Abstract
Background:
The choice between heterologous expression versus chemical synthesis for synthesizing short cysteine-rich insecticidal peptides from arthropods may impact the obtainment of yields and well-folded bioactive molecules for scientific research. Therefore, two recombinant expression systems were compared to that of chemical synthesis for producing Ba1, a cysteine-rich spider neurotoxin.
Methods:
The transcription of the insecticidal neurotoxin Ba1 was obtained from a cDNA library of venom glands of the spider Brachypelma albiceps.It was cloned into the pCR®2.1-TOPO® cloning vector and then introduced in two different expression vectors, pQE40 and pET28a+. Each vector was transfected into E. coli M15 and BL21 cells, respectively, and expressed under induction with isopropyl thiogalactoside (IPTG). The chemical synthesis of Ba1 was performed in an Applied Biosystems 433A peptide synthesizer.
Results:
Both expression systems pQE40 and pET28a+ expressed the His-tagged recombinant protein products, HisrDFHRBa1 and HisrBa1, respectively, as inclusion bodies. The recombinant proteins HisrDFHRBa1 and HisrBa1 presented respective molecular masses of 28,289 and 8274.6 Da, and were not biologically active. These results suggested that both HisrDFHRBa1 and HisrBa1 were oxidized after cell extraction, and that their insecticidal activities were affected by their N-terminal pro-peptides and different disulfide bridge arrangements. The respective protein expression yields for HisrDFHRBa1 and HisrBa1 were 100 μg/L and 900 μg/L of culture medium. HisrBa1 was reduced and folded under in vitroconditions. The in vitro folding of HisrBa1 produced several isoforms, one of which, after removing its N-terminal pro-peptide by enzymatic cleavage, presented elevated insecticidal activities compared to the native Ba1. Furthermore, the His-tagged protein HisrDFHRBa1 underwent enzymatic cleavage to obtain recombinant Ba1 (rBa1). As expected, the molecular mass of rBa1 was 4406.4 Da. On the other hand, Ba1 was chemically synthesized (sBa1) with a yield of 11 mg per 0.1 mmol of amino acid assembly.
Conclusions:
The two recombinant insecticidal peptides and the one synthesized chemically were as active as the native Ba1; however, toxin yields differed drastically.
Keywords:
Insecticidal peptides; Protein expression; Bacteria; Theraphosid spider; Chemical synthesis; Cysteine-rich peptides
Background
Insecticidal spider peptides contain unique properties such as small size, resistance to proteolysis and strong affinities for cell receptors; therefore, they are essential tools for finding and studying new insecticidal receptors with the aim of designing novel and specific biopesticides or conducting structure-function studies [11. Corzo G, Escoubas P, Stankiewicz M, Pelhate M, Kristensen CP, Nakajima T Isolation, synthesis and pharmacological characterization of delta-palutoxins IT, novel insecticidal toxins from the spider Paracoelotes luctuosus (Amaurobiidae). Eur J Biochem. 2000;267(18):5783-95., 22. Corzo G, Escoubas P, Villegas E, Karbat I, Gordon D, Gurevitz M, et al. A spider toxin that induces a typical effect of scorpion alpha-toxins but competes with beta-toxins on binding to insect sodium channels. Biochemistry. 2005;44(5):1542-9.]. Ba1 is an insecticidal toxin obtained from the venom of the theraphosid spider Brachypelma albiceps. The primary structure of Ba1 comprises 39 residues and 6 half-cystines that form three disulfide bridges. The three dimensional structure of Ba1 was solved by NMR, and it contains three antiparallel β-sheets and belongs to the DDH family of spider neurotoxins [33. Corzo G, Bernard C, Clement H, Villegas E, Bosmans F, Tytgat J, et al. Insecticidal peptides from the theraposid spider Brachypelma albiceps: an NMR-based model of Ba2. Biochim Biophys Acta. 2009;1794(8):1190-6.]. Although the cell receptor of Ba1 has not been found, it is thought that its target is the voltage-gated sodium channel (Nav) of insects because of its rapid and strong insecticidal activity, which resembles that of the known Nav neurotoxins from the venom of spiders and scorpions [44. Cestèle S, Stankiewicz M, Mansuelle P, de Waard M, Dargent B, Gilles N, et al. Scorpion alpha-like toxins, toxic to both mammals and insects, differentially interact with receptor site 3 on voltage-gated sodium channels in mammals and insects. Eur J Neurosci. 1999;11(3):975–85., 55. Gordon D, Martin-Eauclaire MF, Cestèle S, Kopeyan C, Carlier E, Khalifa RB, et al. Scorpion toxins affecting sodium current inactivation bind to distinct homologous receptor sites on rat brain and insect sodium channels. J Biol Chem. 1996;271(14):8034–45.].
Since appreciable amounts of Ba1 are needed to continue the search for insect cell receptors and structure-function studies, and because its natural extraction is quite laborious and limited, the recombinant expression or chemical synthesis of Ba1 could provide an alternative for obtaining larger quantities of this insecticidal molecule for further biochemical studies. Herein, we report the production of a recombinant Ba1 using two expression systems as well as the production of Ba1 by chemical synthesis. The two expression systems selected used the plasmids pQE40 and pET28, which contain N-terminal propeptides of 190 and 22 residues, respectively. The two plasmids were se lected according to the manufacturer's recommendations and to scientific references for short peptide recombinant expression [66. Kumar GS, Sarma SP. Cloning, overexpression, folding and purification of a biosynthetically derived three disulfide scorpion toxin (BTK-2) from Mesobuthus tamulus. Protein Expr Purif. 2010;70(2):137–42.–99. Novagen: pET System Manual. 10th edition. http://richsingiser.com/4402/Novagen%20pET%20system%20manual.pdf. 2003.
http://richsingiser.com/4402/Novagen%20p...
]. Thus, we report the advantages and disadvantages of two expression systems as well as that of the chemical synthesis for obtaining active cysteine-rich spider peptides. This work may be of interest for researchers in the field that face the decision to synthesize, biologically or chemically, cysteine-rich peptide ligands from arthropods.
