Resumos
Relatar um caso de sobrecarga de ferro secundária à xerocitose, uma doença rara, em uma adolescente, diagnosticada por meio de ressonância magnética em T2*. Relatamos o caso de uma paciente sintomática com xerocitose, nível de ferritina de 350ng/mL e sobrecarga de ferro cardíaca significativa. Ela foi diagnosticada por ressonância magnética em T2* e recebeu terapia de quelação. Análise por ectacitometria confirmou o diagnóstico de xerocitose hereditária. Na sequência, a ressonância magnética em T2* demonstrou resolução completa da sobrecarga de ferro em vários órgãos e novo ecocardiograma revelou resolução completa das alterações cardíacas anteriores. A paciente permanece em terapia de quelação. Xerocitose é uma desordem genética autossômica dominante rara, caracterizada por estomatocitose desidratada. O paciente pode apresentar fadiga intensa e sobrecarga de ferro. Sugerimos o uso regular de ressonância magnética em T2* para o diagnóstico e controle da resposta à quelação de ferro em xerocitose e acreditamos que o exame pode ser útil também em outras anemias hemolíticas que necessitam de transfusões.
Anemia; Sobrecarga de ferro; Imagem por ressonância magnética; Terapia por quelação; Relatos de casos
To report a case of iron overload secondary to xerocytosis, a rare disease in a teenager, diagnosed, by T2* magnetic resonance imaging. We report the case of a symptomatic patient with xerocytosis, a ferritin level of 350ng/mL and a significant cardiac iron overload. She was diagnosed by T2* magnetic resonance imaging and received chelation therapy Ektacytometric analysis confirmed the diagnosis of hereditary xerocytosis. Subsequent T2* magnetic resonance imaging demonstrated complete resolution of the iron overload in various organs, as a new echocardiography revealed a complete resolution of previous cardiac alterations. The patient remains in chelation therapy. Xerocytosis is a rare autosomal dominant genetic disorder characterized by dehydrated stomatocytosis. The patient may present with intense fatigue and iron overload. We suggest the regular use of T2* magnetic resonance imaging for the diagnosis and control of the response to iron chelation in xerocytosis, and we believe it can be used also in other hemolytic anemia requiring transfusions.
Anemia; Iron overload; Magnetic resonance imaging; Chelation therapy; Case reports
RELATO DE CASO
Sobrecarga de ferro em adolescente com xerocitose: a importância da ressonância nuclear magnética
Reijâne Alves de AssisI; Carolina KassabI; Fernanda Salles SeguroI; Fernando Ferreira CostaII; Paulo Augusto Achucarro SilveiraI; John WoodIII; Nelson HamerschlakI
IHospital Israelita Albert Einstein, São Paulo, SP, Brasil
IIUniversidade Estadual de Campinas, Campinas, SP, Brasil
IIIUniversity of Southern California, Califórnia, Estados Unidos
Endereço para correspondência Endereço para correspondência: Nelson Hamerschlak Hospital Israelita Albert Einstein Centro de Pesquisa Clínica do Instituto Israelita Albert Einstein Avenida Albert Einstein, 627/701 - Morumbi CEP: 05652-900 - São Paulo, SP - Brasil Tel.: (11) 3773-6590 e 2151-3203 - Fax: (11) 2151-3522 E-mail: hamer@einstein.br
RESUMO
Relatar um caso de sobrecarga de ferro secundária à xerocitose, uma doença rara, em uma adolescente, diagnosticada por meio de ressonância magnética em T2*. Relatamos o caso de uma paciente sintomática com xerocitose, nível de ferritina de 350ng/mL e sobrecarga de ferro cardíaca significativa. Ela foi diagnosticada por ressonância magnética em T2* e recebeu terapia de quelação. Análise por ectacitometria confirmou o diagnóstico de xerocitose hereditária. Na sequência, a ressonância magnética em T2* demonstrou resolução completa da sobrecarga de ferro em vários órgãos e novo ecocardiograma revelou resolução completa das alterações cardíacas anteriores. A paciente permanece em terapia de quelação. Xerocitose é uma desordem genética autossômica dominante rara, caracterizada por estomatocitose desidratada. O paciente pode apresentar fadiga intensa e sobrecarga de ferro. Sugerimos o uso regular de ressonância magnética em T2* para o diagnóstico e controle da resposta à quelação de ferro em xerocitose e acreditamos que o exame pode ser útil também em outras anemias hemolíticas que necessitam de transfusões.
