ABSTRACT
Clupeiformes (herring, sardines, shad, anchovies and allies) are a globally distributed clade with nearly 400 marine, freshwater, and diadromous species. Although best known as filter feeding fishes that form large schools, this group occupies a diverse array of trophic guilds and habitats. Theory suggests that species richness in clades is modulated by ecological limits, which results in diversity-dependent clade growth, a pattern that most clades exhibit. As a trans-marine/freshwater clade that has undergone repeated transitions between marine and freshwaters, Clupeiformes are an excellent system for investigating the interplay between ecological diversity and macroevolutionary dynamics. In this study we review the systematics of Clupeiformes and explore discordance in phylogenetic relationships and divergence times between mitochondrial and nuclear loci. We then use comparative methods to test whether ecological limits regulate diversity in Clupeiformes. We find discordance in phylogenetic relationships at various taxonomic scales, but also considerable agreement between genomes. Our results suggest that trans-marine/freshwater clades are able to circumvent ecological limits on clade growth at regional, but not on local scales. Our study demonstrates that phylogenies are a critical link between ecology and macroevolutionary dynamics, and suggests habitat transitions can play a key role in shaping diversity patterns, particularly in the neotropics.
Keywords:
Congruence; Comparative methods; Discordance; Diversification; Diversity-dependent
RESUMO
Clupeiformes (apapás, sardinhas e manjubas) são um clado globalmente distribuído com quase 400 espécies marinhas, de água doce e diádromas. Embora mais conhecida pela presença de peixes filtradores formadores de cardumes, este grupo apresenta uma diversidade de guilda e habitats tróficos. A teoria sugere que a riqueza de espécies em clados é modulada por limites ecológicos, o que resulta em um crescimento dependente da diversidade, um padrão que a maioria dos clados exibem. Como um clado que sofreu repetidas transições entre águas marinhas e as águas doces, os clupeiformes são um excelente grupo para investigar a interação entre a diversidade ecológica e a dinâmica macroevolutiva. Neste estudo, revisamos a sistemática de Clupeiformes e exploramos a discordância nas relações filogenéticas e os tempos de divergência entre loci mitocondriais e nucleares. Em seguida, utilizamos métodos comparativos para testar se os limites ecológicos regulam a diversidade em Clupeiformes. Encontramos discordância nas relações filogenéticas em várias escalas taxonômicas, mas também considerável concordância entre os genomas. Nossos resultados sugerem que clados que sofreram sucessivas transições entre águas marinhas e águas doces são capazes de contornar os limites ecológicos do crescimento durante a sua diversificação em escala global, mas não localmente. Nosso estudo demonstra que as filogenias apresentam um vínculo crítico entre a ecologia e a dinâmica macroevolutiva, e sugere que as transições de hábitats podem desempenhar um papel fundamental na modelagem dos padrões de diversidade, particularmente no neotrópico.
Palavras-chave:
Congruência; Discordância; Diversificação; Diversidade-dependente; Métodos comparativos
Introduction
Understanding the processes that determine spatial and phylogenetic diversity patterns is a fundamental goal of ecology and evolutionary biology. The rise of phylogenetic comparative methods has led to the recognition that diversity patterns are the outcome of the interplay between ecology and evolutionary dynamics. A central idea that has emerged from the integration of ecology and evolutionary biology is the concept of ecological limits on clade diversity (Rabosky, Lovette, 2008Rabosky DL, Lovette IJ. Density-dependent diversification in North American wood warblers. Proc R Soc B [serial on the Internet]. 2008; 275(1649):2363-71. Available from: http://dx.doi.org/10.1098/rspb.2008.0630
http://dx.doi.org/10.1098/rspb.2008.0630...
; Rabosky, 2013Rabosky DL. Diversity-dependence, ecological speciation, and the role of competition in macroevolution. Annu Rev Ecol Evol Syst [serial on the Internet]. 2013; 44:481-502. Available from: https://doi.org/10.1146/annurev-ecolsys-110512-135800
https://doi.org/10.1146/annurev-ecolsys-...
). The ecological limits hypothesis suggests that equilibrium processes, such as availability of resources and competition, determine clade richness (Rabosky, 2009aRabosky DL. Ecological limits and diversification rate: alternative paradigms to explain the variation in species richness among clades and regions. Ecol Lett [serial on the Internet]. 2009a; 12(8):735-43. Available from: http://dx.doi.org/10.1111/j.1461-0248.2009.01333.x
http://dx.doi.org/10.1111/j.1461-0248.20...
; Moen, Morlon, 2014Moen D, Morlon H. Why does diversification slow down? Trends Ecol Evol [serial on the Internet]. 2014; 29(4):190-97. Available from: https://doi.org/10.1016/j.tree.2014.01.010
https://doi.org/10.1016/j.tree.2014.01.0...
). A prediction of the ecological limits hypothesis is that as a clade diversifies resource availability decreases due to competition, and clade growth slows over time. If a clade is bounded by ecological limits it is best explained by a diversity-dependent model and the resulting pattern is a logistic curve in a lineage through time plot (LTT). Alternatively, diversity may be unregulated and determined by non-equilibrium processes, such as vicariance and dispersal (Harmon, Harrison, 2015Harmon LJ, Harrison S. Species Diversity Is Dynamic and unbounded at local and continental scales. Am Nat [serial on the Internet]. 2015; 185(5):584-93. Available from: http://dx.doi.org/10.1086/680859
http://dx.doi.org/10.1086/680859...
). Under this scenario clade growth is constant and unbounded over time, resulting in an exponential curve in a LTT plot. Clade dynamics are not static and may shift from diversity-dependent to diversity-independent when environmental conditions change (Moen, Morlon, 2014Moen D, Morlon H. Why does diversification slow down? Trends Ecol Evol [serial on the Internet]. 2014; 29(4):190-97. Available from: https://doi.org/10.1016/j.tree.2014.01.010
https://doi.org/10.1016/j.tree.2014.01.0...
). In some cases, the environmental conditions may change because a lineage undergoes a habitat transition, which can alter clade dynamics by shifting rates of lineage diversification (speciation and extinction) and morphological evolution, which in turn can help shape diversity patterns (Collar et al., 2010Collar DC, Schulte JA, O’Meara BC, Losos JB. Habitat use affects morphological diversification in dragon lizards. J Evol Biol [serial on the Internet]. 2010; 23(5):1033-49. Available from: http://dx.doi.org/10.1111/j.1420-9101.2010.01971.x
http://dx.doi.org/10.1111/j.1420-9101.20...
; Betancur-R et al., 2012Betancur-R R, Ortí G, Stein AM, Marceniuk AP, Pyron AR. Apparent signal of competition limiting diversification after ecological transitions from marine to freshwater habitats. Ecol Lett [serial on the Internet]. 2012; 15(8):822-30. Available from: http://dx.doi.org/10.1111/j.1461-0248.2012.01802.x
http://dx.doi.org/10.1111/j.1461-0248.20...
; Bloom et al., 2013Bloom DD, Weir JT, Piller KR, Lovejoy NR. Do freshwater fishes diversify faster than marine fishes? A test using state-dependent diversification analyses and molecular phylogenetics of New World silversides (Atherinopsidae). Evolution [serial on the Internet]. 2013; 67(7):2040-57. Available from: https://doi.org/10.1111/evo.12074
https://doi.org/10.1111/evo.12074...
).
Habitat transitions from marine to freshwaters represent a profound shift in environmental conditions (Lee, Bell, 1999Lee CE, Bell MA. Causes and consequences of recent freshwater invasions by saltwater animals. Trends Ecol Evol [serial on the Internet]. 1999; 14(7):284-88. Available from: https://doi.org/10.1016/S0169-5347(99)01596-7
https://doi.org/10.1016/S0169-5347(99)01...
; Vermeij, Dudley, 2000Vermeij GJ, Dudley R. Why are there so few evolutionary transitions between aquatic and terrestrial ecosystems? Biol J Linn Soc [serial on the Internet]. 2000; 70(4):541-54. Available from: https://doi.org/10.1111/j.1095-8312.2000.tb00216.x
https://doi.org/10.1111/j.1095-8312.2000...
). There is generally a mutually exclusive aquatic community composition between adjacent marine and freshwaters, likely due to strong biotic and abiotic barriers. Thus a lineage that transitions between marine and freshwater habitats may be released from diversity-dependent ecological limits on clade growth (Schluter, 1996Schluter D. Ecological causes of adaptive radiation. Am Nat . 1996; 148:40-64.; Yoder et al., 2010Yoder JB, Clancey E, Roches SD, Eastman JM, Gentry L, Godsoe W et al. Ecological opportunity and the origin of adaptive radiations. J Evol Biol [serial on the Internet]. 2010; 23(8):1581-96. Available from: https://doi.org/10.1111/j.1420-9101.2010.02029.x
https://doi.org/10.1111/j.1420-9101.2010...
). If habitat transitions circumvent ecological limits, then clades that repeatedly undergo transitions would continue to experience exponential clade growth. The Neotropics harbor the greatest freshwater fish diversity in the world (Lundberg et al., 2000Lundberg JG, Kottelat M, Smith GR, Stiassny MLJ, Gill AC. So many fishes, so little time: An overview of recent ichthyological discovery in continental waters. Ann Missouri Bot Gard. 2000; 87(1):26-62.; Albert, Reis, 2011Albert JS, Reis RE. Historical biogeography of neotropical freshwater fishes. Berkeley and Los Angeles, California. University of California Press; 2011.), which would predict this fauna may have reached diversity equilibrium (López-Fernández et al., 2013López-Fernández H, Arbour JH, Winemiller KO, Honeycutt RL. Testing for ancient adaptive radiations in neotropical cichilid fishes. Evolution [serial on the Internet]. 2013; 67(5):1321-37. Available from: https://doi.org/10.1111/evo.12038
https://doi.org/10.1111/evo.12038...
). Yet, Neotropical freshwaters are a hotspot for trans-marine/freshwater lineages - clades that repeatedly transitioned between marine and freshwaters over macroevolutionary timescales - some of which have invaded freshwaters in the recent past (Bloom, Lovejoy, 2017Bloom DD, Lovejoy NR. On the origins of marine-derived freshwater fishes in South America. J Biogeogr [serial on the Internet]. 2017; 44(9):1927-38. Available from: https://doi.org/10.1111/jbi.12954
https://doi.org/10.1111/jbi.12954...
). The repeated invasions of Neotropical freshwaters, and reversals to marine waters (Betancur-R, 2010Betancur-R R. Molecular phylogenetics supports multiple evolu tionary transitions from marine to freshwater habitats in ariid catfishes. Mol Phylogenet Evol [serial on the Internet]. 2010; 55(1):249-58. Available from: https://doi.org/10.1016/j.ympev.2009.12.018
https://doi.org/10.1016/j.ympev.2009.12....
; Bloom, Lovejoy, 2012Bloom DD, Lovejoy NR. Molecular phylogenetics reveals a pattern of biome conservatism in New World anchovies (family Engraulidae). J Evol Biol [serial on the Internet]. 2012; 25(4):701-15. Available from: http://dx.doi.org/10.1111/j.1420-9101.2012.02464.x
http://dx.doi.org/10.1111/j.1420-9101.20...
, 2017Bloom DD, Lovejoy NR. On the origins of marine-derived freshwater fishes in South America. J Biogeogr [serial on the Internet]. 2017; 44(9):1927-38. Available from: https://doi.org/10.1111/jbi.12954
https://doi.org/10.1111/jbi.12954...
) suggest that ecological limits have not been reached. This raises a number of questions about the genesis of diversity in the Neotropics. Are there ecological limits on clade diversity in trans/marine-freshwater fishes in general? Do Neotropical lineages exhibit different lineage accumulation patterns than clades in other biogeographic areas? Understanding the role of ecological limits in structuring Neotropical fish diversity will help provide answers to these urgent questions.
Clupeiformes (herring, sardines, shad, anchovies and allies) are best known as near-shore, schooling and filter-feeding fishes (Whitehead, 1985bWhitehead PJ. FAO species catalogue. Clupeoid fishes of the world (suborder Clupeioidei). An annotated and illustrated catalogue of the herrings, sardines, pilchards, sprats, shads, anchovies and wolf-herrings. FAO Fish Synop. 1985b; 125(7):1-303.). However, the ecological diversity of the group is often underappreciated (Whitehead, 1985aWhitehead P. King Herring: His place amongst the clupeoids. Can J Fish Aquat Sci. 1985a; 42(1):3-20.). Clupeiformes display a wide range phenotypic diversity, including miniaturized species that achieve a maximum size of 2 mm and large species reaching 300 mm TL (Whitehead, 1985bWhitehead PJ. FAO species catalogue. Clupeoid fishes of the world (suborder Clupeioidei). An annotated and illustrated catalogue of the herrings, sardines, pilchards, sprats, shads, anchovies and wolf-herrings. FAO Fish Synop. 1985b; 125(7):1-303.; Bloom et al. in press). The group occupies an array of trophic guilds, such as planktivores, algivores, piscivores, molluscivores, and more (Egan et al., 2018Egan JP, Bloom DD, Kuo CH, Hammer MP, Tongnunui P, Iglésias SP, Sheaves M, Grudpan C, Simons AM. Phylogenetic analysis of trophic niche evolution reveals a latitudinal herbivory gradient in Clupeoidei (herrins, anchovies and allies). Mol Phylogenet Evol [serial on the Internet]. 2018; 124(July 2018):151-61. Available from: http://dx.doi.org/10.1016/j.ympev.2018.03.011
http://dx.doi.org/10.1016/j.ympev.2018.0...
). Clupeiformes also inhabit a broad spectrum of habitats, including open ocean, coastal areas, estuaries, and freshwater rivers and lakes. While most vertebrate clades are restricted to either marine or continental waters, Clupeiformes are a trans-marine/freshwater clade, yielding nearly 400 marine, freshwater, and diadromous species recognized today (Bloom, Lovejoy, 2014Bloom DD, Lovejoy NR. The evolutionary origins of diadromy inferred from a time-calibrated phylogeny for Clupeiformes (herring and allies). Proc R Soc B [serial on the Internet]. 2014; 281(1778):20132081. Available from: http://dx.doi.org/10.1098/rspb.2013.2081
http://dx.doi.org/10.1098/rspb.2013.2081...
). The ability to traverse the marine/freshwater boundary is shared with groups such as stingrays, needlefishes, silversides, drums and pufferfishes (Lovejoy et al., 2006Lovejoy NR, Albert JS, Crampton WGR. Miocene marine incursions and marine/freshwater transitions: Evidence from Neotropical fishes. J South Am Earth Sci [serial on the Internet]. 2006; 21(1-2):5-13. Available from: https://doi.org/10.1016/j.jsames.2005.07.009
https://doi.org/10.1016/j.jsames.2005.07...