Methods
Bacterial strains, enzymes and plasmids
E. coli XL1 Blue was used for plasmid propagation. M15 and BL21 were used for the expression of the proteins. Plasmids pCR®2.1-TOPO® (Invitrogen), pQE40 (Qiagen) and pET28a+ (Novagen) were used for cloning and production of the proteins linked to a 6His-tag. Restriction enzymes, Taq polymerase, factor Xa and T4 DNA ligase were purchased from New England Biolabs.
Gene cloning
Based on the information obtained from direct peptide sequencing of Ba1, a specific oligonucleotide was designed and used for the PCR reaction employing as a template the cDNA material obtained previously from a venom gland cDNA library. The PCR reaction was performed in Taq DNA polymerase buffer, 200 μM dNTPs, 0.25 μM forward primer (5′-ATTCTCGAATGTGTTTTCTCATG-3′) 0.25 μM; reverse CDS3'primer (5′-AATCTTCAGACACAACTTGATC-3′) and two units of Taq DNA polymer-ase in a final volume of 50 μL in a Perkin Elmer 9600 in strument. The reaction was incubated at 94 °C for three minutes and, after the initial cycle, the mixture was incubated at 94 °C for 30 s, 60 °C for one minute and 72 °C for one minute for 30 cycles, followed by a final seven-minute step at 72 °C. PCR products were purified using the High Pure Plasmid Isolation Kit (Roche) following the manufacturer's instructions, and then ligated into a pCR®2.1-TOPO® + EcoR1-digested plasmid. The ligation reaction was used to transform competent E. coli XL1 Blue cells. Positive clones were sequenced from both ends using the Thermo sequence radiolabeled terminator cycle sequencing kit (Amersham).
Plasmid construction
The DNA fragment encoding the Ba1 sequence, preceded by factor Xa recognition site was amplified by PCR from a cDNA clone obtained from the library previously described.
Appropriate oligonucleotides were designed for introdu cing the enzymatic restriction sites Bgl II and PstI for direc tional insertion into pQE40. The PCR-amplified product was cloned into pCR®2.1-TOPO®, digested with Eco RI and subcloned using standard methods. The PCR amplifica tions were carried out using the following oligonucleotides: Ba1Bgl corresponds to 5′-CGCAGATCTATCGAGGGAAGGATTCTCGAATGTGTTTTCTCATG-3′, and Ba1P: 5′-CGCCTGCAGTTACTAAATCTTCAGACACAACTTGATC-3′. The PCR conditions were: three minutes at 94 °C, 30 cycles for 30 s at 94 °C, one minute at 60 °C, one minute at 72 °C, and a final extension of seven minutes at 72 °C. The PCR products and the corresponding plasmids were digested with the corresponding restriction enzymes at 37 °C overnight. PCR products were purified using the High Pure Plasmid Isolation Kit (Roche) before ligation. The ligation reaction (20 μL) was carried out with T4 DNA ligase with a 10-fold insert excess over plasmid for 16 h at 16 °C; ten microliters of the ligation reaction were used to transfect competent E. coliXL1 Blue cells. Positive clones with the expected insert were grown in LB ampi-cillin medium. The plasmids of positive colonies were puri fied by means of the High Pure Plasmid Isolation Kit (Roche). Plasmid constructs were verified by sequencing from both sites, the insert boundaries to confirm the reading frame and conservation of restriction sites. M15 strains were transfected with the corresponding plasmid for 30 s at 42 °C, followed by five minutes in ice and 30-min recov ery at 37 °C in LB medium. Plates of LB contained 100 μg/mL of ampicillin and 30 μg/mL of kanamycin.
Similarly, appropriate oligonucleotides were designed for introducing the enzymatic restriction sites BamH1 and Sal1 for directional insertion into pET28a+. The PCR-amplified product was cloned into pCR®2.1-TOPO®, digested with Eco RI and subcloned using standard methods. The PCR amplifications were carried out using the following oligonucleotides: Ba1Bam corresponds to 5′-GGATCCATCGAGGGAAGGATTCTCGAATGTGTTTTCTCATGC-3′, and Ba1S to 5′-GTCGACTTACTAAATCTTCAGACACAACTTGATC-3′. The PCR conditions were: three minutes at 94 °C, 30 cycles for 30 s at 94 °C, one minute at 60 °C, one minute at 72 °C, and a final extension of seven minutes at 72 °C. The PCR products and the corresponding plasmids were digested with the corresponding restriction enzymes at 37 °C overnight. PCR products were purified using the High Pure Plasmid Isolation kit (Roche) before ligation. The ligation reaction (20 μL) was carried out via T4 DNA ligase with a 10-fold insert excess over plasmid for 16 h at 16 °C; ten microliters of the ligation reaction was used to transfect competent E. coli XL1 Blue cells. Positive clones with the expected insert were grown in LB ampicillin medium. The plasmids of positive colonies were purified by means of the same plasmid isolation kit (Roche). Plasmid constructs were verified by sequencing from both sites, the insert boundaries to confirm the reading frame and conservation of restriction sites. BL21 (DE3) strains were transfected with the corresponding plasmid for 30 s at 42 °C, followed by five minutes in ice and 30-min recovery at 37 °C in LB medium. Plates of LB contained 30 μg/mL of kanamycin.
