaniasis worldwide. Trans R Soc Trop Med Hyg. 2004;95(3):239-43.
- 4. WHO. World Health Organization. Health topics: leishmaniasis [Internet]. Geneva: Special Programme for Research & Training in Tropical Diseases (TDR). [cited 2010 Jan 26]. Available from: http://apps.who.int/tdr/svc/diseases/leishmaniasis
- 5. Lainson R, Shaw JJ. Evolution, classification and geographical distribution. In: Peters W, Killick-Kendrick R, editors. The leishmaniasis in biology and medicine. London: Academic Press; 1987. p. 1-120.
- 6. Dantas-Torres F. Canine leishmaniosis in South America. Parasit Vectors. 2009;2(Suppl 1):S1.
- 7. Singh S. New developments in diagnosis of leishmaniasis. Indian J Med Res. 2006;123(1):311-30.
- 8. Chappuis F, Sundar S, Hailu A, Ghalib H, Rijal S, Peeling RW et al. Visceral leishmaniasis: what are the needs for diagnosis, treatment and control? Nat Rev Microbiol. 2007;5(11):873-82.
- 9. Singh S, Dey A, Sivakumar R. Applications of molecular methods for Leishmania control. Expert Rev Mol Diagn. 2005;5(1):251-65.
- 10. Schallig HD, Oskam L. Molecular biological applications in the diagnosis and control of leishmaniasis and parasite identification. Trop Med Int Health. 2002;7(8):641-51.
- 11. Camargo JB, Langoni H, Troncarelli MZ, Machado JG, Lucheis SB, Padovani CR. Performance of IFAT, ELISA, direct parasitological examination and PCR on lymph node aspirates for canine visceral leishmaniasis diagnosis. J Venom Anim Toxins incl Trop Dis. 2010;16(3):414-20.
- 12. Piarroux R, Gambarelli F, Dumon H, Fontes M, Dunan S, Mary C, et al. Comparison of PCR with direct examination of bone marrow aspiration, myeloculture, and serology for diagnosis of visceral leishmaniasis in inmunocompromised patients. J Clin Microbiol. 1994;32(1):746-9.
- 13. Wilson SM. DNA-based methods in the detection of Leishmania parasites: fields applications and practicalities. Ann Trop Med Parasitol.1995;89(Suppl 1):95-100.
- 14. Ozensoy S, Ozbel Y, Turgay N, Alkan MZ, Gul K, Gilman-Sachs A, et al. Serodiagnosis and epidemiology of visceral leishmaniasis in Turkey. Am J Trop Med Hyg. 1998;59(3):363-9.
- 15. Hu XS, Yang WT, Lu HG, Yan HP, Cheng JP, Ma Y, et al. Sequencing a specific kinetoplast DNA fragment of Leishmania donovani for polymerase chain reaction amplification in diagnosis of leishmaniasis in bone marrow and blood samples. J Parasitol. 2000;86(4):822-6.
- 16. Ikonomopoulos J, Kokotas S, Gazouli M, Zavras A, Stoitsiou M, Gorgoulis VG. Molecular diagnosis of leishmaniosis in dogs. Comparative application of traditional diagnostic methods and the propose assay on clinical samples. Vet Parasitol. 2003;113(2):99-113.
- 17. Paiva-Cavalcanti M, Régis-da-Silva CG, Gomes YM. Comparison of real-time PCR and conventional PCR for detection of Leishmania (Leishmania) infantum infection: a mini-review. J Venom Anim Toxins incl Trop Dis. 2010;16(4):537-42.
- 18. Santamaría E, Ponce N, Puerta C, Ferro C. Validación de la PCR en la detección de parásitos de Leishmania (Viaña) spp. en Lutzomyia (Diptera:Psychodidae) como herramienta en la definición de especies vectores. Biomedica. 2005;25(1):271-9.
- 19. Schwartz E, Hatz C, Blum J. New world cutaneous leishmaniasis in travellers. Lancet Infect Dis. 2006;6(6):342-9.
- 20. Paiva-Cavalcanti M, Felinto de Brito ME, de Souza WV, de Miranda Gomes Y, Abath FC. The development of a real-time PCR assay for the quantification of Leishmania infantum DNA in canine blood. Vet J. 2009;182(2):356-8.
- 21. Fisa R, Riera C, Gállego M, Manubens J, Portús M. Nested PCR for diagnosis of canine leishmaniasis in peripheral blood, lymph node and bone marrow aspirates. Vet Parasitol. 2001;99(2):105-11.
- 22. Akhavan AA, Mirhendi H, Khamesipour A, Alimohammadia MH, Rassi Y, Bates P, et al. Leishmania species: detection and identification by nested PCR assay from skin samples of rodent reservoirs. Exp Parasitol. 2010;126(4):552-6.