; Bloom, Lovejoy, 2017Bloom DD, Lovejoy NR. On the origins of marine-derived freshwater fishes in South America. J Biogeogr [serial on the Internet]. 2017; 44(9):1927-38. Available from: https://doi.org/10.1111/jbi.12954
https://doi.org/10.1111/jbi.12954...
). While most trans-marine/freshwater groups have colonized the freshwaters of two or fewer continents, freshwater Clupeiforms are found on every continent except Antarctica and in many cases have colonized freshwaters multiple times in the same geographic area (Bloom, Lovejoy, 2012Bloom DD, Lovejoy NR. Molecular phylogenetics reveals a pattern of biome conservatism in New World anchovies (family Engraulidae). J Evol Biol [serial on the Internet]. 2012; 25(4):701-15. Available from: http://dx.doi.org/10.1111/j.1420-9101.2012.02464.x
http://dx.doi.org/10.1111/j.1420-9101.20...
; 2014Bloom DD, Lovejoy NR. The evolutionary origins of diadromy inferred from a time-calibrated phylogeny for Clupeiformes (herring and allies). Proc R Soc B [serial on the Internet]. 2014; 281(1778):20132081. Available from: http://dx.doi.org/10.1098/rspb.2013.2081
http://dx.doi.org/10.1098/rspb.2013.2081...
).
The systematic relationships among Clupeiformes have received increasing attention, but higher-level relationships remain unresolved. Clupeiformes is divided into two suborders, Denticipitoidei, which includes a single species, Denticeps clupeoides, and Clupeoidei, which is comprised of all other extant Clupeiforms. Clupeoidei has traditionally been divided into five families: Clupeidae (herrings, shads, sardines), Engraulidae (anchovies), Pristigasteridae (longfin herrings), Chirocentridae (wolf herrings) and Denticipitidae (denticle herrings). A sixth family, Sundasalangidae, was described in 1981 and later placed in Clupeiformes by Siebert (1997Siebert, DJ. Notes on the anatomy and relationships of Sundasalanx Roberts (Teleostei, Clupeidae), with descriptions of four new species from Borneo. Bull Br Mus Nat His Zool 1997; 63: 13-26.); Sundasalangids have since been included in Clupeidae (Lavoué et al., 2014Lavoué S, Konstantinidis P, Chen WJ. Progress in clupeiform systematics. In: Ganias K, editor. Biology and ecology of sardines and anchovies. Boca Raton, FL: CRC Press; 2014. p.3-42.). Early morphological work by Grande (1985Grande L. Recent and fossil clupeomorph fishes with materials for revision of the subgroups of clupeoids. Bull Am Museum Nat Hist. 1985; 181(2):231-372.) hypothesized Clupeidae as the sister to Chirocentridae, with the position of Engraulidae and Pristigasteridae unresolved. More recently, Di Dario (2009Di Dario F. 2009. Chirocentrids as engrauloids: evidence from suspensorium, branchial arches, and infraorbital bones (Clupeomorpha, Teleostei). Zool J Linn Soc. 2009; 156:363-383.) found evidence for Chirocentridae as sister to Engraulidae. Di Dario (2009Di Dario F. 2009. Chirocentrids as engrauloids: evidence from suspensorium, branchial arches, and infraorbital bones (Clupeomorpha, Teleostei). Zool J Linn Soc. 2009; 156:363-383.) also supported Clupeidae as the closest relative to the Chirocentridae+Engraulidae clade, with Pristigasteridae the earliest branching lineage within Clupeoidei. Using mitogenomics Lavoué et al. (2013Lavoué S, Miya M, Musikasinthorn P, Chen WJ, Nishida M. Mitogenomic evidence for an Indo-West Pacific origin of the Clupeoidei (Teleostei: Clupeiformes). PLoS One [serial on the Internet]. 2013; 8(2):e56485. Available from: https://doi.org/10.1371/journal.pone.0056485
https://doi.org/10.1371/journal.pone.005...
) supported Clupeidae+Pristigasteridae, but also found that Clupeidae was not monophyletic because Spratelloidiini fell outside of a clade that included Clupeidae, Pristigasteridae and Chirocentridae. Studies that incorporated both mitochondrial (mtDNA) and nuclear DNA (nDNA) also failed to recover a monophyletic Clupeidae, but differed from mitogenomic results because Chirocentridae was sister to Engraulidae (Li, Ortí, 2007Li CH, Ortí G. Molecular phylogeny of clupeiformes (Actinopterygii) inferred from nuclear and mitochondrial DNA sequences. Mol Phylogenet Evol [serial on the Internet]. 2007; 44(1):386-98. Available from: http://dx.doi.org/10.1016/j.ympev.2006.10.030
http://dx.doi.org/10.1016/j.ympev.2006.1...
; Bloom, Lovejoy, 2014Bloom DD, Lovejoy NR. The evolutionary origins of diadromy inferred from a time-calibrated phylogeny for Clupeiformes (herring and allies). Proc R Soc B [serial on the Internet]. 2014; 281(1778):20132081. Available from: http://dx.doi.org/10.1098/rspb.2013.2081
http://dx.doi.org/10.1098/rspb.2013.2081...
). Combined mtDNA and nDNA analyses suggested Pristigasteridae was sister to the Chirocentridae+Engraulidae, but with no statistical support (Bloom, Lovejoy, 2014Bloom DD, Lovejoy NR. The evolutionary origins of diadromy inferred from a time-calibrated phylogeny for Clupeiformes (herring and allies). Proc R Soc B [serial on the Internet]. 2014; 281(1778):20132081. Available from: http://dx.doi.org/10.1098/rspb.2013.2081
http://dx.doi.org/10.1098/rspb.2013.2081...
). Most recently, using six nuclear genes, Egan et al. (2018Egan JP, Bloom DD, Kuo CH, Hammer MP, Tongnunui P, Iglésias SP, Sheaves M, Grudpan C, Simons AM. Phylogenetic analysis of trophic niche evolution reveals a latitudinal herbivory gradient in Clupeoidei (herrins, anchovies and allies). Mol Phylogenet Evol [serial on the Internet]. 2018; 124(July 2018):151-61. Available from: http://dx.doi.org/10.1016/j.ympev.2018.03.011
http://dx.doi.org/10.1016/j.ympev.2018.0...
) supported Chirocentridae as sister to Engraulidae, but also recovered Pristigasteridae as nested within Clupeidae.
The phylogenetic uncertainty at higher taxonomic levels is also prevalent at lower taxonomic levels. Grande (1985Grande L. Recent and fossil clupeomorph fishes with materials for revision of the subgroups of clupeoids. Bull Am Museum Nat Hist. 1985; 181(2):231-372.) proposed five subfamilies within Clupeidae (Alosinae, Clupeinae, Pellonulinae, Dorosomatinae and Dussumieriinae), but noted these taxa were mostly out of convenience rather than supported by data. Indeed molecular studies have demonstrated that none of the five subfamilies of Clupeidae proposed by Grande (1985Grande L. Recent and fossil clupeomorph fishes with materials for revision of the subgroups of clupeoids. Bull Am Museum Nat Hist. 1985; 181(2):231-372.) are monophyletic. Lavoué et al. (2014Lavoué S, Konstantinidis P, Chen WJ. Progress in clupeiform systematics. In: Ganias K, editor. Biology and ecology of sardines and anchovies. Boca Raton, FL: CRC Press; 2014. p.3-42.) revised the composition of these five subfamilies based in part on recent molecular studies (Li, Orti, 2007Li CH, Ortí G. Molecular phylogeny of clupeiformes (Actinopterygii) inferred from nuclear and mitochondrial DNA sequences. Mol Phylogenet Evol [serial on the Internet]. 2007; 44(1):386-98. Available from: http://dx.doi.org/10.1016/j.ympev.2006.10.030
http://dx.doi.org/10.1016/j.ympev.2006.1...
; Lavoué et al., 2010Lavoué S, Miya M, Nishida M. Mitochondrial phylogenomics of anchovies (family Engraulidae) and recurrent origins of pronounced miniaturization in the order Clupeiformes. Mol Phylogenet Evol [serial on the Internet]. 2010; 56(1):480-85. Available from: https://doi.org/10.1016/j.ympev.2009.11.022
https://doi.org/10.1016/j.ympev.2009.11....
, 2013Lavoué S, Miya M, Musikasinthorn P, Chen WJ, Nishida M. Mitogenomic evidence for an Indo-West Pacific origin of the Clupeoidei (Teleostei: Clupeiformes). PLoS One [serial on the Internet]. 2013; 8(2):e56485. Available from: https://doi.org/10.1371/journal.pone.0056485
https://doi.org/10.1371/journal.pone.005...
; Bloom, Lovejoy, 2012Bloom DD, Lovejoy NR. Molecular phylogenetics reveals a pattern of biome conservatism in New World anchovies (family Engraulidae). J Evol Biol [serial on the Internet]. 2012; 25(4):701-15. Available from: http://dx.doi.org/10.1111/j.1420-9101.2012.02464.x
http://dx.doi.org/10.1111/j.1420-9101.20...
, 2014Bloom DD, Lovejoy NR. The evolutionary origins of diadromy inferred from a time-calibrated phylogeny for Clupeiformes (herring and allies). Proc R Soc B [serial on the Internet]. 2014; 281(1778):20132081. Available from: http://dx.doi.org/10.1098/rspb.2013.2081
http://dx.doi.org/10.1098/rspb.2013.2081...
); herein we used this revised taxonomy. Engraulidae (anchovies) is divided into a two subfamilies: Engraulinae is a largely New World clade, and Coilinae is restricted to the Old World. Engraulinae is comprised of eight genera, six of which are polytypic and two are monotypic. Of the six polytypic New World anchovy genera, only Lycengraulis and Cetengraulis have been recovered as monophyletic (Bloom, Lovejoy, 2012Bloom DD, Lovejoy NR. Molecular phylogenetics reveals a pattern of biome conservatism in New World anchovies (family Engraulidae). J Evol Biol [serial on the Internet]. 2012; 25(4):701-15. Available from: http://dx.doi.org/10.1111/j.1420-9101.2012.02464.x
http://dx.doi.org/10.1111/j.1420-9101.20...
, 2014Bloom DD, Lovejoy NR. The evolutionary origins of diadromy inferred from a time-calibrated phylogeny for Clupeiformes (herring and allies). Proc R Soc B [serial on the Internet]. 2014; 281(1778):20132081. Available from: http://dx.doi.org/10.1098/rspb.2013.2081
http://dx.doi.org/10.1098/rspb.2013.2081...
). However, the most taxon rich study (Bloom, Lovejoy, 2014Bloom DD, Lovejoy NR. The evolutionary origins of diadromy inferred from a time-calibrated phylogeny for Clupeiformes (herring and allies). Proc R Soc B [serial on the Internet]. 2014; 281(1778):20132081. Available from: http://dx.doi.org/10.1098/rspb.2013.2081
http://dx.doi.org/10.1098/rspb.2013.2081...
) had a large amount of missing data, and relied heavily on mtDNA data. Both missing data and mtDNA are known to mislead both tree topology (Leaché, 2010Leaché AD. Species trees for spiny lizards (Genus Sceloporus): Identifying points of concordance and conflict between nuclear and mitochondrial data. Mol Phylogenet Evol [serial on the Internet]. 2010; 54(1):162-71. Available from: http://dx.doi.org/10.1016/j.ympev.2009.09.006
http://dx.doi.org/10.1016/j.ympev.2009.0...
; Wiens et al., 2010Wiens JJ, Kuczynski CA, Stephens PR. Discordant mitochondrial and nuclear gene phylogenies in emydid turtles: Implications for speciation and conservation. Biol J Linn Soc [serial on the Internet]. 2010; 99(2):445-61. Available from: https://doi.org/10.1111/j.1095-8312.2009.01342.x
https://doi.org/10.1111/j.1095-8312.2009...
; Platt et al., 2018Platt RN, Faircloth BC, Sullivan KAM, Kieran TJ, Glenn TC, Vandewege MW et al. Conflicting evolutionary histories of the mitochondrial and nuclear genomes in New World myotis bats. Syst Biol [serial on the Internet]. 2018; 67(2):236-49. Available from: http://dx.doi.org/10.1093/sysbio/syx070
http://dx.doi.org/10.1093/sysbio/syx070...
) and divergence time estimates (Zheng et al., 2011Zheng Y, Peng R, Kuro-O M, Zeng X. Exploring patterns and extent of bias in estimating divergence time from mitochondrial DNA sequence data in a particular lineage: A case study of salamanders (Order Caudata). Mol Biol Evol [serial on the Internet]. 2011; 28(9):2521-35. Available from: http://dx.doi.org/10.1093/molbev/msr072
http://dx.doi.org/10.1093/molbev/msr072...
; Dornburg et al., 2014Dornburg A, Townsend JP, Friedman M, Near TJ. Phylogenetic informativeness reconciles ray-finned fish molecular divergence times. BMC Evol Biol [serial on the Internet]. 2014; 14(169):1-14. Available from: https://doi.org/10.1186/s12862-014-0169-0
https://doi.org/10.1186/s12862-014-0169-...
). The widespread taxonomic incongruence with phylogeny and dependence on mitochondrial data calls for an evaluation of discordance between mitochondrial and nuclear topologies and divergence times in Clupeiformes.
In this study we investigated clupeiform phylogenetics and patterns of lineage accumulation across different phylogenetic scales in Clupeiformes. Our objectives were 1) to evaluate differences in tree topology and divergence times between mitochondrial and nuclear datasets and 2) test the hypothesis that habitat transitions have allowed Clupeiformes to circumvent ecological limits on clade growth. We reviewed the state of systematics in Clupeiformes and proposed that this group is good model for investigating how habitat transitions can influence clade dynamics. More generally, we demonstrated that clades with unresolved systematics can be used to understand the genesis of diversity in Neotropical fishes.
Material and Methods
Taxon sampling. Previous studies that employed both mtDNA and nDNA had large amounts of missing nDNA, which can be problematic for phylogenetic inference (Thomson, Shaffer, 2010Thomson RC, Shaffer HB. Sparse supermatrices for phylogenetic inference: taxonomy, alignment, rogue taxa, and the phylogeny of living turtles. Syst Biol [serial on the Internet]. 2010; 59(1):42-58. Available from: http://dx.doi.org/10.1093/sysbio/syp075
http://dx.doi.org/10.1093/sysbio/syp075...