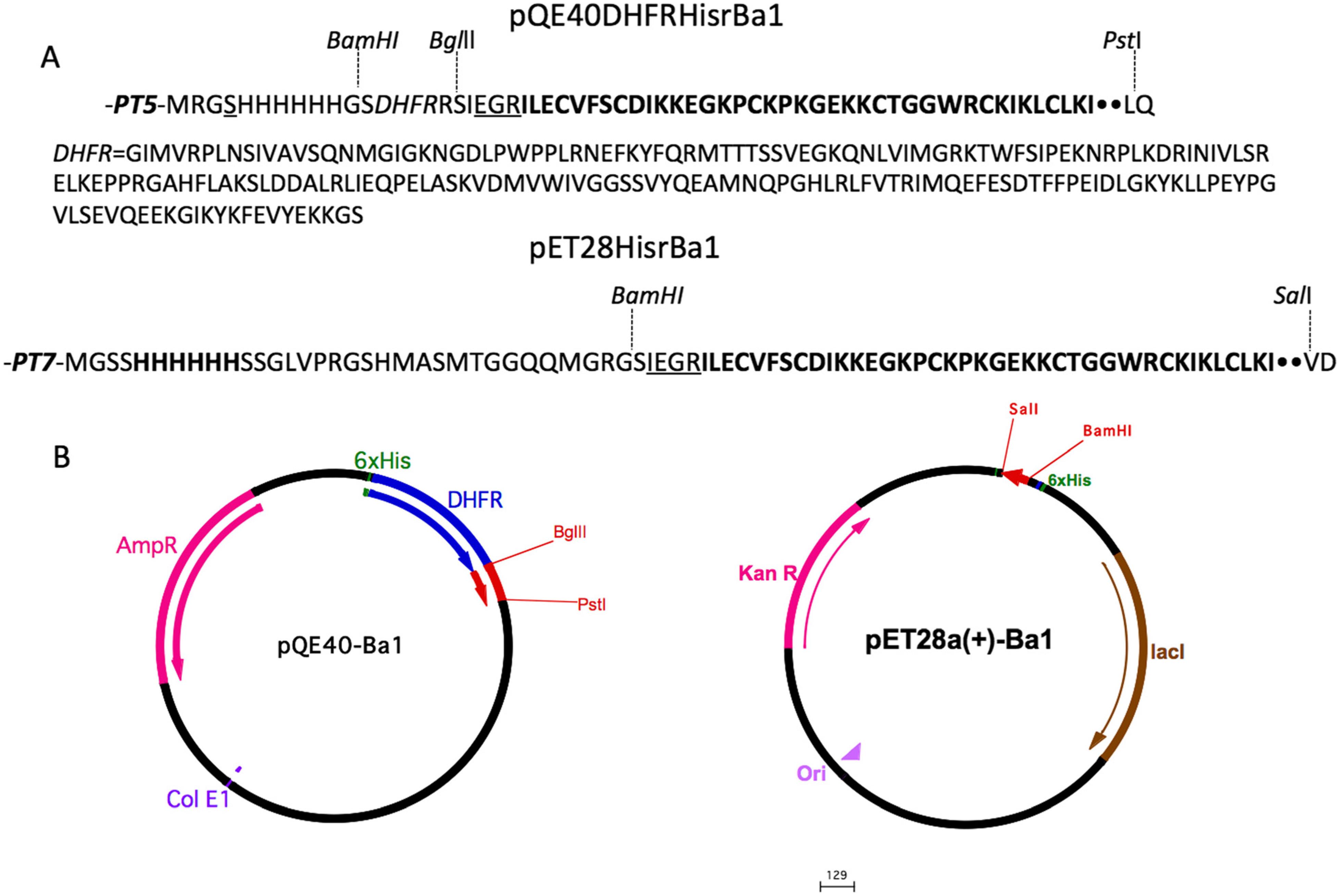
Thus, the plasmids essentially contained a pro-peptide, a 6His-tag, a sequence coding for the amino acids recognized by the protease (factor Xa) and the gene of Ba1. The constructions were named pQE40HisrDHFRBa1 and pET28HisrBa1, and its products were abbreviated HisrDFHRBa1 and HisrBa1, respectively (Fig. 1).
Gene construction of the plasmids carrying either pQE40HisrDHFRBa1 or pET28HisrBa1. a The organization of the gene and amino acid sequences in pQE40HisrDHFRBa1 and pET28HisrBa1. b Schematic representation of the expression vectors
Production and purification of HisrDFHRBa1 and HisrBa1
The E. coli strains M15 and BL21 (DE3), cells expressing the respective plasmids pQE40HisrDHFRBa1 and pET28 HisrBa1, were grown in LB medium. After the absorbance at 600 nm reached 0.6 absorption units, the cultures were induced with 0.1 mM IPTG (isopropyl-β-D-thiogalactopyranoside) for 14 h at 21 °C. Cells were harvested by centrifugation (9820 × g for 20 min) using a Beckman centrifuge, model JA-14, recovered in washing buffer (50 mM Tris–HCl, pH 8.0) and lysed with a BugBuster® protein extraction reagent (Novagen, Germany). This material was centrifuged again (17,400 × g for 20 min) and the supernatant was recovered. Moreover, the insoluble fraction was resuspended twice with the washing buffer and centrifuged under the same conditions. The recombinant toxin was extracted from the soluble part and from the inclusion bodies using 6 M guanidinium chloride (GndHCl) in a 50 mM Tris-base, buffer, pH 8.0 and centrifuged for 20 min in a Beckman centrifuge, model JA-20 (17,400 × g for 20 min). The supernatant was purified by affinity column. Purification of the recombinant Ba1 by Ni-NTA (Ni-nitrilotriacetic acid) affinity column chromatography was performed according to the instructions of the manufacturer (Qiagen, USA) using denaturing conditions with buffer A (6 M GndHCl in a 50 mM Tris-base buffer, pH 8.0) and buffer B (6 M GndHCl in 50 mM Tris-base buffer, containing 400 mM imidazole, pH 8.0). Buffer B was eliminated by a second purification step under a high-performance liquid chromatography system (Agilent, series 1100), equipped with a binary pump and diode array detector systems. An analytic C18reverse-phase column (Vydac 218 TP54) was run from solvent A (0.1 % trifluoroacetic acid – TFA in water) to solvent B (0.1 % TFA in acetonitrile) at 1 mL/min flow rate. The absorbance was registered at 280 and 230 nm, and the fractions were manually collected and freeze-dried. The HPLC system previously described [1010. Rodríguez A, Villegas E, Satake H, Possani LD, Corzo G. Amino acid substitutions in an alpha-helical antimicrobial arachnid peptide affect its chemical properties and biological activity towards pathogenic bacteria but improves its therapeutic index. Amino Acids. 2011;40(1):61–88.] was used for this separation, and the gradient varied according to type of protein to purified. For instance, for HisrD FHRBa1 it was run from 0-60 % solvent B, during 60 min, and for HisrBa1 it was run from 20 % to 70 % solvent B, for 50 min.