Descritores: Anemia; Sobrecarga de ferro; Imagem por ressonância magnética; Terapia por quelação; Relatos de casos
INTRODUÇÃO
A forma hereditária de estomatocitose desidratada (xerocitose) é um distúrbio genético autossômico dominante caracterizado pelo aumento da permeabilidade (extravasamento) da membrana eritrocitária da bomba de potássio, produzindo desidratação de eritrócitos. O termo "estomatocitose", inicialmente descrito em 1974(1,2), deriva da morfologia dos eritrócitos no esfregaço de sangue periférico, que se assemelham a uma boca ou lábios. Outras situações clínicas podem apresentar essa morfologia celular, tais como doenças do fígado ou outras anemias hemolíticas, porém esses distúrbios não apresentam mudanças na permeabilidade da membrana do eritrócito(3).
A xerocitose é um distúrbio raro, com incidência de cerca de 1 caso/50.000 nascidos vivos, com ocorrência de 10 a 20 vezes menor do que a esferocitose hereditária. As mudanças mais frequentes na xerocitose foram mapeadas como alterações do alelo do cromossomo 16q23-qter(3-5). A xerocitose pode ser considerada uma síndrome pleiotrópica, no sentido de que o defeito no gene afeta mais do que os eritrócitos(6). Existem relatos de casos de xerocitose com abortos recorrentes e hidropsia fetal sem resposta ao uso de transfusões intrauterinas, que é resolvida espontaneamente após o nascimento(1,6). Outra complicação clínica frequente é o aumento da tendência de eventos tromboembólicos em pacientes com xerocitose após esplenectomia. O mecanismo fisiopatológico desses eventos não está bem definido, porém estudos mostram que os eritrócitos desses indivíduos são mais aderentes ao endotélio. O diagnóstico correto desses distúrbios de membrana pode modificar a evolução clínica, porque o diagnóstico errôneo de esferócito se leva a esplenectomia, que é formalmente contraindicada na xerocitose(1,3,5).
A análise laboratorial pode revelar macrocitose, concentração de hemoglobina corpuscular média (CHCM) elevada, reticulocitose, diminuição da fragilidade osmótica (aumento da resistência à lise osmótica) e altos níveis de potássio sérico nas medidas sequenciais em intervalos regulares (pseudo-hipercalemia)(1,4). As células mostram maior resistência à fragmentação quando submetidas a altas temperaturas (46 a 49oC) quando comparadas com eritrócitos normais(1). Em contraste à forma hiper-hidratada de estomatocitose hereditária (hidrocitose), o esfregaço de sangue periférico na xerocitose apresenta células espiculadas e poucos estomatócitos. O número de estomatócitos presentes na xerocitose é geralmente menor do que 10% dos glóbulos vermelhos(6). A ectacitometria e as medidas seriadas de potássio em uma mesma amostra de sangue mostrando pseudo-hipercalemia, podem confirmar o diagnóstico(4). A ectacitometria de gradiente osmótico mostra uma curva que é absolutamente específica (o índice máximo de deformabilidade é normal; o "ponto hiposmótico", o "ponto hiperosmótico", a fragilidade osmótica e a hidratação celular são reduzidos)(5).