; Roure et al., 2013Roure B, Baurain D, Philippe H. Impact of missing data on phylogenies inferred from empirical phylogenomic data sets. Mol Biol Evol [serial on the Internet]. 2013; 30(1):197-214. Available from: http://dx.doi.org/10.1093/molbev/mss208
http://dx.doi.org/10.1093/molbev/mss208...
). Because we wanted to focus our efforts on identifying incongruence between loci we used the Bloom and Lovejoy data matrix as a starting point and pruned the dataset to remove all taxa missing more than a single locus. The original Bloom and Lovejoy dataset included DNA sequences from two nuclear (rag1, rag2) and two mitochondrial (cytb, 16s) genes and 152 terminals. Our pruned data set included 98 in-group taxa representing all clupeiform families (sensuLavoué et al., 2014Lavoué S, Konstantinidis P, Chen WJ. Progress in clupeiform systematics. In: Ganias K, editor. Biology and ecology of sardines and anchovies. Boca Raton, FL: CRC Press; 2014. p.3-42.) and 36 clupeiform genera (S1 - available only as online supplementary file accessed with the online version of the article at http://www.scielo.br/ni). Sequences were aligned using the MUSCLE algorithm (Edgar, 2004Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004; 32(5):1792-97.) in Geneious v. 6.0.3 (www.geneious.com; Biomatters Ltd., Auckland, New Zealand). We verified the quality of alignments by visual inspection of sequences and their amino acid translation and comparison to previously published alignments (Bloom, Lovejoy, 2014Bloom DD, Lovejoy NR. The evolutionary origins of diadromy inferred from a time-calibrated phylogeny for Clupeiformes (herring and allies). Proc R Soc B [serial on the Internet]. 2014; 281(1778):20132081. Available from: http://dx.doi.org/10.1098/rspb.2013.2081
http://dx.doi.org/10.1098/rspb.2013.2081...
), resulting in sequences of the following lengths (in base pairs): rag1 1491, rag2 1237, cytb 1130, and 16s 1353.
Phylogenetic analyses. We assembled three concatenated datasets for phylogenetic analysis: (1) mitochondrial genes, (2) nuclear genes, (3) and a combined dataset containing all four loci. We used PartitionFinder v.1.01 (Lanfear et al., 2012Lanfear R, Calcott B, Ho SYW, Guindon S. PartitionFinder: Combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol [serial on the Internet]. 2012; 29(6):1695-701. Available from: http://dx.doi.org/10.1093/molbev/mss020
http://dx.doi.org/10.1093/molbev/mss020...
) to select partitioning schemes and nucleotide substitution models using Bayesian information criterion (BIC) scores. We did not implement the invariant sites parameter because it is redundant with the gamma distribution parameter (Yang, 2006Yang Z. Computational Molecular Evolution. Oxford: Oxford University Press; 2006.). The best fitting partitioning scheme for all datasets partitioned by gene and codon position and assigned GTR + gamma nucleotide substitution models to all partitions.
We inferred maximum likelihood phylogenies using all three datasets with RAxML v.8.2.4 (Stamatakis, 2014Stamatakis A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics [serial on the Internet]. 2014; 30(9):1312-13. Available from: http://dx.doi.org/10.1093/bioinformatics/btu033
http://dx.doi.org/10.1093/bioinformatics...
) via CIPRES. Tree searching and non-parametric bootstrap estimation of node support was conducted simultaneously using the rapid bootstrapping algorithm. We used the bootstopping option, which determines the number of bootstrap replicates required to obtain stable support values and stops analyses automatically.
To time-calibrate our phylogeny we used six exponential calibration priors based upon priors previously used in clupeiform systematics (Bloom, Lovejoy, 2014Bloom DD, Lovejoy NR. The evolutionary origins of diadromy inferred from a time-calibrated phylogeny for Clupeiformes (herring and allies). Proc R Soc B [serial on the Internet]. 2014; 281(1778):20132081. Available from: http://dx.doi.org/10.1098/rspb.2013.2081
http://dx.doi.org/10.1098/rspb.2013.2081...
; Lavoué et al., 2017bLavoué S, Bertrand JAM, Wang H, Chen W, Ho HC, Motomura H et al. Molecular systematics of the anchovy genus Encrasicholina in the Northwest Pacific. PLoS One [serial on the Internet]. 2017b; 12(7):e0181329. Available from: https://doi.org/10.1371/journal.pone.0181329
https://doi.org/10.1371/journal.pone.018...
):
(1) Most recent common ancestor (MRCA) of Clupeoidei: The crown clupeoid †Cynoclupea nelsoni (Malabarba, Di Dario, 2017Malabarba MC, Di Dario F. A new predatory herring-like fish (Teleostei: Clupeiformes) from the early Cretaceous of Brazil, and implications for relationships in the Clupeoidei. Zool J Linn Soc . 2017; 180:175-94.) set a minimum age of 125 Ma and the absence of Jurassic Clupeomorpha fossils set a soft 95% maximum age of 145 Ma.
(2) MRCA of Dorosoma: A Dorosoma petenense fossil (Miller, 1982Miller RR. First Fossil record (Plio-Pleistocene) of Threadfin Shad, Dorosoma petenense, from the Gatuna Formation of southeastern New Mexico. J Paleontol. 1982; 56(2):423-25.) set a minimum age of 2.5 Ma for the MRCA of Dorosoma. We set a soft 95% maximum MRCA age of 86.3 because most crown clupeoid fossils are younger.
(3)-(5) MRCA of three sister pairs of anchovies separated by the Isthmus of Panama (Cetengraulis edentulus/C. mysticetus, Anchovia macrolepidota/A. clupeoides, and Lycengraulis grossidens/L. poeyi): We set a minimum age of 3.0 Ma and soft 95% maximum age of 86.3 (Bloom, Lovejoy, 2014Bloom DD, Lovejoy NR. The evolutionary origins of diadromy inferred from a time-calibrated phylogeny for Clupeiformes (herring and allies). Proc R Soc B [serial on the Internet]. 2014; 281(1778):20132081. Available from: http://dx.doi.org/10.1098/rspb.2013.2081
http://dx.doi.org/10.1098/rspb.2013.2081...
). Our maximum age calibration does not exclude a later separation of the Isthmus of Panama (Montes et al., 2015Montes C, Cardona A, Jaramillo C, Pardo A, Silva JC, Valencia V, Ayala C, Pérez-Angel LC, Rodriguez-Parra LA, Ramirez V, Niño H. Middle Miocene closure of the Central American Seaway. Science [serial on the Internet]. 2015; 348(6231):226-29. Available from: http://dx.doi.org/10.1126/science.aaa2815
http://dx.doi.org/10.1126/science.aaa281...
).
(6) MRCA of Engraulidae: †Eoengraulis fasolo (Marramà, Carnevale, 2017Marramà G, Carnevale G. The relationships of Gasteroclupea branisai Signeux, 1964, a freshwater double-armored herring (Clupeomorpha, Ellimmichthyiformes) from the Late Cretaceous-Paleocene of South America. Hist Biol [serial on the Internet]. 2017; 29(7):904-17. Available from: https://doi.org/10.1080/08912963.2016.1262855
https://doi.org/10.1080/08912963.2016.12...
) set a minimum age of 50 Ma and we set a soft 95% maximum age of 86.3 Ma (Bloom, Lovejoy, 2014Bloom DD, Lovejoy NR. The evolutionary origins of diadromy inferred from a time-calibrated phylogeny for Clupeiformes (herring and allies). Proc R Soc B [serial on the Internet]. 2014; 281(1778):20132081. Available from: http://dx.doi.org/10.1098/rspb.2013.2081
http://dx.doi.org/10.1098/rspb.2013.2081...
).
We generated two time-calibrated phylogenies using the nuclear and mitochondrial datasets via Bayesian phylogenetic analyses in BEAST v.2.4.5 (Bouckaert et al., 2014Bouckaert R, Heled J, Kühnert D, Vaughan T, Wu CH, Xie D et al. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comput Biol [serial on the Internet]. 2014; 10(4):e1003537. Available from: https://doi.org/10.1371/journal.pcbi.1003537
https://doi.org/10.1371/journal.pcbi.100...
) via the CIPRES Science Gateway portal (Miller et al., 2010Miller MA, Pfeiffer W, Schwartz T. Creating the CIPRES science gateway for inference of large phylogenetic trees. In: Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, 14 November 2010, pp. 1-8.). For all analyses we implemented a birth-death speciation prior because extinction has occurred in clupeiformes (Grande, 1985Grande L. Recent and fossil clupeomorph fishes with materials for revision of the subgroups of clupeoids. Bull Am Museum Nat Hist. 1985; 181(2):231-372.; Grande, Nelson, 1985Grande L, Nelson G. Interrelationships of fossil and recent anchovies (Teleostei: Engrauloidea) and description of a new species from the Miocene of Cyprus. Am Museum Novit. 1985; 2826:1-16.), an uncorrelated lognormal clock model of molecular evolution, set Markov chain Monte Carlo (MCMC) lengths of 100 million generations, and logged results every 5,000th generation. We conducted five independent BEAST runs for both datasets. We visualized results in Tracer v.1.6.0 (Rambaut et al., 2014Rambaut A, Suchard MA, Xie D, Drummond AJ. Tracer v1.6 [Inter net]. 2014. Available from: http://tree.bio.ed.ac.uk/softwar/tracer
http://tree.bio.ed.ac.uk/softwar/tracer...
) to confirm that MCMC runs reached stationarity, sufficient effective sample sizes of parameters (>200), and convergence of independent MCMC runs. We combined trees and removed burnin in LogCombiner v.2.4.5 and used TreeAnnotator v.2.4.5 to generate maximum clade credibility trees (Bouckaert et al., 2014Bouckaert R, Heled J, Kühnert D, Vaughan T, Wu CH, Xie D et al. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comput Biol [serial on the Internet]. 2014; 10(4):e1003537. Available from: https://doi.org/10.1371/journal.pcbi.1003537
https://doi.org/10.1371/journal.pcbi.100...
).
Comparative methods. Testing for slow-downs in diversification rates requires a time-calibrated phylogeny and is sensitive to taxon sampling. For our comparative analyses we used the phylogeny from Bloom, Lovejoy (2014Bloom DD, Lovejoy NR. The evolutionary origins of diadromy inferred from a time-calibrated phylogeny for Clupeiformes (herring and allies). Proc R Soc B [serial on the Internet]. 2014; 281(1778):20132081. Available from: http://dx.doi.org/10.1098/rspb.2013.2081
http://dx.doi.org/10.1098/rspb.2013.2081...
), which represents the most taxon rich time-calibrated phylogeny available for this group. The Bloom, Lovejoy (2014Bloom DD, Lovejoy NR. The evolutionary origins of diadromy inferred from a time-calibrated phylogeny for Clupeiformes (herring and allies). Proc R Soc B [serial on the Internet]. 2014; 281(1778):20132081. Available from: http://dx.doi.org/10.1098/rspb.2013.2081
http://dx.doi.org/10.1098/rspb.2013.2081...
) phylogeny was a multi-gene tree that included 152 of the approximately 400 species of Clupeiformes. We restrict our comparative analyses to Clupeoidei, which includes all Clupeiformes except Denticeps clupeoides because this species was recovered as sister to Ostariophysi rather than a member of Clupeiformes. Habitat transitions were previously estimated using maximum likelihood ancestral character reconstruction (Bloom, Lovejoy, 2014Bloom DD, Lovejoy NR. The evolutionary origins of diadromy inferred from a time-calibrated phylogeny for Clupeiformes (herring and allies). Proc R Soc B [serial on the Internet]. 2014; 281(1778):20132081. Available from: http://dx.doi.org/10.1098/rspb.2013.2081
http://dx.doi.org/10.1098/rspb.2013.2081...
).
We tested for a slow-down in cladogenesis rates using models of speciation and lineages through time within Clupeoidei and several subclades that we selected according to biogeographic region and across multiple phylogenetic scales. We visualized lineage accumulation patterns by generating lineage through time plots (LTT), which plot the log number of lineages through time (Nee et al., 1992Nee S, Mooers AO, Harvey PH. Tempo and mode of evolution revealed from molecular phylogenies. Proc Natl Acad Sci. 1992. 89(17):8322-26.). Lineages that experience a slow-down in diversification over time are expected to show a downturn (logistic curve), while lineages that accumulate constantly are expected to show a linear pattern (exponential). We computed the gamma-statistic (Pybus, Harvey, 2000Pybus OG, Harvey PH. Testing macro-evolutionary models using incomplete molecular phylogenies. Proc R Soc B [serial on the Internet]. 2000; 267(1459):2267-72. Available from: http://dx.doi.org/10.1098/rspb.2000.1278
http://dx.doi.org/10.1098/rspb.2000.1278...
) to test for a signal of decreased diversification over time for all Clupeoidei and several subclades, respectively. A gamma value <0 indicates decelerating diversification through time and a gamma value >0 indicates increasing diversification rates through time. Incomplete taxon sampling can incorrectly infer a decline in diversification rates, and even completely sampled phylogenies may fail to include cryptic or undescribed species. To incorporate known missing taxa we used the Monte Carlo Constant Rates (MCCR) test, which examines gamma values for simulated trees that include a known percentage of missing taxa to generate a critical value (0.05) for rejecting constants rates. We implemented the MCCR test for all clades that returned a negative gamma value without accounting for missing species. Because the portion of missing species is unknown, we tested for a significant slow-down using the MCCR test for a proportion missing taxa from 0.25 to 1.0.
If habitat transitions allow a lineage to circumvent ecological limits we expect clades that have experienced numerous transitions between marine and freshwaters will show patterns of constant diversification (Rabosky, 2013Rabosky DL. Diversity-dependence, ecological speciation, and the role of competition in macroevolution. Annu Rev Ecol Evol Syst [serial on the Internet]. 2013; 44:481-502. Available from: https://doi.org/10.1146/annurev-ecolsys-110512-135800
https://doi.org/10.1146/annurev-ecolsys-...
; Moen, Morlon, 2014Moen D, Morlon H. Why does diversification slow down? Trends Ecol Evol [serial on the Internet]. 2014; 29(4):190-97. Available from: https://doi.org/10.1016/j.tree.2014.01.010
https://doi.org/10.1016/j.tree.2014.01.0...
). We fitted five diversification rate models, including two constant rate and three rate variable models of lineage diversification. These models included: exponential (DDX) and linear (DDL) diversity dependent models, a nondiversity dependent variable rate model (Yule-2-rate), and constant rate models (pure-birth and birth-death). Diversity dependent models are consistent with ecological limits on clade growth because speciation rates depend on the number of extant lineages at a give time (Rabosky, Lovette, 2008Rabosky DL, Lovette IJ. Density-dependent diversification in North American wood warblers. Proc R Soc B [serial on the Internet]. 2008; 275(1649):2363-71. Available from: http://dx.doi.org/10.1098/rspb.2008.0630
http://dx.doi.org/10.1098/rspb.2008.0630...