In vitro folding conditions and enzymatic cleavage
When needed, the recombinant proteins, HisrDFHRBa1 and HisrBa1, were vacuum dried and the cystines reduced using 50 mM DTT in 50 mM Tris-base buffer, pH 8.0. The folding conditions were 2 M GndHCl in 200 mM Tris-base buffer, pH 8.0, containing 1 mM reduced glutathione (GSH)/0.1 mM oxidized glutathione (GSSG). The folding reaction was carried out at room temperature for 18 h, after which the products were separated by rpHPLC. The folded recombinant products, HisrDFHRBa1 and HisrBa1, were cleaved by FXa using a ratio 50:1 in 20 mM Tris-base buffer, pH 8.0, at 37 °C for five hours, and the recombinant Ba1 (rBa1) eluted from a reverse-phase column, vacuum dried and stored at-20 °C. The identity of the rBa1 was confirmed by both automatic Edman degradation and mass spectrometry analysis using a Finnigan LCQDUOion trap mass spectrometer (San Jose, USA). These techniques are currently employed in our laboratory and have been reported elsewhere [1111. Estrada G, Garcia BI, Schiavon E, Ortiz E, Cestele S, Wanke E, et al. Four disulfide-bridged scorpion beta neurotoxin CssII: heterologous expression and proper folding in vitro. Biochim Biophys Acta. 2007;1770(8):1161–8.].
Ba1 chemical synthesis
Ba1 was chemically synthesized by a solid-phase method using the Fmoc methodology on an Applied Biosystems 433A peptide synthesizer. Fmoc-Ile-Wang resin (ChemPep Inc.) was used to provide the first residue with a free carboxyl at the C-terminal of synthetic sBa1 (sBa1). Chemical synthesis, cleavage and deprotection of peptide resins were performed as previously described [22. Corzo G, Escoubas P, Villegas E, Karbat I, Gordon D, Gurevitz M, et al. A spider toxin that induces a typical effect of scorpion alpha-toxins but competes with beta-toxins on binding to insect sodium channels. Biochemistry. 2005;44(5):1542-9.]. The crude synthetic peptide was dissolved in 20 % aqueous acetonitrile, and the reduced insecticidal toxin separated by rpHPLC on a semi-preparative C18 column (Vydac 218 TP54) using a gradient from solvent A (0.1 % TFA in water) to solvent B (0.1 % TFA in acetonitrile) at 2 mL/min flow rate. The six free cysteine residues were allowed to oxidize by air exposure for 18 h at 5 °C in a 2 M aqueous ammonium acetate solution containing 1 mM reduced glutathione/0.1 mM oxidized glutathione. The biologically active synthetic toxin was purified on an analytical C18rpHPLC column using a gradient from solvent A (0.1 % TFA in water) to solvent B (0.1 % TFA in acetonitrile) at 1 mL/min flow rate. The structural identity between synthetic and natural peptides was verified by co-elution experiments and by mass spectrometry.
Biological activity
The protocol employed for assaying the in vivo activity of HisrDFHRBa1, HisrBa1, rBa1 (pQE40), Ba1 (pET28) and sBa1 utilized house crickets (Acheta domesticus). Crickets were injected intrathoracically between the second and third pair of legs, with 5 μL of five different doses of each toxin previously dissolved in distillated water. Groups of five individuals were injected with at least an equivalent to produce the 100 % mortality of the house crickets based on the native Ba1.
Committee approval
The present study was approved by the Committee of Animal Welfare of the Institute of Biotechnology, UNAM. The number of animals was kept to a minimum required to validate the experiments.
Results and discussion
Gene cloning and recombinant expression
As described in the Methods section, a gene coding for the Ba1 toxin was obtained from a cDNA library. The PCR products with the expected size (~164 bp) were purified using the High Pure Plasmid Isolation Kit (Roche), cloned into pCR®2.1-TOPO® (Invitrogen) and its nucleotide sequence was verified. The Ba1 gene was cloned in two plasmids harboring long pro-peptide structures to facilitate the expression of the recombinant proteins. In the expression vector pQE40 that coded for 247 residues (four for initial protein translation – MRGS, six for the His-tag, 192 for the dihydrofolate reductase – DHFR, two for the restriction site, four for the Factor Xa proteolytic cleavage region and 39 for the mature Ba1 peptide, pQE40HisrDHFRBa1), and in the expression vector pET28a+ that coded for 77 residues (four for initial protein translation – MRGS, six for the His-tag, 24 extra residues, four for the Factor Xa proteo-lytic cleavage region before the 39 residues of the mature Ba1 peptide – pET28HisrBa1). In brief, the gene product of the plasmid pQE40 encloses a 26 kDa DHFR pro-peptide (HisrDHFRBa1), while the gene product of the plasmid pET28 encloses a 8 kDa pro-peptide (HisrBa1) (Fig. 1).
Expression, purification, in vitro folding and enzymatic cleavage
The E. coli strains M15 and BL21 were transformed, respectively, with plasmids pQE40HisrDHFRBa1 and pET28HisrBa1. Cells of either M15 or BL21 obtained after induction with IPTG were decanted by centrifugation, resuspended and ruptured with the BugBuster® protein extraction reagent as described in the Methods section. After solubilization with GndHCl the supernatant was purified by affinity column. Figure 2 shows the SDS-PAGE of the expressed proteins obtained in the presence or absence of IPTG, and before and after purification by affinity column. For protein expression in M15 cells, lane 3 clearly shows the presence of an intense protein band (indicated by an arrow) with the expected molecular weight for the protein HisrDHFRBa1, which is more intense in lane 5 (cell inclusion bodies); lanes 6 and 7 show the result of the purification of HisrDHFRBa1 using agarose nickel affinity columns, where a minor increase in purity was observed (Fig. 2a). For protein expression in BL21 cells, lanes 2 to 7 shows almost imperceptible amounts of HisrBa1 (indicated by an arrow), but lanes 8 and 9 show an intense band of the histidine-tagged HisrBa1 (8 kDa) purified using nickel affinity agarose columns (Fig. 2b). Although an intense band of a 25 kDa protein was observed in lanes 8 and 9, this protein was not observed during purification by rpHPLC. Therefore, it was assumed that HisrBa1 might form a tetramer under SDS-PAGE conditions.