- 23. de Pita-Pereira D, Cardoso MA, Alves CR, Brazil RP, Britto C. Detection of natural infection in Lutzomyia cruzi and Lutzomyia forattinii (Diptera: Psychodidae: Phlebotominae) by Leishmania infantum chagasi in an endemic area of visceral leishmaniasis in Brazil using a PCR multiplex assay. Acta Trop. 2008;107(1):66-9.
- 24. Rodríguez-González I, Marín C, Logoni SS, Mateo H, Alunda JM, Minaya G, et al. Identification of New World Leishmania species from Peru by biochemical techniques and multiplex PCR assay. FEMS Microbiol Lett. 2007;267(1):9-16.
- 25. Jorquera A, González R, Marchán-Marcano E, Oviedo M, Matos M. Multiplex-PCR for detection of natural Leishmania infection in Lutzomyia spp. captured in an endemic region for cutaneous leishmaniasis in state of Sucre, Venezuela. Mem Inst Oswaldo Cruz. 2005;100(1):45-8.
- 26. Harris E, Kropp G, Belli A, Rodriguez B, Agabian N. Single-step multiplex PCR assay for characterization of new world Leishmania complexes. J Clin Microbiol. 1998;36(7):1989-95.
- 27. de Pita-Pereira D, Souza GD, Pereira T de A, Zwetsch A, Britto C, Rangel EF. Lutzomyia (Pintomyia) fischeri (Diptera: Psychodidae: Phlebotominae), a probable vector of American cutaneous leishmaniasis: detection of natural infection by Leishmania (Viannia) DNA in specimens from the municipality of Porto Alegre (RS), Brazil, using multiplex PCR assay. Acta Trop. 2011;120(3):273-5.
- 28. Oliveira DM, Reinhold-Castro KR, Bernal MV, Legriffon CM, Lonardoni MV, Teodoro U, et al. Natural infection of Nyssomyia neivai by Leishmania (Viannia) spp. in the state of Paraná, southern Brazil, detected by multiplex polymerase chain reaction. Vector Borne Zoonotic Dis. 2011;11(2):137-43.
- 29. Bruijn MH, Barker DC. Diagnosis of New World leishmaniasis: specific detection of species of the Leishmania braziliensis complex by amplification of kinetoplast DNA. Acta Trop. 1992;52(1):45-58.
- 30. Lopez M, Inga R, Cangalaya M, Echevarria J, Llanos-Cuentas A, Orrego C et al. Diagnosis of leishmania using the polymerase chain reaction: a simplified procedure for field work. Am J Trop Med Hyg. 1993;49(3):348-56.
- 31. Uliana SR, Nelson K, Beverley SM, Camargo EP, Floeter-Winter LM. Discrimination amongst Leishmania by polymerase chain reaction and hybridization with small subunit ribosomal DNA derived oligonucleotides. J Eukaryot Microbiol. 1994;41(4):324-30.
- 32. Castilho TM, Shaw JJ, Floeter-Winter LM. New PCR assay using glucose-6-phosphate dehydrogenase for identification of Leishmania species. J Clin Microbiol. 2003;41(2):540-6.
- 33. Castilho TM, Camargo LMA, McMahon-Pratt D, Shaw JJ, Floeter-Winter LM. A real-time polymerase chain reaction assay for the identification and quantification of American Leishmania species on the basis of glucose-6-phosphate dehydrogenase. Am J Trop Med Hyg. 2008;78(1):122-32.
- 34. Gilsbach R, Kouta M, Bönisch H, Brüss M. Comparison of in vitro and in vivo reference genes for internal standardization of real-time PCR data. Biotechniques. 2006;40(2):173-7.
- 35. Solano-Gallego L, Rodriguez-Cortes A, Trotta M, Zampieron C, Razia L, Furlanello T, et al. Detection of Leishmania infantum DNA by fret-based realtime PCR in urine from dogs with natural clinical leishmaniasis. Vet Parasitol. 2007;147(3-4):315-9.
- 36. Lachaud L, Chabbert E, Dubessay P, Dereure J, Lamothe J, Dedet JP, et al. Value of two PCR methods for the diagnosis of canine visceral leishmaniasis and the detection of asymptomatic carriers. Parasitology. 2002;125(Pt 3):197-207.
- 37. Ferreira AW, Ávila SLM. Sorologia: importância e parâmetros. In: Ávila SLM, Ferreira AW, editors. Diagnósticos de laboratório das principais doenças infecciosas e auto-imunes. 2nd ed. Rio de Janeiro: Guanabara Koogan Press; 2001. 3-8 p.
- 38. Jackson DP, Haden JD, Quirke P. Extraction of nucleic acid from fresh and archival material. In: McPherson MJ, Quirke P, Taylor GR, editors. PCR: a pratical approach. New York: Oxford University Press; 1991. 29-50 p.
- 39. Degrave W, Fernandes O, Campbell D, Bozza M, Lopes U. Use of molecular probes and PCR for detection and typing of Leishmania - a mini-review. Mem Inst Oswaldo Cruz. 1994;89(3):463-9.