). We estimated the maximum likelihood of model parameters using the R package LASER (Rabosky, 2006Rabosky DL. LASER: a maximum likelihood toolkit for detecting temporal shifts in diversification rates from molecular phylogenies. Evol Bioinform [serial on the Internet]. 2006; 2(Januray 1):247-250. Available from: https://doi.org/10.1177%2F117693430600200024.
https://doi.org/10.1177%2F11769343060020...
) and determined the best-fit model for each clade using sample-size corrected Akaike Information Criterion (AICc), where the best model has the lowest AIC values.
Results
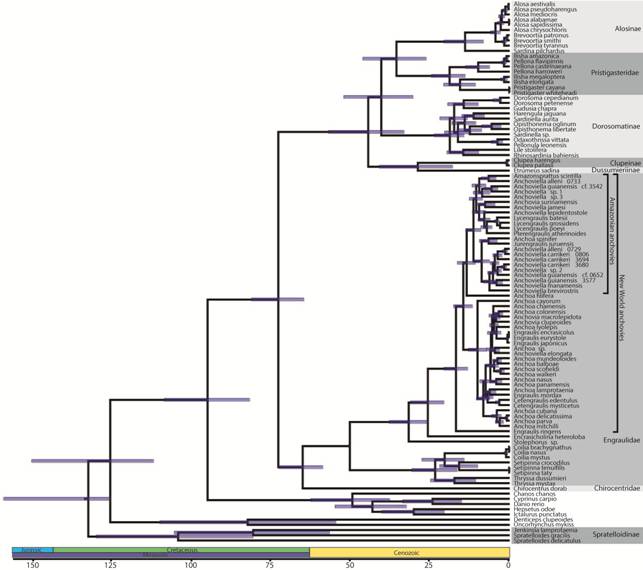
Phylogenetic relationships. Our concatenated Bayesian and maximum likelihood analyses based upon the combined nDNA and mtDNA dataset resulted in largely congruent topologies (Fig. 1; S2 - available only as online supplementary file accessed with the online version of the article at http://www.scielo.br/ni). Both analyses produced phylogenies with poor support for higher-level relationships. In both phylogenies, Clupeoidei (all clupeiforms except the monotypic clupeiform family Denticipitidae containing Denticeps clupeoides) was monophyletic. Denticeps clupeoides was consistently placed among the outgroup taxa, rendering Clupeiformes paraphyletic. Anchovies (Engraulidae) and longfin herrings (Pristigasteridae) were recovered as monophyletic. The herring and sardine family (Clupeidae) was not recovered as monophyletic in either our Bayesian or likelihood analyses. Our Bayesian analysis placed Clupeinae in a lineage with Dussumieriinae that was sister to a lineage containing Pristigasteridae+Chirocentridae+Engraulidae+the remaining clupeid lineages. Our maximum likelihood phylogeny placed Clupeinae as sister to a lineage containing Pristigasteridae+Chirocentridae+Engraulidae in the maximum likelihood phylogeny. Both analyses recovered Spratelloidinae, a subfamily of Dussumieriidae (sensuLavoué et al., 2014Lavoué S, Konstantinidis P, Chen WJ. Progress in clupeiform systematics. In: Ganias K, editor. Biology and ecology of sardines and anchovies. Boca Raton, FL: CRC Press; 2014. p.3-42.) as sister to all remaining clupeoids.

Clupeiform phylogenies pruned to only show major lineages estimated using concatenated Bayesian analysis of the mtDNA dataset (left) and nDNA dataset (right) in BEAST v.2.4.5. Red lines illustrate similarities and differences in the placement of major lineages by mtDNA versus nDNA. Time, in millions of years, is shown along the x-axis. Line drawings depict representative species from clupeiform lineages: Brevoortia tyrannus, Ilisha elongata, Dorosoma cepedianum, Etrumeus sadina, Clupea harengus, Pterengraulis atherinoides, Cetengraulis edentulus, Encrasicholina heteroloba, Stolephorus sp., Coilia dussumieri, Chirocentrus dorab, and Spratelloides gracilis (from top to bottom).
We found discordance between mtDNA and nDNA in tree topology, particularly at the highest taxonomic levels (Figs. 1-2). We only discuss maximum likelihood and Bayesian results separately if they produced conflicting results. The nDNA dataset was largely consistent with the combined nDNA + mtDNA topology with a few notable exceptions. The nDNA Bayesian phylogeny resolved Chirocentridae as sister to Pristigasteridae (PP = 0.70) and the maximum likelihood analysis resolved Chirocentridae as sister to a lineage containing Pristigasteridae, Clupeinae, and Dussumieriinae (BS = 0.82). Bayesian analyses of nDNA placed Clupeinae sister to Pristigasteridae + Chirocentridae (PP = 1.0) and maximum likelihood analyses of nDNA placed Clupeinae sister to Dussumieriinae (BS = 0.67). mtDNA placed Pristigasteridae within Clupeidae sister to Alosinae (PP = 1.0, BS = 0.33), but analyses of nDNA recovered prisigasteridae as sister to a clade including Engraulidae, Chirocentridae, Clupeinae, and Dussumieriinae. In contrast to the nDNA, our mtDNA phylogeny placed Alosinae, Pristigasteridae, Dorosomatinae, Clupeinae, and Dussumierinae in a single clade. nDNA placed Spratelloidinae sister to all remaining Clupeoidei, but mtDNA placed Spratelloidinae outside of Clupeiformes. There was also incongruence between nDNA and mtDNA at lower taxonomic levels. For example, in New World anchovies Anchoa spinifer+Jurengraulis juruensis were either sister to all other members of the Amazonian anchovy clade (nDNA) or nested well within this clade (mtDNA). The placement of a clade that included Engraulis encrasicolus, E. eurystole, and E. japonicas was resolved as sister to all other New World marine anchovies except Anchoa filifera (nDNA) or nested deeply within New World marine anchovies (mtDNA).

Time-calibrated clupeoid phylogeny resulting from Bayesian analysis of the mtDNA dataset in BEAST v.2.4.5. Time, in millions of years, is shown along the x-axis. Node bars show the 95% highest posterior density interval of divergence time estimates.
Divergence times. The combined nDNA+mtDNA, the nDNA, and the mtDNA datasets all estimated an early to middle Cretaceous MRCA of Clupeoidei (Figs. 1 - 4). Branching events between major clupeiform lineages were estimated to occur during the late Cretaceous and early Cenozoic: Spratelloidinae (nDNA: MRCA = 100 Ma; mtDNA: MRCA = 109 Ma), Clupeidae (excluding Clupeinae) (nDNA: MRCA = 79 Ma; mtDNA: MRCA = 33 Ma), Engraulidae (nDNA: MRCA = 50 Ma; mtDNA: MRCA = 51 Ma), and Pristigasteridae (nDNA: MRCA = 42 Ma; mtDNA: MRCA = 21 Ma).

Time-calibrated clupeoid phylogeny resulting from Bayesian analysis of the nDNA dataset in BEAST v.2.4.5. Time, in millions of years, is shown along the x-axis. Node bars show the 95% highest posterior density interval of divergence time estimates.

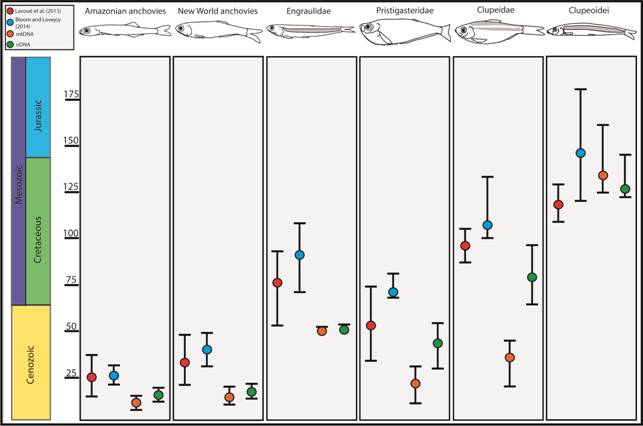
Divergence time estimates for major clupeiform lineages estimated using nDNA and mtDNA separately by this study, mitochondrial genomes by Lavoué et al. (2013Lavoué S, Miya M, Musikasinthorn P, Chen WJ, Nishida M. Mitogenomic evidence for an Indo-West Pacific origin of the Clupeoidei (Teleostei: Clupeiformes). PLoS One [serial on the Internet]. 2013; 8(2):e56485. Available from: https://doi.org/10.1371/journal.pone.0056485
https://doi.org/10.1371/journal.pone.005... ) and a combined mtDNA + nDNA dataset by Bloom, Lovejoy (2014Bloom DD, Lovejoy NR. The evolutionary origins of diadromy inferred from a time-calibrated phylogeny for Clupeiformes (herring and allies). Proc R Soc B [serial on the Internet]. 2014; 281(1778):20132081. Available from: http://dx.doi.org/10.1098/rspb.2013.2081
http://dx.doi.org/10.1098/rspb.2013.2081... ). Time, in millions of years, is shown along the y-axis. Circles represent mean age estimates and whiskers delineate the 95% highest posterior density interval of divergence time estimates.
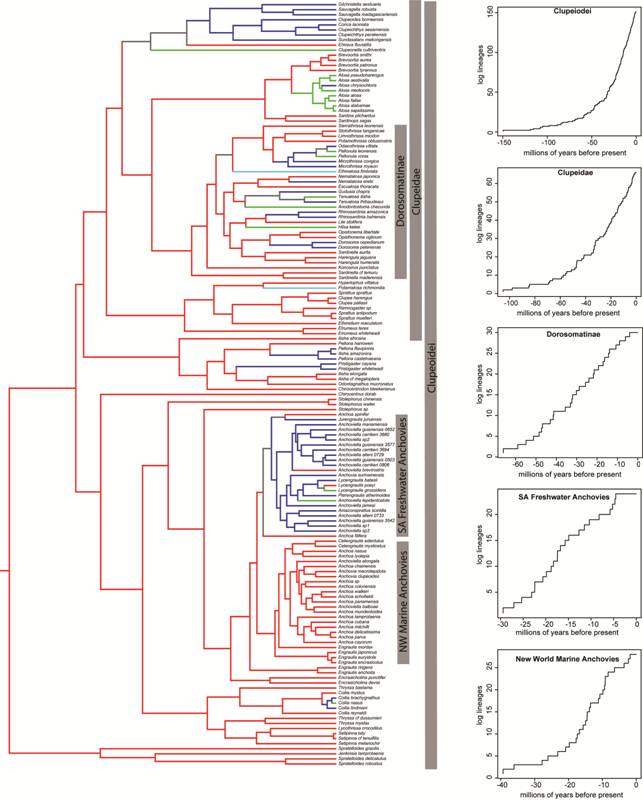
Comparative analyses. The LTT plot for Clupeoidei shows constant lineage accumulation through time (Fig. 5). The calculated gamma value was -0.61 but not significant if missing taxon sampling is as low as 25% (P = 0.89); the Bloom, Lovejoy (2014Bloom DD, Lovejoy NR. The evolutionary origins of diadromy inferred from a time-calibrated phylogeny for Clupeiformes (herring and allies). Proc R Soc B [serial on the Internet]. 2014; 281(1778):20132081. Available from: http://dx.doi.org/10.1098/rspb.2013.2081
http://dx.doi.org/10.1098/rspb.2013.2081...
) dataset included approximately 37% of described Clupeoidei. The best-supported model of diversification was a pure birth model, in which lineages accumulate constantly over time regardless of the number of species in the clade. Clupeidae LTT plots show a constant accumulation of lineages over time. The MCCR test showed the negative gamma value of -1.34 for this clade was not significant (P = 0.93) at threshold of 67% missing data, which represents described clupeid diversity and is likely an underestimate of the actual number of species in this clade. The chosen diversification model was a pure birth model, followed closely by a diversity-dependent model. The LTT plot for the subclade Dorosomatinae (sensuLavoué et al., 2014Lavoué S, Konstantinidis P, Chen WJ. Progress in clupeiform systematics. In: Ganias K, editor. Biology and ecology of sardines and anchovies. Boca Raton, FL: CRC Press; 2014. p.3-42.) showed a slow-down in lineage accumulation and a corresponding highly negative gamma value (-3.08). The negative gamma value for Dorosomatinae was significant at a threshold of 50% missing data (P = 0.01) but not when missing data was set at 75% (P = 0.13). The best-fit model for Dorosomatinae was a diversity dependent logistic growth model in which lineages accumulate as a function of clade diversity. The LTT plot of the New World marine anchovy clade showed a slow-down in lineage accumulation rate and had a gamma value of -2.51. The negative gamma is significant with 25% missing taxa (P = 0.02), but only marginally significant with 50% missing data (P = 0.51). Our taxon sampling (28 species) likely represents more than 50% of the species in this clade, thus we interpret this clade as experiencing a slow-down. The chosen diversification model for New World marine anchovies was diversity dependent growth. The LTT plot for South American freshwater anchovies reveals a slow-down in lineage accumulation over time and the gamma value of -3.59 is highly negative and significant at a threshold of 75% missing taxa (P = 0.01). The best-fit model for New World freshwater anchovies was a diversity dependent model.

The left panel shows a phylogeny of Clupeiformes showing ancestral reconstructions of marine (red), freshwater (blue), anadromous (green) and catadromous (light blue) lineages from Bloom, Lovejoy (2014Bloom DD, Lovejoy NR. The evolutionary origins of diadromy inferred from a time-calibrated phylogeny for Clupeiformes (herring and allies). Proc R Soc B [serial on the Internet]. 2014; 281(1778):20132081. Available from: http://dx.doi.org/10.1098/rspb.2013.2081
http://dx.doi.org/10.1098/rspb.2013.2081... ). The right panel shows lineage through time plots for select clades, which are indicated by grey bars.
Discussion
Concordant and conflicting phylogenetic relationships. The phylogenies resulting from our concatenated Bayesian and maximum likelihood analyses based upon the combined nDNA+mtDNA dataset yielded topologies largely congruent with previous molecular studies (Lavoué et al., 2007Lavoué S, Miya M, Saitoh K, Ishiguro NB, Nishida M. Phylogenetic relationships among anchovies, sardines, herrings and their relatives (Clupeiformes), inferred from whole mitogenome sequences. Mol Phylogenet Evol [serial on the Internet]. 2007; 43(3):1096-105. Available from: http://dx.doi.org/10.1016/j.ympev.2006.09.018
http://dx.doi.org/10.1016/j.ympev.2006.0...