SDS-PAGE showing the expression of pQE40HisrDHFRBa1 or pET28HisrBa1. a Gel of pQE40HisrDHFRBa1 expression. Left lane shows the molecular weight markers (MWM). Lane 1, cells with vector pQE40; lane 2, cells without IPTG induction; lane 3, cells with IPTG induction; lane 4, supernatant; lane 5, inclusion bodies; lanes 6 and 7, DHFR-Ba after affinity chromatography. b Gel of pET28HisrBa1 expression. Left lane shows the molecular weight markers (MWM). Lane 1, cells without IPTG induction; lane 2, cells with IPTG induction; lane 3, supernatant; lane 4, inclusion bodies; lanes 5, first affinity chromatography elution; lanes 6 and 7, first and second wash with 20 mM Imidazol; l 8 and 9, HisrBa1 after affinity chromatography. The protein content applied in each lane was between 5 and 10 μg
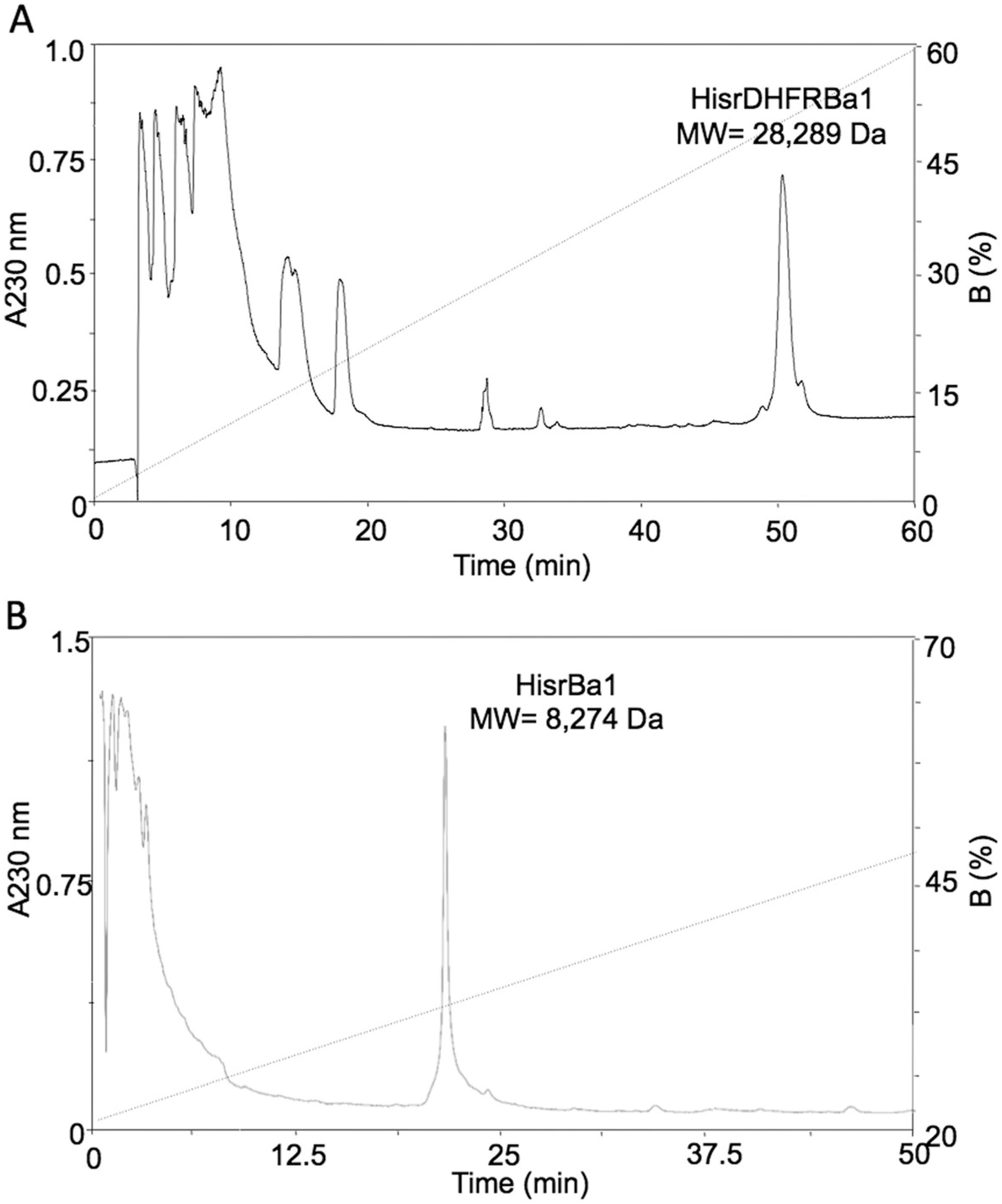
Imidazole eluates from the affinity chromatography columns were directly loaded into a C18 column by rpHPLC separation (Fig. 3). The rpHPLC profile of the separation of HisrDHFRBa1 shows a single broad fraction with a molecular mass of 28,289.0 Da (Fig. 3a). Although the expected theoretical molecular mass was 28,274.0 Da, the chromatographic peak for this fraction showed 15 amu higher than the expected, which could be the consequence of the oxidation of one of its seven methionines to methionine sulfoxide (Δ15.9 amu). On the other hand, the rpHPLC profile of HisrBa1 separation shows a single sharp peak (Fig. 3b) with an unexpected experimental molecular mass of 8274.8 Da that represents a difference of 131 Da below the expected theoretical mass of 8405.8 Da. The difference of 131 Da represents the molecular weight of a methionine, which has been reported to undergo a post-translational processing in E. coli when the second residue of a nascent peptide is a short one [1212. Ben-Bassat A, Bauer K, Chang SY, Myambo K, Boosman A, Chang S. Processing of the initiation methionine from proteins: properties of the Escherichia coli methionine aminopeptidase and its gene structure. J Bacteriol. 1987;169(2):751–7.]. In this case, the second residue of HisrBa1 is glycine (see Fig. 1). Therefore, the experimental mass found of 8274.8 Da corroborates this post-translational processing. The yields of the recombinant insecticidal toxins HisrDHFRBa1 and HisrBa1, after rpHPLC purification, starting with 1 L of cell culture, were 100 μg/L and 900 μg/L, respectively.