Clinicamente, a xerocitose se manifesta por anemia hemolítica leve a moderada. O paciente pode apresentar fadiga desproporcional aos graus de anemia, que possivelmente deriva de níveis baixos de 2,3-difosfoglicerato (2,3 DPG) e consumo elevado de trifosfato de adenosina (ATP) como resultado do aumento da atividade da bomba de Na/K, que pode afetar a afinidade para o oxigênio(3). Os indivíduos afetados geralmente não precisam de transfusões frequentes. Apesar disso, eles tendem a apresentar sobrecarga de ferro, e o mecanismo para tal ainda não foi devidamente esclarecido(4,5). Percebe-se também uma falta de correlação entre os níveis de ferritina sérica e outros resultados diagnósticos de sobrecarga de ferro, dentre eles a ressonância magnética em T2* (RM T2*), muitas vezes demonstrando altos níveis de sobrecarga de ferro orgânica(7,8).
RELATO DE CASO
Paciente branca do gênero feminino de 17 anos de idade foi admitida em nosso serviço em março de 1999, queixando-se de anemia sintomática e apresentando quadro de hemólise, com icterícia e colúria, tendo precisado de transfusões esporádicas desde a infância. Também relatou histórico de esplenectomia e colecistectomia realizadas em outra instituição, e biópsia hepática (realizada em março de 1996) que revelou hemossiderose. A paciente negou histórico familiar semelhante e informou histórico de ascite neonatal.
Exames laboratoriais mostraram concentração de hemoglobina de 9,8/dL, volume corpuscular médio (VCM) de 113,2fl e CHCM de 31,7g/dL. O teste de eletroforese de hemoglobina e glicose 6-fosfato desidrogenase (G6PD) estavam normais. A análise molecular para talassemia alfa não mostrou mutações e nenhuma hemoglobina instável foi detectada. A hemoglobinúria paroxística noturna examinada por meio do teste de Ham (lises por soro acidificado), a triagem de sucrose e a subsequente citometria de fluxo foram todas negativas. Os achados da medula óssea mostraram apenas hipercelularidade na série eritroide. A curva de fragilidade osmótica sugeriu aumento da resistência globular. A saturação de transferrina foi de 73% e o nível de ferritina sérica foi de 350ng/mL. Não houve mutações para hemocromatose (C282Y e H63D) avaliadas por exame de reação em cadeia da polimerase (PCR).
Em abril de 2001, a paciente teve arritmia e apresentou sinais de insuficiência cardíaca sistólica, confirmada por ecocardiografia. Ela também apresentava sinais de hipertensão pulmonar. Como estava sintomática, o tratamento com aldactone (25mg/dia), amiodarona (200mg/dia) e digoxina (0,25mg/dia, com controle do soro em doses regulares) foi iniciado e obteve boa resposta. O sangue coletado após o episódio de insuficiência cardíaca mostrou trombocitose, que foi atribuída à esplenectomia anterior.
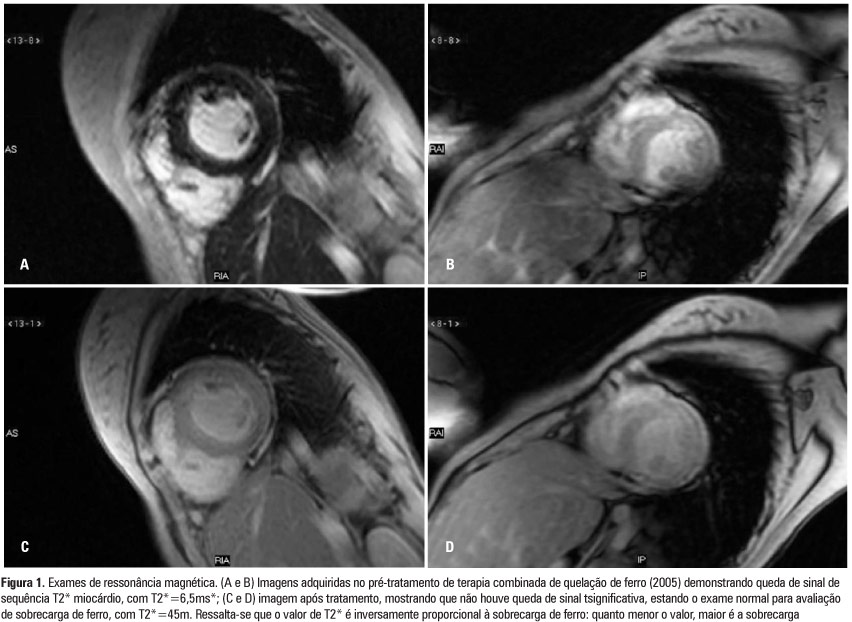
Em abril de 2005, quase 4 anos após o início dos sintomas cardíacos e tratamento específico para insuficiência cardíaca, a RM T2* foi solicitada para quantificação de depósitos de ferro no coração, fígado e pâncreas. Os índices foram: para o coração, índice de 6,5ms; fígado, 6,7ms (4,46mg Fe/g fígado); e pâncreas, 9,1ms, com resultados positivos para sobrecarga de ferro nos 3 órgãos (Figura 1) e nível de ferritina sérica de 267ng/mL.