, 2010Lavoué S, Miya M, Nishida M. Mitochondrial phylogenomics of anchovies (family Engraulidae) and recurrent origins of pronounced miniaturization in the order Clupeiformes. Mol Phylogenet Evol [serial on the Internet]. 2010; 56(1):480-85. Available from: https://doi.org/10.1016/j.ympev.2009.11.022
https://doi.org/10.1016/j.ympev.2009.11....
, 2013Lavoué S, Miya M, Musikasinthorn P, Chen WJ, Nishida M. Mitogenomic evidence for an Indo-West Pacific origin of the Clupeoidei (Teleostei: Clupeiformes). PLoS One [serial on the Internet]. 2013; 8(2):e56485. Available from: https://doi.org/10.1371/journal.pone.0056485
https://doi.org/10.1371/journal.pone.005...
, 2017aLavoué S, Bertrand JAM, Chen WJ, Ho HC, Motomura H, Sado T et al. Phylogenetic position of the rainbow sardine Dussumieria (Dussumieriidae) and its bearing on the early evolution of the Clupeoidei. Gene [serial on the Internet]. 2017a; 623:41-47. Available from: http://dx.doi.org/10.1016/j.gene.2017.04.032
http://dx.doi.org/10.1016/j.gene.2017.04...
,bLavoué S, Bertrand JAM, Wang H, Chen W, Ho HC, Motomura H et al. Molecular systematics of the anchovy genus Encrasicholina in the Northwest Pacific. PLoS One [serial on the Internet]. 2017b; 12(7):e0181329. Available from: https://doi.org/10.1371/journal.pone.0181329
https://doi.org/10.1371/journal.pone.018...
; Li, Orti, 2007Li CH, Ortí G. Molecular phylogeny of clupeiformes (Actinopterygii) inferred from nuclear and mitochondrial DNA sequences. Mol Phylogenet Evol [serial on the Internet]. 2007; 44(1):386-98. Available from: http://dx.doi.org/10.1016/j.ympev.2006.10.030
http://dx.doi.org/10.1016/j.ympev.2006.1...
; Bloom, Lovejoy, 2012Bloom DD, Lovejoy NR. Molecular phylogenetics reveals a pattern of biome conservatism in New World anchovies (family Engraulidae). J Evol Biol [serial on the Internet]. 2012; 25(4):701-15. Available from: http://dx.doi.org/10.1111/j.1420-9101.2012.02464.x
http://dx.doi.org/10.1111/j.1420-9101.20...
, 2014Bloom DD, Lovejoy NR. The evolutionary origins of diadromy inferred from a time-calibrated phylogeny for Clupeiformes (herring and allies). Proc R Soc B [serial on the Internet]. 2014; 281(1778):20132081. Available from: http://dx.doi.org/10.1098/rspb.2013.2081
http://dx.doi.org/10.1098/rspb.2013.2081...
), and the Lavoué et al. (2014Lavoué S, Konstantinidis P, Chen WJ. Progress in clupeiform systematics. In: Ganias K, editor. Biology and ecology of sardines and anchovies. Boca Raton, FL: CRC Press; 2014. p.3-42.) classification of Clupeoidei. However, relationships among major clupeiform lineages remain unresolved. Our separate analyses of the nDNA and mtDNA datasets revealed discordance between the mitochondrial and nuclear genomes among major clupeiform lineages.
Genome and gene discordance has long been recognized as a challenge for inferring phylogenetic relationships, including in ray-finned fishes (Waters et al., 2010Waters JM, Rowe DL, Burridge CP, Wallis GP. Gene trees versus species trees: Reassessing life-history evolution in a freshwater fish radiation. Syst Biol [serial on the Internet]. 2010; 59(5):504-17. Available from: https://doi.org/10.1093/sysbio/syq031
https://doi.org/10.1093/sysbio/syq031...
; Toews, Brelsford, 2012Toews DPL, Brelsford A. The biogeography of mitochondrial and nuclear discordance in animals. Mol Ecol [serial on the Internet]. 2012; 21(16):3907-30. Available from: http://dx.doi.org/10.1111/j.1365-294X.2012.05664.x
http://dx.doi.org/10.1111/j.1365-294X.20...
; Betancur-R et al., 2013Betancur-R R, Li C, Munroe TA, Ballesteros JA, Ortí G. Addressing gene tree discordance and non-stationarity to resolve a multi-locus phylogeny of the flatfishes (Teleostei: Pleuronectiformes). Syst Biol [serial on the Internet]. 2013; 62(5):763-85. Available from: http://dx.doi.org/10.1093/sysbio/syt039
http://dx.doi.org/10.1093/sysbio/syt039...
), and can be caused by interspecific hybridization and incomplete lineage sorting (Pamilo, Nei, 1988Pamilo P, Nei M. Relationships between gene trees and species trees. Mol Biol Evol . 1988; 5(5):568-83.; Maddison et al., 2006Maddison WP, Knowles LL. Inferring phylogeny despite incomplete lineage sorting. Syst Biol [serial on the Internet]. 2006; 55(1):21-30. Available from: http://dx.doi.org/10.1080/10635150500354928
http://dx.doi.org/10.1080/10635150500354...
; Wallis et al., 2017Wallis GP, Cameron-Christie SR, Kennedy HL, Palmer G, Sanders TR, Winter DJ. Interspecific hybridization causes long-term phylogenetic discordance between nuclear and mitochondrial genomes in freshwater fishes. Mol Ecol [serial on the Internet]. 2017; 26(12):3116-27. Available from: http://dx.doi.org/10.1111/mec.14096
http://dx.doi.org/10.1111/mec.14096...
). Therefore, it is not surprising that our separate analyses of the nDNA and mtDNA datasets revealed discordance between the mitochondrial and nuclear genomes. In our dataset, most disagreement between genomes occurred in early branching events, with notable conflicts concerning the placement of Spratelloidinae, Chirocentridae, Pristigasteridae, Clupeinae, and Dussumieriinae. Both genomes found Clupeidae and Dussumieriidae (sensuLavoué et al., 2014Lavoué S, Konstantinidis P, Chen WJ. Progress in clupeiform systematics. In: Ganias K, editor. Biology and ecology of sardines and anchovies. Boca Raton, FL: CRC Press; 2014. p.3-42.) to be non-monophyletic because they placed Clupeinae sister to Dussumieriinae and did not place Spratelloidinae sister to Dussumieriinae. Like previous mitogenomic studies (Lavoué et al., 2013Lavoué S, Miya M, Musikasinthorn P, Chen WJ, Nishida M. Mitogenomic evidence for an Indo-West Pacific origin of the Clupeoidei (Teleostei: Clupeiformes). PLoS One [serial on the Internet]. 2013; 8(2):e56485. Available from: https://doi.org/10.1371/journal.pone.0056485
https://doi.org/10.1371/journal.pone.005...
), our mtDNA analyses suggest a close affiliation between Pristigasteridae and Clupeidae, although our mtDNA tree recovered Pristigasteridae nested within Clupeidae, a result that seems tenuous. Previous studies that included nDNA support Pristigasteridae, Engraulidae, and Chirocentridae as a clade, however our analysis of nDNA does not support this topology. Previous mitogenomic studies placed Chirocentridae in a clade sister to Pristigasteridae+Clupeidae (Lavoué et al., 2013Lavoué S, Miya M, Musikasinthorn P, Chen WJ, Nishida M. Mitogenomic evidence for an Indo-West Pacific origin of the Clupeoidei (Teleostei: Clupeiformes). PLoS One [serial on the Internet]. 2013; 8(2):e56485. Available from: https://doi.org/10.1371/journal.pone.0056485
https://doi.org/10.1371/journal.pone.005...
), but previous morphological and molecular studies that included nuclear data have inferred Chirocentridae as sister to Engraulidae (Bloom, Lovejoy, 2014Bloom DD, Lovejoy NR. The evolutionary origins of diadromy inferred from a time-calibrated phylogeny for Clupeiformes (herring and allies). Proc R Soc B [serial on the Internet]. 2014; 281(1778):20132081. Available from: http://dx.doi.org/10.1098/rspb.2013.2081
http://dx.doi.org/10.1098/rspb.2013.2081...
). Therefore, it is surprising that our nDNA dataset placed Chirocentridae sister to Pristigasteridae and our mtDNA dataset placed Chirocentridae sister to Engraulidae. Reconciling incongruence between mitogenomic (Lavoué et al., 2013Lavoué S, Miya M, Musikasinthorn P, Chen WJ, Nishida M. Mitogenomic evidence for an Indo-West Pacific origin of the Clupeoidei (Teleostei: Clupeiformes). PLoS One [serial on the Internet]. 2013; 8(2):e56485. Available from: https://doi.org/10.1371/journal.pone.0056485
https://doi.org/10.1371/journal.pone.005...
) and combined mtDNA+nDNA (Li, Orti, 2007Li CH, Ortí G. Molecular phylogeny of clupeiformes (Actinopterygii) inferred from nuclear and mitochondrial DNA sequences. Mol Phylogenet Evol [serial on the Internet]. 2007; 44(1):386-98. Available from: http://dx.doi.org/10.1016/j.ympev.2006.10.030
http://dx.doi.org/10.1016/j.ympev.2006.1...
; Bloom, Lovejoy, 2014Bloom DD, Lovejoy NR. The evolutionary origins of diadromy inferred from a time-calibrated phylogeny for Clupeiformes (herring and allies). Proc R Soc B [serial on the Internet]. 2014; 281(1778):20132081. Available from: http://dx.doi.org/10.1098/rspb.2013.2081
http://dx.doi.org/10.1098/rspb.2013.2081...
) has been challenging because the combined datasets relied heavily on mtDNA. The short branches and lack of statistical support for any of these relationships suggest lineage sorting might make resolving these relationships challenging. Unfortunately our pruned dataset, which removed nearly all missing data, does not clarify the unstable higher level relationships of Clupeiformes.
This study and previous studies on clupeiform systematics and biology suggest gene tree and genome discordance result from both incomplete lineage sorting and hybridization (Anderson, Karel, 2007Anderson JD, Karel WJ. Genetic evidence for asymmetric hybridization between menhadens (Brevoortia spp.) from peninsular Florida. J Fish Biol [serial on the Internet]. 2007; 71(SB):235-49. Available from: https://doi.org/10.1111/j.1095-8649.2007.01597.x
https://doi.org/10.1111/j.1095-8649.2007...
; Jolly et al., 2011Jolly MT, Maitland PS, Genner MJ. Genetic monitoring of two decades of hybridization between allis shad (Alosa alosa) and twaite shad (Alosa fallax). Conserv Genet. 2011; 12(4):1087-100.; Bloom, Lovejoy, 2012Bloom DD, Lovejoy NR. Molecular phylogenetics reveals a pattern of biome conservatism in New World anchovies (family Engraulidae). J Evol Biol [serial on the Internet]. 2012; 25(4):701-15. Available from: http://dx.doi.org/10.1111/j.1420-9101.2012.02464.x
http://dx.doi.org/10.1111/j.1420-9101.20...
). Hybridization can be difficult to detect (Holder et al., 2001Holder MT, Anderson JA, Holloway AK. Difficulties in detecting hybridization. Syst Biol [serial on the Internet]. 2001; 50(6):978-82. Available from: https://doi.org/10.1080/106351501753462911
https://doi.org/10.1080/1063515017534629...
), and the extent that it has occurred in Clupeiformes over macroevolutionary time is unclear. However, hybridization is common in fishes and there are instances of contemporary interspecific hybridization within marine and freshwater clupeiforms (Anderson, Karel, 2007Anderson JD, Karel WJ. Genetic evidence for asymmetric hybridization between menhadens (Brevoortia spp.) from peninsular Florida. J Fish Biol [serial on the Internet]. 2007; 71(SB):235-49. Available from: https://doi.org/10.1111/j.1095-8649.2007.01597.x
https://doi.org/10.1111/j.1095-8649.2007...
; Jolly et al., 2011Jolly MT, Maitland PS, Genner MJ. Genetic monitoring of two decades of hybridization between allis shad (Alosa alosa) and twaite shad (Alosa fallax). Conserv Genet. 2011; 12(4):1087-100.; McBride et al., 2014McBride MC, Willis TV, Bradford RG, Bentzen P. Genetic diversity and structure of two hybridizing anadromous fishes (Alosa pseudoharengus, Alosa aestivalis) across the northern portion of their ranges. Conserv Genet . 2014; 15(6):1281-98.). Incomplete lineage sorting is most likely when there are large effective population sizes and short branch lengths (Pamilo, Nei, 1988Pamilo P, Nei M. Relationships between gene trees and species trees. Mol Biol Evol . 1988; 5(5):568-83.). There are several regions of the clupeiform phylogeny with very short branches such as New World anchovies and shads (Alosa spp.). Consequently, future clupeiform molecular phylogenetics research should use large, multi-locus datasets to build upon our existing knowledge of clupeiform systematics that is based upon mtDNA datasets (Lavoué et al., 2007Lavoué S, Miya M, Saitoh K, Ishiguro NB, Nishida M. Phylogenetic relationships among anchovies, sardines, herrings and their relatives (Clupeiformes), inferred from whole mitogenome sequences. Mol Phylogenet Evol [serial on the Internet]. 2007; 43(3):1096-105. Available from: http://dx.doi.org/10.1016/j.ympev.2006.09.018
http://dx.doi.org/10.1016/j.ympev.2006.0...
, 2010Lavoué S, Miya M, Nishida M. Mitochondrial phylogenomics of anchovies (family Engraulidae) and recurrent origins of pronounced miniaturization in the order Clupeiformes. Mol Phylogenet Evol [serial on the Internet]. 2010; 56(1):480-85. Available from: https://doi.org/10.1016/j.ympev.2009.11.022
https://doi.org/10.1016/j.ympev.2009.11....
, 2013Lavoué S, Miya M, Musikasinthorn P, Chen WJ, Nishida M. Mitogenomic evidence for an Indo-West Pacific origin of the Clupeoidei (Teleostei: Clupeiformes). PLoS One [serial on the Internet]. 2013; 8(2):e56485. Available from: https://doi.org/10.1371/journal.pone.0056485
https://doi.org/10.1371/journal.pone.005...