rpHPLC separation of oxidized pQE40HisrDHFRBa1 or pET28HisrBa1 from affinity chromatography. a The fractions from NiNta were separated employing rpHPLC (C18 column) using a gradient from 0 to 60 % B in 60 min at 1 mL/min. b The fractions from NiNta were separated using rpHPLC (C18 column) using a gradient from 0 to 50 % B in 50 min at 1 mL/min
The purified protein fractions HisrDHFRBa1 and HisrBa1 were vacuum dried, resuspended and injected into house crickets, but they were not lethal to the insects (Table 1). These results suggested missing folded proteins and prompted us to conduct in vitro reduction-oxidation steps for each protein; therefore, they were reduced and folded under in vitro conditions in the presence of reduced and oxidized glutathione as a redox pair to improve the correct folding of the cystines [1313. Estrada G, Restano-Cassulini R, Ortiz E, Possani LD, Corzo G. Addition of positive charges at the C-terminal peptide region of CssII, a mammalian scorpion peptide toxin, improves its affinity for sodium channels Nav1.6. Peptides. 2011;32(1):75–9.].
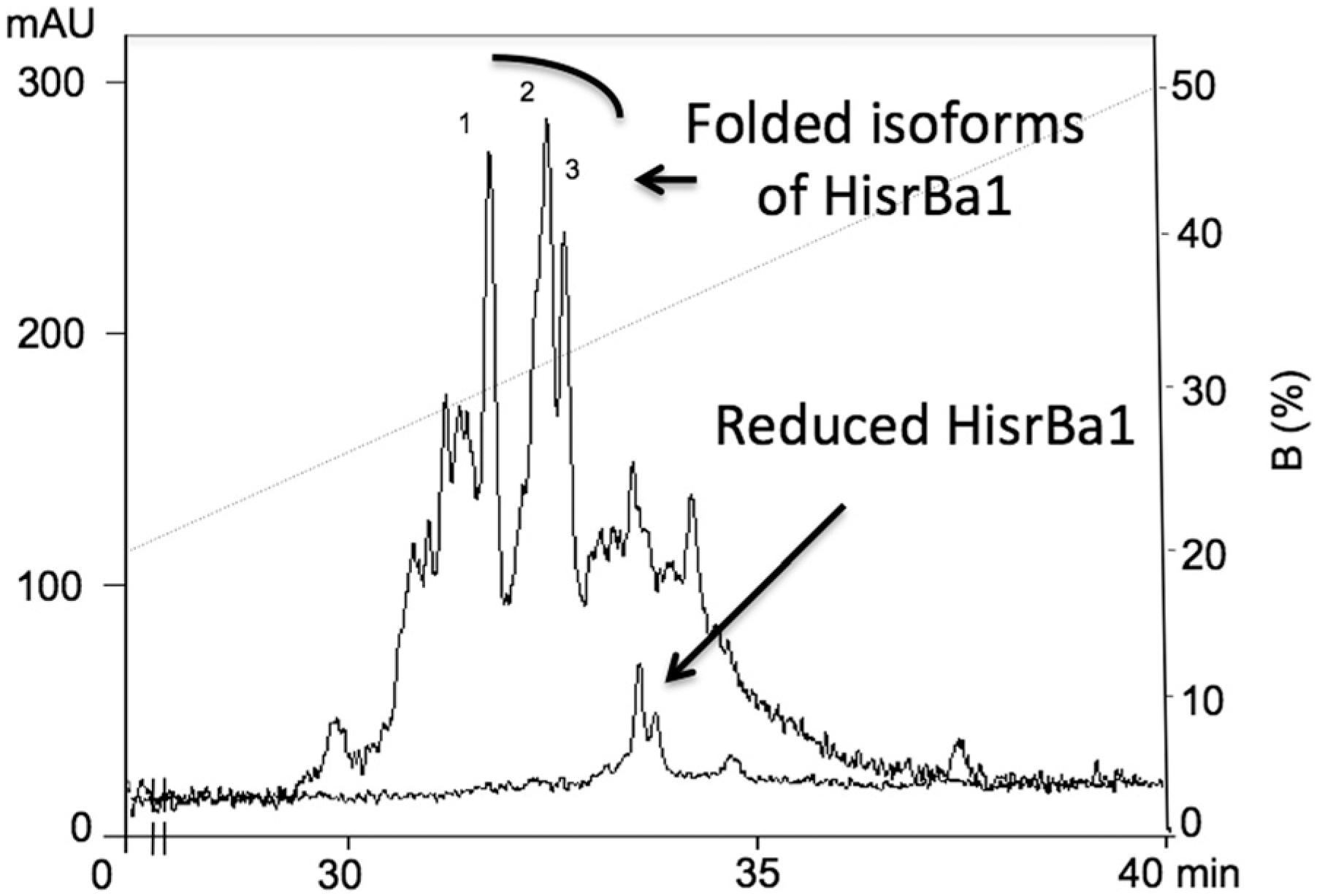
The rpHPLC profile of the separation of HisrDHFRBa1 after folding conditions also showed a single broad fraction without insecticidal activity. However, the rpHPLC profile of the separation of HisrBa1 displayed three main chromatographic peaks (Fig. 4), labeled from 1 to 3, with the expected molecular mass of 8274.8 Da. Thus, three main isoforms from HisrBa1 were generated during oxi dation of the thiol groups for formation of the disulfide bridges; here only the chromatographic peak number 1 produced paralytic activity when injected in house crickets (Table 1). In Fig. 4, we have superimposed both the results of the rpHPLC separation of the multiple oxidized forms and the single reduced form of HisrBa1, later eluted in this chromatogram.