- 40. Yang S, Rothman RE. PCR-based diagnostics for infectious diseases: uses, limitations, and future applications in acute-care settings. Lancet Infect Dis. 2004;4(6):337-48.
- 41. Thoreson AC, Borre M, Andersen LP, Jorgensen F, Kiilerich S, Scheibel J, et al. Helicobacter pylori detection in human biopsies: a competitive PCR assay with internal control reveals false results. FEMS Immunol Med Microbiol. 1999;24(2):201-8.
- 42. LeBlanc-Maridor M, Garénaux A, Beaudeau F, Chidaine B, Seegers H, Denis M, et al. Quantification of Campylobacter spp. in pig feces by direct realtime PCR with an internal control of extraction and amplification. J Microbiol Methods. 2011;85(1):53-61.
- 43. Bezold G, Volkenandt M, Gottlöber P, Peter RU. Detection of herpes simplex virus and varicellazoster virus in clinical swabs: frequent inhibition of PCR as determined by internal controls. Mol Diagn. 2000;5(4):279-84.
- 44. Lund M, Madsen M. Strategies for the inclusion of an internal amplification control in conventional and real time PCR detection of Campylobacter spp. in chicken fecal samples. Mol Cell Probes. 2006;20(2):92-9.
- 45. Murphy NM, McLauchlin J, Ohai C, Grant KA. Construction and evaluation of a microbiological positive process internal control for PCRbased examination of food samples for Listeria monocytogenes and Salmonella enterica. Int J Food Microbiol. 2007;120(1-2):110-9.
- 46. Cubero J, van der Wolf J, van Beckhoven J, López MM. An internal control for the diagnosis of crown gall by PCR. J Microbiol Methods. 2002;51(3):387-92.
- 47. Mary C, Faraut F, Lascombe L, Dumon H. Quantification of Leishmania infantum DNA by a realtime PCR assay with high sensitivity. J Clin Microbiol. 2004;42(11):5249-55.
- 48. Kompalic-Cristo A, Frotta C, Suárez-Mutis M, Fernandes O, Britto C. Evaluation of a real-time PCR assay based on the repetitive B1 gene for the detection of Toxoplasma gondii in human peripheral blood. Parasitol Res. 2007;101(3):619-25.
- 49. Müller N, Zimmermann V, Forster U, Bienz M, Gottstein B, Welle M. PCR-based detection of canine Leishmania infections in formalinfixed and paraffin-embedded skin biopsies: elaboration of a protocol for quality assessment of the diagnostic amplification reaction. Vet Parasitol. 2003;114(3):223-9.
- 50. Palaniappan RU, Chang YF, Chang CF, Pan MJ, Yang CW, Harpending P, et al. Evaluation of ligbased conventional and real time PCR for the detection of pathogenic leptospires. Mol Cell Probes. 2005;19(2):111-7.
- 51. Ferrari HF, Luvizotto MC, Rahal P, Cardoso TC. Detection of bovine Herpesvirus type 5 in formalinfixed, paraffin-embedded bovine brain by PCR: a useful adjunct to conventional tissue-based diagnostic test of bovine encephalitis. J Virol Methods. 2007;146(12):335-40.
- 52. Miyamoto CT, Gomes ML, Marangon AV, de Araújo SM, Bahia MT, Martins-Filho OA, et al. Usefulness of the polymerase chain reaction for monitoring cure of mice infected with different Trypanosoma cruzi clonal genotypes following treatment with benznidazole. Exp Parasitol. 2008;120(1):45-9.
- 53. Montoya A, Miró G, Mateo M, Ramírez C, Fuentes I. Detection of Toxoplasma gondii in cats by comparing bioassay in mice and polymerase chain reaction (PCR). Vet Parasitol. 2009;160(1-2):159-62.
- 54. Disch J, Caligiorne RB, Maciel F, Oliveira MC, Orsini M, Dias-Neto E, et al. Single-step duplex kDNA-PCR for detection of Leishmania donovani complex in human peripheral blood samples. Diagn Microbiol Infect Dis. 2006;56(4):395-400.
- 55. Marcussi VM, Marcussi LM, Barbosa-Tessmann IP, Lonardoni MV, Silveira TG. Leishmania (Viannia) braziliensis: new primers for identification using polymerase chain reaction. Exp Parasitol. 2008;120(4):300-5.
- 56. Gomes AH, Armelin IM, Menon SZ, Pereira-Chioccola VL. Leishmania (V.) braziliensis: detection by PCR in biopsies from patients with cutaneous leishmaniasis. Exp Parasitol. 2008;119(3):319-24.
Correspondence to:
Publication Dates
-
Publication in this collection
21 June 2012 -
Date of issue
2012
History
-
Received
17 Nov 2011 -
Accepted
24 Feb 2012