, 2017aLavoué S, Bertrand JAM, Chen WJ, Ho HC, Motomura H, Sado T et al. Phylogenetic position of the rainbow sardine Dussumieria (Dussumieriidae) and its bearing on the early evolution of the Clupeoidei. Gene [serial on the Internet]. 2017a; 623:41-47. Available from: http://dx.doi.org/10.1016/j.gene.2017.04.032
http://dx.doi.org/10.1016/j.gene.2017.04...
,bLavoué S, Bertrand JAM, Wang H, Chen W, Ho HC, Motomura H et al. Molecular systematics of the anchovy genus Encrasicholina in the Northwest Pacific. PLoS One [serial on the Internet]. 2017b; 12(7):e0181329. Available from: https://doi.org/10.1371/journal.pone.0181329
https://doi.org/10.1371/journal.pone.018...
) and datasets containing small numbers of loci (Li, Orti, 2007Li CH, Ortí G. Molecular phylogeny of clupeiformes (Actinopterygii) inferred from nuclear and mitochondrial DNA sequences. Mol Phylogenet Evol [serial on the Internet]. 2007; 44(1):386-98. Available from: http://dx.doi.org/10.1016/j.ympev.2006.10.030
http://dx.doi.org/10.1016/j.ympev.2006.1...
; Bloom, Lovejoy, 2012Bloom DD, Lovejoy NR. Molecular phylogenetics reveals a pattern of biome conservatism in New World anchovies (family Engraulidae). J Evol Biol [serial on the Internet]. 2012; 25(4):701-15. Available from: http://dx.doi.org/10.1111/j.1420-9101.2012.02464.x
http://dx.doi.org/10.1111/j.1420-9101.20...
, 2014Bloom DD, Lovejoy NR. The evolutionary origins of diadromy inferred from a time-calibrated phylogeny for Clupeiformes (herring and allies). Proc R Soc B [serial on the Internet]. 2014; 281(1778):20132081. Available from: http://dx.doi.org/10.1098/rspb.2013.2081
http://dx.doi.org/10.1098/rspb.2013.2081...
). We conclude that analyzing individual data partitions is a necessary step in phylogenetic analyses, and that while relying only on mtDNA can be problematic, in combination with nDNA it remains a useful source of data for phylogenetic inference.
Spratelloidinae is currently is either recognized as a subfamily of either Dussumieriidae or Clupeidae (Lavoué et al., 2014Lavoué S, Konstantinidis P, Chen WJ. Progress in clupeiform systematics. In: Ganias K, editor. Biology and ecology of sardines and anchovies. Boca Raton, FL: CRC Press; 2014. p.3-42.). Our analyses found strong support for the recognition of Spratelloidinae as a distinct lineage. Spratelloidinae did not form a monophyletic group with Clupeidae or the either of the two lineages of Dussumieriidae in any of our analyses. Bayesian mtDNA and nDNA analyses placed Spratelloidinae sister to all remaining Clupeidae and mtDNA analyses placed Spratelloidinae with our outgroup. Previous molecular studies have also recovered Spratelloidinae as a distinct group and have most consistently placed this lineage sister to all remaining Clupeoidei (Lavoué et al., 2013Lavoué S, Miya M, Musikasinthorn P, Chen WJ, Nishida M. Mitogenomic evidence for an Indo-West Pacific origin of the Clupeoidei (Teleostei: Clupeiformes). PLoS One [serial on the Internet]. 2013; 8(2):e56485. Available from: https://doi.org/10.1371/journal.pone.0056485
https://doi.org/10.1371/journal.pone.005...
, 2014Lavoué S, Konstantinidis P, Chen WJ. Progress in clupeiform systematics. In: Ganias K, editor. Biology and ecology of sardines and anchovies. Boca Raton, FL: CRC Press; 2014. p.3-42.; Bloom, Lovejoy, 2014Bloom DD, Lovejoy NR. The evolutionary origins of diadromy inferred from a time-calibrated phylogeny for Clupeiformes (herring and allies). Proc R Soc B [serial on the Internet]. 2014; 281(1778):20132081. Available from: http://dx.doi.org/10.1098/rspb.2013.2081
http://dx.doi.org/10.1098/rspb.2013.2081...
; Egan et al., 2018Egan JP, Bloom DD, Kuo CH, Hammer MP, Tongnunui P, Iglésias SP, Sheaves M, Grudpan C, Simons AM. Phylogenetic analysis of trophic niche evolution reveals a latitudinal herbivory gradient in Clupeoidei (herrins, anchovies and allies). Mol Phylogenet Evol [serial on the Internet]. 2018; 124(July 2018):151-61. Available from: http://dx.doi.org/10.1016/j.ympev.2018.03.011
http://dx.doi.org/10.1016/j.ympev.2018.0...
). As such, including Spratelloidinae in either Dussumieriidae or Clupeidae renders these taxa as non-monophyletic. To resolve this taxonomic issue, we propose elevating Spratelloidinae from a subfamily to the level of family and changing the taxon name to Spratelloididae.
Divergence times. Our separate mtDNA- and nDNA-based estimates of divergence times suggest that conflicting results reported by previous mtDNA (Lavoué et al., 2013Lavoué S, Miya M, Musikasinthorn P, Chen WJ, Nishida M. Mitogenomic evidence for an Indo-West Pacific origin of the Clupeoidei (Teleostei: Clupeiformes). PLoS One [serial on the Internet]. 2013; 8(2):e56485. Available from: https://doi.org/10.1371/journal.pone.0056485
https://doi.org/10.1371/journal.pone.005...
, 2017bLavoué S, Bertrand JAM, Wang H, Chen W, Ho HC, Motomura H et al. Molecular systematics of the anchovy genus Encrasicholina in the Northwest Pacific. PLoS One [serial on the Internet]. 2017b; 12(7):e0181329. Available from: https://doi.org/10.1371/journal.pone.0181329
https://doi.org/10.1371/journal.pone.018...
) and mtDNA+nDNA (Bloom, Lovejoy, 2014Bloom DD, Lovejoy NR. The evolutionary origins of diadromy inferred from a time-calibrated phylogeny for Clupeiformes (herring and allies). Proc R Soc B [serial on the Internet]. 2014; 281(1778):20132081. Available from: http://dx.doi.org/10.1098/rspb.2013.2081
http://dx.doi.org/10.1098/rspb.2013.2081...
) studies on clupeiform divergence times are partially due to the different genes in these datasets. We estimated divergence times for major clupeiform clades that were nearly always younger than previous studies. In contrast with previous studies (Zheng et al., 2011Zheng Y, Peng R, Kuro-O M, Zeng X. Exploring patterns and extent of bias in estimating divergence time from mitochondrial DNA sequence data in a particular lineage: A case study of salamanders (Order Caudata). Mol Biol Evol [serial on the Internet]. 2011; 28(9):2521-35. Available from: http://dx.doi.org/10.1093/molbev/msr072
http://dx.doi.org/10.1093/molbev/msr072...
; Mulcahy et al., 2012Mulcahy DG, Noonan BP, Moss T, Townsend TM, Reeder TW, Sites JW et al. Estimating divergence dates and evaluating dating methods using phylogenomic and mitochondrial data in squamate reptiles. Mol Phylogenet Evol [serial on the Internet]. 2012; 65(3):974-91. Available from: http://dx.doi.org/10.1016/j.ympev.2012.08.018
http://dx.doi.org/10.1016/j.ympev.2012.0...
; Dornburg et al., 2014Dornburg A, Townsend JP, Friedman M, Near TJ. Phylogenetic informativeness reconciles ray-finned fish molecular divergence times. BMC Evol Biol [serial on the Internet]. 2014; 14(169):1-14. Available from: https://doi.org/10.1186/s12862-014-0169-0
https://doi.org/10.1186/s12862-014-0169-...
) our mtDNA dataset did not systematically estimate older divergence times than the nDNA dataset (Fig. 4). This result and may be because the nuclear genes in our dataset appear to evolve at a rate similar to the mitochondrial loci. This finding is encouraging because it suggests that including mtDNA in divergence time analyses does not always lead to spurious age estimates. Age estimates for the MRCA of Pristigasteridae and Clupeidae were particularly different between the mtDNA and nDNA datasets. Differences between in our age estimates from previous studies are likely also due to the exclusion of fossils used in previous studies because of uncertainty regarding their placement (Lavoué et al., 2017bLavoué S, Bertrand JAM, Wang H, Chen W, Ho HC, Motomura H et al. Molecular systematics of the anchovy genus Encrasicholina in the Northwest Pacific. PLoS One [serial on the Internet]. 2017b; 12(7):e0181329. Available from: https://doi.org/10.1371/journal.pone.0181329
https://doi.org/10.1371/journal.pone.018...
): †Gasteroclupea branisai, previously used to set a minimum age for the MRCA of Pristigasteridae, †Nolfia riachuelensis, previously used to set a minimum age for the MRCA of Clupeidae (Bloom, Lovejoy, 2014Bloom DD, Lovejoy NR. The evolutionary origins of diadromy inferred from a time-calibrated phylogeny for Clupeiformes (herring and allies). Proc R Soc B [serial on the Internet]. 2014; 281(1778):20132081. Available from: http://dx.doi.org/10.1098/rspb.2013.2081
http://dx.doi.org/10.1098/rspb.2013.2081...
), and †Lecceclupea ehiravaensis to set a minimum age for the MRCA of the clupeid lineage Ehiravini (Gilchristella + Clupeichthys; Lavoué et al., 2013Lavoué S, Miya M, Musikasinthorn P, Chen WJ, Nishida M. Mitogenomic evidence for an Indo-West Pacific origin of the Clupeoidei (Teleostei: Clupeiformes). PLoS One [serial on the Internet]. 2013; 8(2):e56485. Available from: https://doi.org/10.1371/journal.pone.0056485
https://doi.org/10.1371/journal.pone.005...
).
Habitat transitions circumvent ecological limits across large phylogenetic scales. Over time, ecological limits are thought to slow net diversification rates within clades via resource competition (Rabosky, 2009aRabosky DL. Ecological limits and diversification rate: alternative paradigms to explain the variation in species richness among clades and regions. Ecol Lett [serial on the Internet]. 2009a; 12(8):735-43. Available from: http://dx.doi.org/10.1111/j.1461-0248.2009.01333.x
http://dx.doi.org/10.1111/j.1461-0248.20...
,bRabosky DL. Ecological limits on clade diversification in higher taxa. Am Nat . 2009b; 173(5):662-74., 2013Rabosky DL. Diversity-dependence, ecological speciation, and the role of competition in macroevolution. Annu Rev Ecol Evol Syst [serial on the Internet]. 2013; 44:481-502. Available from: https://doi.org/10.1146/annurev-ecolsys-110512-135800
https://doi.org/10.1146/annurev-ecolsys-...
; Moen, Morlon, 2014Moen D, Morlon H. Why does diversification slow down? Trends Ecol Evol [serial on the Internet]. 2014; 29(4):190-97. Available from: https://doi.org/10.1016/j.tree.2014.01.010
https://doi.org/10.1016/j.tree.2014.01.0...
). Our results show that some trans-marine/freshwater lineages have likely circumvented ecological limits on clade diversity, while ecological limits have potentially slowed net diversification rates within several other clupeiform clades. For example, diversification patterns across all Clupeoidei and Clupeidae, which are widespread clades containing dozens of marine/freshwater transitions, showed constant diversification rates over time. However, both geographically restricted trans-marine/freshwater clades and entirely marine or freshwater clades showed diversity dependent diversification. For example, two neotropical clades, the new world marine anchovies and South American freshwater anchovies (largely freshwater, but includes at least two reversals to marine) showed diversity dependent diversification. Dorosomatinae, a largely tropical and subtropical trans-marine/freshwater clade showed declining lineage accumulation and the best-fit model was diversity dependent diversification. This suggests that on regional or local scales ecological limits are regulating diversity patterns in Clupeoidei, even in trans-marine/freshwater clades. A large study on diversification in mammals found that medium to large clades (hundreds of species) experienced diversification slow-downs, while small clades experienced constant rates (Machac et al., 2018Machac A, Graham CH, Storch D. Ecological controls of mammalian diversification vary with phylogenetic scale. Glob Ecol Biogeogr [serial on the Internet]. 2018; 27(1):32-46. Available from: https://doi.org/10.1111/geb.12642
https://doi.org/10.1111/geb.12642...
). The relationship between clade size and diversification dynamics detected in mammals is consistent with theory on ecological limits (Rabosky, 2009bRabosky DL. Ecological limits on clade diversification in higher taxa. Am Nat . 2009b; 173(5):662-74., 2013Rabosky DL. Diversity-dependence, ecological speciation, and the role of competition in macroevolution. Annu Rev Ecol Evol Syst [serial on the Internet]. 2013; 44:481-502. Available from: https://doi.org/10.1146/annurev-ecolsys-110512-135800
https://doi.org/10.1146/annurev-ecolsys-...
), and numerous empirical studies (Rabosky, Glor, 2010Rabosky DL, Glor RE. Equilibrium speciation dynamics in a model adaptive radiation of island lizards. Proc Natl Acad Sci [serial on the Internet]. 2010. 107(51):22178-83. Available from: https://doi.org/10.1073/pnas.1007606107
https://doi.org/10.1073/pnas.1007606107...
; Kennedy et al., 2012Kennedy JD, Weir JT, Hooper DM, Tietze DT, Martens J, Price TD. Ecological limits on diversification of the Himalayan core Corvoidea. Evolution [serial on the Internet]. 2012; 66(8):2599-613. Available from: https://doi.org/10.1111/j.1558-5646.2012.01618.x
https://doi.org/10.1111/j.1558-5646.2012...
; López-Fernández et al., 2013López-Fernández H, Arbour JH, Winemiller KO, Honeycutt RL. Testing for ancient adaptive radiations in neotropical cichilid fishes. Evolution [serial on the Internet]. 2013; 67(5):1321-37. Available from: https://doi.org/10.1111/evo.12038
https://doi.org/10.1111/evo.12038...
; Weir, Mursleen, 2013Weir JT, Mursleen S. Diversity-dependent cladogenesis and trait evolution in the adaptive radiation of Auks (Aves: Alcidae). Evolution [serial on the Internet]. 2013; 67(2):403-16. Available from: https://doi.org/10.1111/j.1558-5646.2012.01786.x
https://doi.org/10.1111/j.1558-5646.2012...
), which taken with our findings, suggests that trans-marine/freshwater clades may have unique diversification dynamics. Determining the relationship between phylogenetic and geographic scales and diversification dynamics will be critical step towards understanding the role of ecological limits on diversification (Rabosky, Hurlbert, 2015Rabosky DL, Hurlbert AH. Species richness at continental scales is dominated by ecological limits. Am Nat [serial on the Internet]. 2015; 185(5):572-83. Available from: http://dx.doi.org/10.1086/680850
http://dx.doi.org/10.1086/680850...