rpHPLC separation of the in vitro folding of HisrBa1. The components of the in vitro folding reaction of HisrBa1 were separated employing rpHPLC (C18 column) using a gradient from 0 to 50 % B in 50 min at 1 mL/min. A superimposed chromatographic rpHPLC profile under the same elution conditions shows the retention time of the reduced HisrBa1
Since HisrDHFRBa1, after folding conditions, was eluted in a broad single peak as a high-molecular-weight protein, it was difficult to determine whether it contained multiple oxidized forms as HisrBa1. To prove the existence of multiple forms in HisrDHFRBa1 another type of protein separation may be used. Because the in vitro folded HisrDHFRBa1 and HisrBa1 were not fully active on insects, the enzymatic cleavage with FXa was conducted to release the mature peptide Ba1 from their N-terminal propeptide.
Figure 5 shows the enzymatic cleavage of HisrDHFRBa1 and HisrBa1. Figure 5a shows a multiple unspecific cleav age of HisrDHFRBa1 at different incubation times, and rBa1, from HisrDHFRBa1, which was obtained in small quantities. On the other hand, the digestion of HisrBa1 was straightforward but rBa1 was also found in minute amounts (Fig. 5b).
rpHPLC separation of rBa1 from HisrDHFRBa1 and HisrBa1 using FXa. (a) Enzymatic hydrolysis of HisrDHFRBa1 using FXa where rBa1 (asterisk) was separated by rpHPLC (C18 column) with a gradient from 10 to 60 % B in 50 min at 1 mL/min. (b) Enzymatic hydrolysis of HisrBa1 using FXa, rBa1 was separated by rpHPLC (C18 column) with a gradient from 0 to 60 % B in 60 min at 1 mL/min. A superimposed chromatographic rpHPLC profile under the same elution conditions shows the retention time of the nBa1. The fact that rBa1 and nBa1 eluted at the same retention time suggests similar secondary and tertiary structures
Chemical synthesis of Ba1
Ba1 was synthesized in its free carboxyl-terminus form. The overall assembly and cleavage yield of Ba1 was 55 % according to theoretical values of the peptidyl resin and the calculated mass increase for 0.1 mmol peptide to produce 242 mg of crude synthetic peptide. The yield after chromatographic purification of the linear peptide was 31.4 mg (7.1 %) and after refolding was 11 mg (2.5 %). Because of the hydrophobic exposure in an unstructured peptide, the retention time of the unfolded sBa1 was larger than that of the folded sBa1 (Fig. 6). The structural identity between the synthetic peptide sBa1 and the native one was verified by mass spectrometry and in co-injection experiments, native Ba1 (nBa1) and synthetic Ba1 (sBa1) co-eluted in a single peak, indicating that they have similar retention times and suggesting a similar secondary and 3D structure (data not shown).
Chromatographic purification of the chemically synthesized Ba1. rpHPLC profile of Ba1 before and after folding experiments. Separation was performed using a gradient from 0 to 60 % B in 60 min at 1 mL/min. The dashed line represents the reduced form of Ba1, which eluted later and it is much more hydrophobic than the folded sBa1
Biological activity
The well-folded recombinant and chemically synthesized insecticidal peptides were active against house crickets (Acheta domesticus). Table 1 shows the biological activity of the recombinant and chemically synthetic Ba1, which were tested in groups of five crickets. Although the LD50 of nBa1 was calculated in 10.8 μg/g of cricket, a dose of 12.1 μg/g of cricket was enough to kill all of them. As mentioned, the uncleaved recombinant HisrDHFRBa1 and HisrBa1 were not biological active, a result that may be attributable to the large amount of extra N-terminal residues for Ba1 in HisrDHFRBa1 (84 %) and in HisrBa1 (46.7 %), which may have reduced the molecular diffusion and molecular recognition towards the insect cellular target of Ba1.
Conclusions
Synthesis of cysteinerich peptides by chemical means has recently attracted more interest, as they allow the production of wholly biologically active molecules by either combinations of solid-phase peptide synthesis or native chemical ligation [1414. Yamaji N, Little MJ, Nishio H, Billen B, Villegas E, Nishiuchi Y, et al. Synthesis, solution structure, and phylum selectivity of a spider delta-toxin that slows inactivation of specific voltage-gated sodium channel subtypes. J Biol Chem. 2009;284(36):24568–82.]. However, because of the great amount of β-sheets, and paucity of random and α-helix structures in this molecule type, only peptides containing less than 50 amino acids can be reliably prepared with acceptable yields and purity. Hence, a strategy of peptide segment condensation could be used in which peptide fragments are subjected to chemical ligation methods. A drawback for chemical synthesis could be proteins that require post-translational and conformational modifications in order to render biological activity. Therefore, oxidative folding has to be performed usually in a one-step oxidative folding procedure in the presence of a redox pair that is sufficient for spontaneous peptide folding. Consequently, chemical synthesis could be a less expensive and time-consuming procedure as compared to the heterologous expression in spite of the high cost of chemical reagents.
On the other hand, heterologous expression of cysteine-rich peptides is currently a widely used technique and diverse expression systems have been reported. Protein expression in E. coli is relatively simple and less expensive compared to yeast or eukaryotic systems; however, a major drawback is that the recombinant protein often needs to be fused to a carrier protein to increase its solubility and thus avoid formation of inclusion bodies [1515. Vriens K, Cammue BP, Thevissen K. Antifungal plant defensins: mechanisms of action and production. Molecules. 2014;19(8):12280–303.]. This fusion protein needs to be released during or after purification of the protein of interest via enzymatic or chemical cleavage to render functional proteins, which results in a decreased yield as we have observed (see Table 1). In addition, fusion proteins are not necessarily properly folded and production of cysteine-rich peptides could be inactive as we have shown during the expression of HisrDHFRBa1.