Foi introduzida por via oral quelação com deferiprona (3.500mg/dia) em combinação com deferoxamina subcutânea (2,0g/dia) por períodos de 10 horas por dia por bomba de infusão. A paciente apresentou-se intolerante à quelação de ferro, com urticária, eritema e reações pruriginosas no local da aplicação de deferoxamina (hipersensibilidade local). A terapia com quelação de ferro foi temporariamente suspensa, sendo reintroduzida apenas 1 mês mais tarde, com boa tolerância, com uso de deferoxamina (2,0g/dia) isolada, por aplicação subcutânea em dias alternados. A dosagem de ferritina sérica durante a retomada do uso de deferoxamina foi de 240ng/mL.
A paciente manteve-se sem diagnóstico etiológico. O quadro clínico de anemia hemolítica, hemocromatose e os achados morfológicos em sangue periférico (que revelavam estomatócitos e acantócitos) sugeriram possível estomatocitose. Foi realizada análise ectacitométrica e, com a suspeita de xerocitose, foi solicitada medida seriada do potássio da paciente e de seus pais. A pseudo-hipercalemia foi encontrada na amostra da paciente, como mostra a tabela 1, confirmando o diagnóstico de estomatocitose hereditária, forma desidratada (xerocitose).
O controle com RM T2* foi realizado em fevereiro de 2006, cerca de 8 meses após à terapia por quelação, mostrando reversão dos sinais de sobrecarga hepática (normalização da função) e melhoria nas funções pancreática e cardíaca (coração: 13,8ms; fígado: 23,5ms; pâncreas: 14ms, tabela 2). A quelação de ferro foi continuada com deferoxamina até abril de 2007, quando foi substituída apenas por deferiprona oral 25mg/kg, três vezes ao dia ou 75mg/kg/dia. A avaliação subsequente por RM T2*, em abril de 2008, revelou normalização das medidas em todos os órgãos, como demonstrado na figura 2.
Desde então, a RM T2* e a ecocardiografia demostraram resolução completa e recuperação da função cardíaca, como mostrado na tabela 2. A paciente permanece em quelação para prevenir novos depósitos de ferro utilizando somente deferiprona.
DISCUSSÃO
Apesar de o mecanismo responsável pelo acúmulo de ferro em pacientes com xerocitose ainda permanecer obscuro, é sabido que os pacientes apresentam depósito considerável de ferro no coração, fígado e pâncreas, levando às mesmas manifestações clínicas de indivíduos com talassemia e anemia falciforme com hemocromatose secundária(3,4). Porém, a falta de correlação entre o nível de ferritina e o acúmulo de ferro pode ser ainda mais deletéria, pois os níveis encontrados são geralmente baixos, retardando o diagnóstico e as medidas terapêuticas, especificamente a quelação(8).
A RM ganhou aceitação como modelo para quantificação de ferro corporal total. Tem bom custo-benefício e realização rápida, é reprodutível e não invasiva. Pode ser utilizada em diversos países devido à boa reprodutibilidade, sendo um teste confiável na conduta clínica diária de diversos distúrbios que levam ao acúmulo de ferro(7-9).