; Graham et al., 2018Graham CH, Storch D, Machac A. Phylogenetic scale in ecology and evolution. Glob Ecol Biogeogr [serial on the Internet]. 2018; 27(2):175-87. Available from: https://doi.org/10.1111/geb.12686
https://doi.org/10.1111/geb.12686...
).
The link between ecological diversity and diversification rates may be widespread (Ezard et al., 2011Ezard THG, Aze T, Pearson PN, Purvis A. Interplay between changing climate and species’ ecology drives macroevolutionary dynamics. Science [serial on the Internet]. 2011; 332(6027):349-51. Available from: http://dx.doi.org/10.1126/science.1203060
http://dx.doi.org/10.1126/science.120306...
). Several other trans-marine/freshwater clades also exhibit constant diversification rates across large phylogenetic scales, such as new world silversides (Atherinopsidae; Bloom et al., 2013Bloom DD, Weir JT, Piller KR, Lovejoy NR. Do freshwater fishes diversify faster than marine fishes? A test using state-dependent diversification analyses and molecular phylogenetics of New World silversides (Atherinopsidae). Evolution [serial on the Internet]. 2013; 67(7):2040-57. Available from: https://doi.org/10.1111/evo.12074
https://doi.org/10.1111/evo.12074...
) and ariid catfishes (Ariidae; Betancur-R et al., 2012Betancur-R R, Ortí G, Stein AM, Marceniuk AP, Pyron AR. Apparent signal of competition limiting diversification after ecological transitions from marine to freshwater habitats. Ecol Lett [serial on the Internet]. 2012; 15(8):822-30. Available from: http://dx.doi.org/10.1111/j.1461-0248.2012.01802.x
http://dx.doi.org/10.1111/j.1461-0248.20...
). Both clades experienced repeated transitions between marine and freshwaters and also show patterns of constant diversification over time. Hydrophilid beetles have transitioned between aquatic and terrestrial habitats repeatedly and have a positive relationship between clade age and species richness, which is evidence for constant diversification rates (Bloom et al., 2014Bloom DD, Fikáček M, Short AEZ. Clade age and diversification rate variation explain disparity in species richness among water scavenger beetle (Hydrophilidae) Lineages. PLoS One [serial on the Internet]. 2014; 9(6):e98430. Available from: https://doi.org/10.1371/journal.pone.0098430
https://doi.org/10.1371/journal.pone.009...
). Derryberry et al. (2011Derryberry EP, Claramunt S, Derryberry G, Chesser RT, Cracraft J, Aleixo A et al. Lineage diversification and morphological evolution in a large-scale continental radiation: The neotropical ovenbirds and woodcreepers (Aves: Furnariidae). Evolution [serial on the Internet]. 2011; 65(10):2973-86. Available from: http://dx.doi.org/10.1111/j.1558-5646.2011.01374.x
http://dx.doi.org/10.1111/j.1558-5646.20...
) found that species rich and ecologically diverse woodcreepers and ovenbirds show constant diversification rates across continental scales and concluded these clades have not reached their ecological limit. Guinot, Cavin (2015Guinot G, Cavin L. Contrasting “fish” diversity dynamics between marine and freswhater environments. Curr Biol [serial on the Internet]. 2015; 25(17):2314-18. Available from: http://dx.doi.org/10.1016/j.cub.2015.07.033
http://dx.doi.org/10.1016/j.cub.2015.07....
) found that freshwater fishes have yet to reach ecological limits, while marine fishes fit an equilibrium model, which suggests in some cases, non-equilibrium dynamics in ecologically diverse clades may be driven by one or few habitats. Together, our results and these studies suggest that clades that undergo evolutionary habitat transitions may avoid ecological limits over deep time.
Most studies to date have detected slowdowns in diversification rates over time (Rabosky, 2013Rabosky DL. Diversity-dependence, ecological speciation, and the role of competition in macroevolution. Annu Rev Ecol Evol Syst [serial on the Internet]. 2013; 44:481-502. Available from: https://doi.org/10.1146/annurev-ecolsys-110512-135800
https://doi.org/10.1146/annurev-ecolsys-...
; Moen, Morlon, 2014Moen D, Morlon H. Why does diversification slow down? Trends Ecol Evol [serial on the Internet]. 2014; 29(4):190-97. Available from: https://doi.org/10.1016/j.tree.2014.01.010
https://doi.org/10.1016/j.tree.2014.01.0...
). Clades revealed to be undergoing constant diversification rates were often young clades that likely have not reached their ecological limit (e.g. Weir, 2006Weir JT. Divergent timing and patterns of species accumulation in lowland and highland neotropical birds. Evolution. 2006; 60(4):842-55.). Assuming ecological limits exist, why do Clupeoidei and other trans-marine/freshwater clades show constant diversification rates across large phylogenetic scales? In aquatic communities ecological limits may operate separately in marine and freshwaters because most organisms are restricted to either marine or freshwater habitats (Lee, Bell, 1999Lee CE, Bell MA. Causes and consequences of recent freshwater invasions by saltwater animals. Trends Ecol Evol [serial on the Internet]. 1999; 14(7):284-88. Available from: https://doi.org/10.1016/S0169-5347(99)01596-7
https://doi.org/10.1016/S0169-5347(99)01...
; Bloom, Lovejoy, 2012Bloom DD, Lovejoy NR. Molecular phylogenetics reveals a pattern of biome conservatism in New World anchovies (family Engraulidae). J Evol Biol [serial on the Internet]. 2012; 25(4):701-15. Available from: http://dx.doi.org/10.1111/j.1420-9101.2012.02464.x
http://dx.doi.org/10.1111/j.1420-9101.20...
; Grosberg et al., 2012Grosberg RK, Vermeij GJ, Wainwright PC. Biodiversity in water and on land. Curr Biol [serial on the Internet]. 2012; 22(21):900-3. Available from: https://doi.org/10.1016/j.cub.2012.09.050
https://doi.org/10.1016/j.cub.2012.09.05...
), with near complete turnover in community composition between adjacent marine and freshwaters (Winemiller, Leslie, 1992Winemiller KO, Leslie MA. Fish assemblages across a complex, tropical freshwater/marine ecotone. Environ Biol Fishes. 1992; 34(1):29-50.). Thus a lineage may reach saturation in either a marine or freshwater habitat, but have minimal influence on the dynamics in the other habitat. If a lineage undergoes a macroevolutionary transition to an adjacent habitat (e.g. marine to freshwater, or vice versa), it may experience reduced competition, release from pathogens, new ecological opportunity for diversification, or newly imposed ecological limits (Rabosky, 2009aRabosky DL. Ecological limits and diversification rate: alternative paradigms to explain the variation in species richness among clades and regions. Ecol Lett [serial on the Internet]. 2009a; 12(8):735-43. Available from: http://dx.doi.org/10.1111/j.1461-0248.2009.01333.x
http://dx.doi.org/10.1111/j.1461-0248.20...
,bRabosky DL. Ecological limits on clade diversification in higher taxa. Am Nat . 2009b; 173(5):662-74., 2013Rabosky DL. Diversity-dependence, ecological speciation, and the role of competition in macroevolution. Annu Rev Ecol Evol Syst [serial on the Internet]. 2013; 44:481-502. Available from: https://doi.org/10.1146/annurev-ecolsys-110512-135800
https://doi.org/10.1146/annurev-ecolsys-...
; Ricklefs, 2011Ricklefs RE. A biogeographical perspective on ecological systems: some personal reflections. J Biogeogr [serial on the Internet]. 2011; 38(11):2045-56. Available from: https://doi.org/10.1111/j.1365-2699.2011.02520.x
https://doi.org/10.1111/j.1365-2699.2011...
; Moen, Morlon, 2014Moen D, Morlon H. Why does diversification slow down? Trends Ecol Evol [serial on the Internet]. 2014; 29(4):190-97. Available from: https://doi.org/10.1016/j.tree.2014.01.010
https://doi.org/10.1016/j.tree.2014.01.0...
). Repeated exposure to ecological opportunity may maintain diversification rates by increasing the probability of speciation (Barraclough et al., 1998Barraclough TG, Vogler AP, Harvey PH. Revealing the factors that promote speciation. Philos Trans Biol Sci. 1998; 353(1366):241-49.). Alternatively, trans-marine/freshwater clades may have an affinity to avoid extinction. Trans-marine/freshwater clades may have an intrinsic ability to respond to dynamic environmental conditions over geological timescales (Bamber, Henderson, 1988Bamber PA, Henderson RN. Pre-adaptive plasticity in atherinids and the estuarine seat of teleost evolution. J Fish Biol . 1988; 33(A):17-23.; Lee, Bell, 1999Lee CE, Bell MA. Causes and consequences of recent freshwater invasions by saltwater animals. Trends Ecol Evol [serial on the Internet]. 1999; 14(7):284-88. Available from: https://doi.org/10.1016/S0169-5347(99)01596-7
https://doi.org/10.1016/S0169-5347(99)01...
), which may cause extinction in lineages that are unable to respond to environmental change (Jablonski, 2008Jablonski D. Species selection: Theory and data. Annu Rev Ecol Evol Syst [serial on the Internet]. 2008; 39(1):501-24. Available from: https://doi.org/10.1146/annurev.ecolsys.39.110707.173510
https://doi.org/10.1146/annurev.ecolsys....
; Ezard et al., 2011Ezard THG, Aze T, Pearson PN, Purvis A. Interplay between changing climate and species’ ecology drives macroevolutionary dynamics. Science [serial on the Internet]. 2011; 332(6027):349-51. Available from: http://dx.doi.org/10.1126/science.1203060
http://dx.doi.org/10.1126/science.120306...
; Moen, Morlon, 2014Moen D, Morlon H. Why does diversification slow down? Trends Ecol Evol [serial on the Internet]. 2014; 29(4):190-97. Available from: https://doi.org/10.1016/j.tree.2014.01.010
https://doi.org/10.1016/j.tree.2014.01.0...
).
Alternatives to ecological limits. We have argued that lineages that undergo habitat transitions avoid ecological limits on species richness and play an important role in structuring diversity patterns. However, this relies on the assumption that ecological limits exist and regulate clade diversity. Recently, Harmon, Harrison (2015Harmon LJ, Harrison S. Species Diversity Is Dynamic and unbounded at local and continental scales. Am Nat [serial on the Internet]. 2015; 185(5):584-93. Available from: http://dx.doi.org/10.1086/680859
http://dx.doi.org/10.1086/680859...
) proposed that species diversity is unbounded by ecological limits at both local and continental scales, and varies dynamically over space and time. It is also possible that diversity patterns are driven by extrinsic factors, such as environmental change and paleogeography rather than diversity dependent interactions (Ezard et al., 2011Ezard THG, Aze T, Pearson PN, Purvis A. Interplay between changing climate and species’ ecology drives macroevolutionary dynamics. Science [serial on the Internet]. 2011; 332(6027):349-51. Available from: http://dx.doi.org/10.1126/science.1203060
http://dx.doi.org/10.1126/science.120306...
; Moen, Morlon, 2014Moen D, Morlon H. Why does diversification slow down? Trends Ecol Evol [serial on the Internet]. 2014; 29(4):190-97. Available from: https://doi.org/10.1016/j.tree.2014.01.010
https://doi.org/10.1016/j.tree.2014.01.0...
). If ecological limits are not regulating species diversity then constant diversification rates are not explained by circumventing ecological limits, but rather driven by neutral processes. Allopatric speciation is the primary mode of speciation in fishes (Coyne, Orr, 2004Coyne JA, Orr HA. Speciation. Sunderland: Sinauer Associates; 2004.). The process of vicariance and subsequent speciation generally divides a widespread ancestor into daughter lineages with smaller ranges sizes. As diversification of a clade proceeds, species with smaller ranges are less likely to experience vicariant events, resulting in a slow-down in diversification (Moen, Morlon, 2014Moen D, Morlon H. Why does diversification slow down? Trends Ecol Evol [serial on the Internet]. 2014; 29(4):190-97. Available from: https://doi.org/10.1016/j.tree.2014.01.010
https://doi.org/10.1016/j.tree.2014.01.0...
). Habitat transitions may result in allopatric speciation; if so, repeated transitions will result in constant diversification over time. Thus if speciation is linked to habitat transitions then ecological limits need not be invoked to explain constant diversification in trans-marine/freshwater clades.
Phylogenetic comparative methods offer a powerful suite of tools to determine the processes underlying diversity patterns. The Neotropics is a biodiversity hotspot, particularly for fishes. However, few studies have tested for ecological limits on clades of Neotropical fishes. This is likely because phylogenetic relationships of many Neotropical clades remain uncertain. However, in this study we demonstrate that Clupeiformes, a clade with unresolved systematics at multiple phylogenetic levels, can be a useful model for testing macroevolutionary hypotheses. Specifically we provide evidence that trans-marine/freshwater clades avoid ecological limits on clade diversity across higher phylogenetic and geographic scales. The neotropics are rich with trans-marine/freshwater clades. If habitat transitions circumvent ecological limits or drive non-diversity dependent processes that yield constant diversification rates, understanding these processes will be important for understanding the origins and maintenance of this region’s tremendous diversity. More generally, habitat transitions across other environmental gradients (e.g. benthic vs. pelagic, riverine vs. lacustrine) may also circumvent ecological limits on clade diversification. We hope this study motivates future studies on lineage diversification and trait evolution to assess the role of ecological limits across Neotropical fishes.
Acknowledgments
DDB. was supported by start up funds and a FRACAA award from Western Michigan University. J.P.E. received financial support from a National Science Foundation Graduate Research Fellowship (00039202). DDB. thanks Hernan Lopez-Fernandez for over a decade of discussing neotropical fish evolution, which greatly improved this manuscript. We thank Ricardo Betancur-R, Marina Loeb and an anonymous reviewer for providing helpful feedback that improved this manuscript. M. Loeb graciously helped us revise our Portuguese abstract. We are grateful for the opportunity to participate in the II International Symposium on Phylogeny and Classification of Neotropical Fishes.
References
- Albert JS, Reis RE. Historical biogeography of neotropical freshwater fishes. Berkeley and Los Angeles, California. University of California Press; 2011.
- Anderson JD, Karel WJ. Genetic evidence for asymmetric hybridization between menhadens (Brevoortia spp) from peninsular Florida. J Fish Biol [serial on the Internet]. 2007; 71(SB):235-49. Available from: https://doi.org/10.1111/j.1095-8649.2007.01597.x
» https://doi.org/10.1111/j.1095-8649.2007.01597.x - Bamber PA, Henderson RN. Pre-adaptive plasticity in atherinids and the estuarine seat of teleost evolution. J Fish Biol . 1988; 33(A):17-23.