In this work, both heterologous expression systems pQE40HisrDHFRBa and pET28HisrBa1 produced fully active toxins but with low yields in relation to time and material efforts. The active fraction rBa1 represented only the 15.6 % of effective peptide in the recombinant HisrDHFRBa1 protein, and it represents 53.3 % in the recombinant HisrBa1 peptide. In this report, the less exigent synthetic process, in time and materials, was the chemical synthesis of Ba1. Although the chemical synthesis of cysteine-rich peptides required an in vitro folding step, it was the same for the recombinant HisrBa1, and perhaps for the HisrDHFRBa1, which may be produced in multiple oxidized forms; so for all of them a folding step may be needed. An alternative to the recombinant efficacy of heterologous expression systems of short cysteine-rich peptides might be recombinant expression in a soluble form with shorter N-terminal pro-peptides, which could avoid folding steps and increase the yield for effective and biological active cysteine-rich neurotoxin peptides. In this work, we compare the biological and chemical synthesis of an insecticidal peptide from spider venom; this comparison may be useful when synthesizing similar cysteine-rich peptides from arthropods.
Abbreviations
TFA: Trifluoroacetic acid; rpHPLC: Reverse-phase HPLC; Ba1: Toxin 1 from the spider Brachypelma albiceps.
Acknowledgments
This work was financed by grants from Dirección General de Asuntos del Personal Académico (DGAPA-UNAM) number IN204415 and from SEP-CONACyT number 240616 to GC. The authors are very grateful for the confirmation of the molecular masses of recombinant Ba1 and laboratory technical assistance by Dr. Fernando Zamudio and Eduardo Abarca, respectively.
References
-
1Corzo G, Escoubas P, Stankiewicz M, Pelhate M, Kristensen CP, Nakajima T Isolation, synthesis and pharmacological characterization of delta-palutoxins IT, novel insecticidal toxins from the spider Paracoelotes luctuosus (Amaurobiidae). Eur J Biochem. 2000;267(18):5783-95.
-
2Corzo G, Escoubas P, Villegas E, Karbat I, Gordon D, Gurevitz M, et al. A spider toxin that induces a typical effect of scorpion alpha-toxins but competes with beta-toxins on binding to insect sodium channels. Biochemistry. 2005;44(5):1542-9.
-
3Corzo G, Bernard C, Clement H, Villegas E, Bosmans F, Tytgat J, et al. Insecticidal peptides from the theraposid spider Brachypelma albiceps: an NMR-based model of Ba2. Biochim Biophys Acta. 2009;1794(8):1190-6.
-
4Cestèle S, Stankiewicz M, Mansuelle P, de Waard M, Dargent B, Gilles N, et al. Scorpion alpha-like toxins, toxic to both mammals and insects, differentially interact with receptor site 3 on voltage-gated sodium channels in mammals and insects. Eur J Neurosci. 1999;11(3):975–85.
-
5Gordon D, Martin-Eauclaire MF, Cestèle S, Kopeyan C, Carlier E, Khalifa RB, et al. Scorpion toxins affecting sodium current inactivation bind to distinct homologous receptor sites on rat brain and insect sodium channels. J Biol Chem. 1996;271(14):8034–45.
-
6Kumar GS, Sarma SP. Cloning, overexpression, folding and purification of a biosynthetically derived three disulfide scorpion toxin (BTK-2) from Mesobuthus tamulus. Protein Expr Purif. 2010;70(2):137–42.
-
7Turkov M, Rashi S, Noam Z, Gordon D, Khalifa RB, Stankiewicz M, et al. In vitro folding and functional analysis of an anti-insect selective scorpion depressant neurotoxin produced in Escherichia coli Protein Expr Purif. 1997;10(1):123–31.
-
8QIAGEN. The QIAexpressionist. A handbook for high-level expression and purification of 6xHis-tagged proteins. 15th ed. Australia: QIAGEN; 2003.
-
9Novagen: pET System Manual. 10th edition. http://richsingiser.com/4402/Novagen%20pET%20system%20manual.pdf. 2003.
» http://richsingiser.com/4402/Novagen%20pET%20system%20manual.pdf -
10Rodríguez A, Villegas E, Satake H, Possani LD, Corzo G. Amino acid substitutions in an alpha-helical antimicrobial arachnid peptide affect its chemical properties and biological activity towards pathogenic bacteria but improves its therapeutic index. Amino Acids. 2011;40(1):61–88.
-
11Estrada G, Garcia BI, Schiavon E, Ortiz E, Cestele S, Wanke E, et al. Four disulfide-bridged scorpion beta neurotoxin CssII: heterologous expression and proper folding in vitro Biochim Biophys Acta. 2007;1770(8):1161–8.
-
12Ben-Bassat A, Bauer K, Chang SY, Myambo K, Boosman A, Chang S. Processing of the initiation methionine from proteins: properties of the Escherichia coli methionine aminopeptidase and its gene structure. J Bacteriol. 1987;169(2):751–7.
-
13Estrada G, Restano-Cassulini R, Ortiz E, Possani LD, Corzo G. Addition of positive charges at the C-terminal peptide region of CssII, a mammalian scorpion peptide toxin, improves its affinity for sodium channels Nav1.6. Peptides. 2011;32(1):75–9.
-
14Yamaji N, Little MJ, Nishio H, Billen B, Villegas E, Nishiuchi Y, et al. Synthesis, solution structure, and phylum selectivity of a spider delta-toxin that slows inactivation of specific voltage-gated sodium channel subtypes. J Biol Chem. 2009;284(36):24568–82.
-
15Vriens K, Cammue BP, Thevissen K. Antifungal plant defensins: mechanisms of action and production. Molecules. 2014;19(8):12280–303.
Publication Dates
-
Publication in this collection
2015
History
-
Received
12 Dec 2014 -
Accepted
18 May 2015