Os níveis séricos de ferritina não se correlacionam com os depósitos cardíacos mensurados pela RM T2*, como já descrito em estudos pregressos e, no coração, a toxicidade intensa do tecido pode ser observada mesmo sem grande depósitos de ferro(8). O ferro cardíaco não pode ser medido pelos níveis de ferritina e nem pelo acúmulo de ferro hepático, e os métodos convencionais de avaliação cardíaca não detectam mudanças até que as disfunções tenham se instalado(7,10). O uso de RM T2* para medir o ferro cardíaco permite a identificação precoce de pacientes que requerem quelação de ferro, mesmo antes do início dos sintomas ou de achados ecocardiográficos consistentes com disfunção sistólica, permitindo redução de mortalidade relacionada à insuficiência cardíaca(10,11).
A RM T2* é considerada atualmente o procedimento padrão para diagnosticar sobrecarga de ferro(7). A importância em se diagnosticar e tratar outras anemias hemolíticas que levam à sobrecarga de ferro, incluindo a xerocitose, deve-se ao fato de que pacientes geralmente apresentam sintomas de insuficiência cardíaca e diabetes, secundários à hemocromatose, mesmo sem um número elevado de transfusões, ou quando os níveis elevados de ferritina ainda estão abaixo de 500ng/mL. Sugerimos o uso regular de RM T2* para diagnóstico, monitoramento, controle de resposta à sobrecarga e quelação de ferro em pacientes com xerocitose, e acreditamos que pode ser utilizada também para acompanhamento de outras anemias hemolíticas que requerem transfusões, mesmo que esporadicamente, permitindo o diagnóstico precoce da sobrecarga de ferro e terapia de quelação.
Data de submissão: 23/5/2012
Aceito em: 28/9/2013
- 1. Aleo Luján E, Gil López C, Ataúlfo González F, Villegas Martínez A, Valverde Moreno F. [Congenital xerocytosis]. An Pediatr (Barc). 2004;61(6):542-5. Spanish.
- 2. Iolascon A, De Falco L, Borgese F, Esposito MR, Avvisati RA, Izzo P, et al. A novel erythroid anion exchange variant (Gly796Arg) of hereditary stomatocytosis associated with dyserythropoiesis. Haematologica. 2009; 94(8):1049-59.
- 3. Stewart GW, Turner EJ. The hereditary stomatocytoses and allied disorders: congenital disorders of erythrocyte membrane permeability to Na and K. Baillieres Best Pract Res Clin Haematol. 1999;12(4):707-27. Review.
- 4. Syfuss PY, Ciupea A, Brahimi S, Cynober T, Stewart GW, Grandchamp B, et al. Mild dehydrated hereditary stomatocytosis revealed by marked hepatosiderosis. Clin Lab Haematol. 2006;28(4):270-4.
- 5. Delaunay J. The molecular basis of hereditary red cell membrane disorders. Blood Rev. 2007;21(1):1-20. Review.
- 6. Bruce LJ. Hereditary stomatocytosis and cation-leaky red cells--recent developments. Blood Cells Mol Dis. 2009;42(3):216-22. Review.
- 7. Wood JC. Magnetic resonance imaging measurement of iron overload. Curr Opin Hematol. 2007;14(3):183-90. Review.
- 8. Wood JC, Ghugre N. Magnetic resonance imaging assessment of excess iron in thalassemia, sickle cell disease and other iron overload diseases. Hemoglobin. 2008;32(1-2):85-96. Review.
- 9. Baur LH. Early detection of iron overload in the heart: a key role for MRI! Int J Cardiovasc Imaging. 2009;25(8):789-90.
- 10. Anderson LJ, Holden S, Davis B, Prescott E, Charrier CC, Bunce NH, et al. Cardiovascular T2-star (T2*) magnetic resonance for the early diagnosis of myocardial iron overload. Eur Heart J. 2001;22(23):2171-9.
- 11. Wood JC. History and current impact of cardiac magnetic resonance imaging on the management of iron overload. Circulation. 2009;120(20):1937-9.
Datas de Publicação
-
Publicação nesta coleção
31 Jan 2014 -
Data do Fascículo
Dez 2013
Histórico
-
Recebido
23 Maio 2012 -
Aceito
28 Set 2013