- Barraclough TG, Vogler AP, Harvey PH. Revealing the factors that promote speciation. Philos Trans Biol Sci. 1998; 353(1366):241-49.
- Betancur-R R. Molecular phylogenetics supports multiple evolu tionary transitions from marine to freshwater habitats in ariid catfishes. Mol Phylogenet Evol [serial on the Internet]. 2010; 55(1):249-58. Available from: https://doi.org/10.1016/j.ympev.2009.12.018
» https://doi.org/10.1016/j.ympev.2009.12.018 - Betancur-R R, Li C, Munroe TA, Ballesteros JA, Ortí G. Addressing gene tree discordance and non-stationarity to resolve a multi-locus phylogeny of the flatfishes (Teleostei: Pleuronectiformes). Syst Biol [serial on the Internet]. 2013; 62(5):763-85. Available from: http://dx.doi.org/10.1093/sysbio/syt039
» http://dx.doi.org/10.1093/sysbio/syt039 - Betancur-R R, Ortí G, Stein AM, Marceniuk AP, Pyron AR. Apparent signal of competition limiting diversification after ecological transitions from marine to freshwater habitats. Ecol Lett [serial on the Internet]. 2012; 15(8):822-30. Available from: http://dx.doi.org/10.1111/j.1461-0248.2012.01802.x
» http://dx.doi.org/10.1111/j.1461-0248.2012.01802.x - Bloom DD, Fikáček M, Short AEZ. Clade age and diversification rate variation explain disparity in species richness among water scavenger beetle (Hydrophilidae) Lineages. PLoS One [serial on the Internet]. 2014; 9(6):e98430. Available from: https://doi.org/10.1371/journal.pone.0098430
» https://doi.org/10.1371/journal.pone.0098430 - Bloom DD, Lovejoy NR. Molecular phylogenetics reveals a pattern of biome conservatism in New World anchovies (family Engraulidae). J Evol Biol [serial on the Internet]. 2012; 25(4):701-15. Available from: http://dx.doi.org/10.1111/j.1420-9101.2012.02464.x
» http://dx.doi.org/10.1111/j.1420-9101.2012.02464.x - Bloom DD, Lovejoy NR. The evolutionary origins of diadromy inferred from a time-calibrated phylogeny for Clupeiformes (herring and allies). Proc R Soc B [serial on the Internet]. 2014; 281(1778):20132081. Available from: http://dx.doi.org/10.1098/rspb.2013.2081
» http://dx.doi.org/10.1098/rspb.2013.2081 - Bloom DD, Lovejoy NR. On the origins of marine-derived freshwater fishes in South America. J Biogeogr [serial on the Internet]. 2017; 44(9):1927-38. Available from: https://doi.org/10.1111/jbi.12954
» https://doi.org/10.1111/jbi.12954 - Bloom DD, Weir JT, Piller KR, Lovejoy NR. Do freshwater fishes diversify faster than marine fishes? A test using state-dependent diversification analyses and molecular phylogenetics of New World silversides (Atherinopsidae). Evolution [serial on the Internet]. 2013; 67(7):2040-57. Available from: https://doi.org/10.1111/evo.12074
» https://doi.org/10.1111/evo.12074 - Bouckaert R, Heled J, Kühnert D, Vaughan T, Wu CH, Xie D et al BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comput Biol [serial on the Internet]. 2014; 10(4):e1003537. Available from: https://doi.org/10.1371/journal.pcbi.1003537
» https://doi.org/10.1371/journal.pcbi.1003537 - Collar DC, Schulte JA, O’Meara BC, Losos JB. Habitat use affects morphological diversification in dragon lizards. J Evol Biol [serial on the Internet]. 2010; 23(5):1033-49. Available from: http://dx.doi.org/10.1111/j.1420-9101.2010.01971.x
» http://dx.doi.org/10.1111/j.1420-9101.2010.01971.x - Coyne JA, Orr HA. Speciation. Sunderland: Sinauer Associates; 2004.
- Derryberry EP, Claramunt S, Derryberry G, Chesser RT, Cracraft J, Aleixo A et al Lineage diversification and morphological evolution in a large-scale continental radiation: The neotropical ovenbirds and woodcreepers (Aves: Furnariidae). Evolution [serial on the Internet]. 2011; 65(10):2973-86. Available from: http://dx.doi.org/10.1111/j.1558-5646.2011.01374.x
» http://dx.doi.org/10.1111/j.1558-5646.2011.01374.x - Di Dario F. 2009. Chirocentrids as engrauloids: evidence from suspensorium, branchial arches, and infraorbital bones (Clupeomorpha, Teleostei). Zool J Linn Soc. 2009; 156:363-383.
- Dornburg A, Townsend JP, Friedman M, Near TJ. Phylogenetic informativeness reconciles ray-finned fish molecular divergence times. BMC Evol Biol [serial on the Internet]. 2014; 14(169):1-14. Available from: https://doi.org/10.1186/s12862-014-0169-0
» https://doi.org/10.1186/s12862-014-0169-0 - Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004; 32(5):1792-97.
- Egan JP, Bloom DD, Kuo CH, Hammer MP, Tongnunui P, Iglésias SP, Sheaves M, Grudpan C, Simons AM. Phylogenetic analysis of trophic niche evolution reveals a latitudinal herbivory gradient in Clupeoidei (herrins, anchovies and allies). Mol Phylogenet Evol [serial on the Internet]. 2018; 124(July 2018):151-61. Available from: http://dx.doi.org/10.1016/j.ympev.2018.03.011
» http://dx.doi.org/10.1016/j.ympev.2018.03.011 - Ezard THG, Aze T, Pearson PN, Purvis A. Interplay between changing climate and species’ ecology drives macroevolutionary dynamics. Science [serial on the Internet]. 2011; 332(6027):349-51. Available from: http://dx.doi.org/10.1126/science.1203060
» http://dx.doi.org/10.1126/science.1203060 - Graham CH, Storch D, Machac A. Phylogenetic scale in ecology and evolution. Glob Ecol Biogeogr [serial on the Internet]. 2018; 27(2):175-87. Available from: https://doi.org/10.1111/geb.12686
» https://doi.org/10.1111/geb.12686 - Grande L. Recent and fossil clupeomorph fishes with materials for revision of the subgroups of clupeoids. Bull Am Museum Nat Hist. 1985; 181(2):231-372.
- Grande L, Nelson G. Interrelationships of fossil and recent anchovies (Teleostei: Engrauloidea) and description of a new species from the Miocene of Cyprus Am Museum Novit. 1985; 2826:1-16.
- Grosberg RK, Vermeij GJ, Wainwright PC. Biodiversity in water and on land. Curr Biol [serial on the Internet]. 2012; 22(21):900-3. Available from: https://doi.org/10.1016/j.cub.2012.09.050
» https://doi.org/10.1016/j.cub.2012.09.050 - Guinot G, Cavin L. Contrasting “fish” diversity dynamics between marine and freswhater environments. Curr Biol [serial on the Internet]. 2015; 25(17):2314-18. Available from: http://dx.doi.org/10.1016/j.cub.2015.07.033
» http://dx.doi.org/10.1016/j.cub.2015.07.033 - Harmon LJ, Harrison S. Species Diversity Is Dynamic and unbounded at local and continental scales. Am Nat [serial on the Internet]. 2015; 185(5):584-93. Available from: http://dx.doi.org/10.1086/680859
» http://dx.doi.org/10.1086/680859 - Holder MT, Anderson JA, Holloway AK. Difficulties in detecting hybridization. Syst Biol [serial on the Internet]. 2001; 50(6):978-82. Available from: https://doi.org/10.1080/106351501753462911
» https://doi.org/10.1080/106351501753462911 - Jablonski D. Species selection: Theory and data. Annu Rev Ecol Evol Syst [serial on the Internet]. 2008; 39(1):501-24. Available from: https://doi.org/10.1146/annurev.ecolsys.39.110707.173510
» https://doi.org/10.1146/annurev.ecolsys.39.110707.173510 - Jolly MT, Maitland PS, Genner MJ. Genetic monitoring of two decades of hybridization between allis shad (Alosa alosa) and twaite shad (Alosa fallax). Conserv Genet. 2011; 12(4):1087-100.
- Kennedy JD, Weir JT, Hooper DM, Tietze DT, Martens J, Price TD. Ecological limits on diversification of the Himalayan core Corvoidea. Evolution [serial on the Internet]. 2012; 66(8):2599-613. Available from: https://doi.org/10.1111/j.1558-5646.2012.01618.x
» https://doi.org/10.1111/j.1558-5646.2012.01618.x - Lanfear R, Calcott B, Ho SYW, Guindon S. PartitionFinder: Combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol [serial on the Internet]. 2012; 29(6):1695-701. Available from: http://dx.doi.org/10.1093/molbev/mss020
» http://dx.doi.org/10.1093/molbev/mss020 - Lavoué S, Bertrand JAM, Chen WJ, Ho HC, Motomura H, Sado T et al Phylogenetic position of the rainbow sardine Dussumieria (Dussumieriidae) and its bearing on the early evolution of the Clupeoidei. Gene [serial on the Internet]. 2017a; 623:41-47. Available from: http://dx.doi.org/10.1016/j.gene.2017.04.032
» http://dx.doi.org/10.1016/j.gene.2017.04.032 - Lavoué S, Bertrand JAM, Wang H, Chen W, Ho HC, Motomura H et al Molecular systematics of the anchovy genus Encrasicholina in the Northwest Pacific. PLoS One [serial on the Internet]. 2017b; 12(7):e0181329. Available from: https://doi.org/10.1371/journal.pone.0181329
» https://doi.org/10.1371/journal.pone.0181329 - Lavoué S, Konstantinidis P, Chen WJ. Progress in clupeiform systematics. In: Ganias K, editor. Biology and ecology of sardines and anchovies. Boca Raton, FL: CRC Press; 2014. p.3-42.
- Lavoué S, Miya M, Musikasinthorn P, Chen WJ, Nishida M. Mitogenomic evidence for an Indo-West Pacific origin of the Clupeoidei (Teleostei: Clupeiformes). PLoS One [serial on the Internet]. 2013; 8(2):e56485. Available from: https://doi.org/10.1371/journal.pone.0056485
» https://doi.org/10.1371/journal.pone.0056485 - Lavoué S, Miya M, Nishida M. Mitochondrial phylogenomics of anchovies (family Engraulidae) and recurrent origins of pronounced miniaturization in the order Clupeiformes. Mol Phylogenet Evol [serial on the Internet]. 2010; 56(1):480-85. Available from: https://doi.org/10.1016/j.ympev.2009.11.022
» https://doi.org/10.1016/j.ympev.2009.11.022 - Lavoué S, Miya M, Saitoh K, Ishiguro NB, Nishida M. Phylogenetic relationships among anchovies, sardines, herrings and their relatives (Clupeiformes), inferred from whole mitogenome sequences. Mol Phylogenet Evol [serial on the Internet]. 2007; 43(3):1096-105. Available from: http://dx.doi.org/10.1016/j.ympev.2006.09.018
» http://dx.doi.org/10.1016/j.ympev.2006.09.018 - Leaché AD. Species trees for spiny lizards (Genus Sceloporus): Identifying points of concordance and conflict between nuclear and mitochondrial data. Mol Phylogenet Evol [serial on the Internet]. 2010; 54(1):162-71. Available from: http://dx.doi.org/10.1016/j.ympev.2009.09.006
» http://dx.doi.org/10.1016/j.ympev.2009.09.006 - Lee CE, Bell MA. Causes and consequences of recent freshwater invasions by saltwater animals. Trends Ecol Evol [serial on the Internet]. 1999; 14(7):284-88. Available from: https://doi.org/10.1016/S0169-5347(99)01596-7
» https://doi.org/10.1016/S0169-5347(99)01596-7 - Li CH, Ortí G. Molecular phylogeny of clupeiformes (Actinopterygii) inferred from nuclear and mitochondrial DNA sequences. Mol Phylogenet Evol [serial on the Internet]. 2007; 44(1):386-98. Available from: http://dx.doi.org/10.1016/j.ympev.2006.10.030
» http://dx.doi.org/10.1016/j.ympev.2006.10.030 - López-Fernández H, Arbour JH, Winemiller KO, Honeycutt RL. Testing for ancient adaptive radiations in neotropical cichilid fishes. Evolution [serial on the Internet]. 2013; 67(5):1321-37. Available from: https://doi.org/10.1111/evo.12038
» https://doi.org/10.1111/evo.12038 - Lovejoy NR, Albert JS, Crampton WGR. Miocene marine incursions and marine/freshwater transitions: Evidence from Neotropical fishes. J South Am Earth Sci [serial on the Internet]. 2006; 21(1-2):5-13. Available from: https://doi.org/10.1016/j.jsames.2005.07.009
» https://doi.org/10.1016/j.jsames.2005.07.009 - Lundberg JG, Kottelat M, Smith GR, Stiassny MLJ, Gill AC. So many fishes, so little time: An overview of recent ichthyological discovery in continental waters. Ann Missouri Bot Gard. 2000; 87(1):26-62.
- Machac A, Graham CH, Storch D. Ecological controls of mammalian diversification vary with phylogenetic scale. Glob Ecol Biogeogr [serial on the Internet]. 2018; 27(1):32-46. Available from: https://doi.org/10.1111/geb.12642
» https://doi.org/10.1111/geb.12642 - Maddison WP, Knowles LL. Inferring phylogeny despite incomplete lineage sorting. Syst Biol [serial on the Internet]. 2006; 55(1):21-30. Available from: http://dx.doi.org/10.1080/10635150500354928
» http://dx.doi.org/10.1080/10635150500354928 - Malabarba MC, Di Dario F. A new predatory herring-like fish (Teleostei: Clupeiformes) from the early Cretaceous of Brazil, and implications for relationships in the Clupeoidei. Zool J Linn Soc . 2017; 180:175-94.
- Marramà G, Carnevale G. The relationships of Gasteroclupea branisai Signeux, 1964, a freshwater double-armored herring (Clupeomorpha, Ellimmichthyiformes) from the Late Cretaceous-Paleocene of South America. Hist Biol [serial on the Internet]. 2017; 29(7):904-17. Available from: https://doi.org/10.1080/08912963.2016.1262855
» https://doi.org/10.1080/08912963.2016.1262855 - McBride MC, Willis TV, Bradford RG, Bentzen P. Genetic diversity and structure of two hybridizing anadromous fishes (Alosa pseudoharengus, Alosa aestivalis) across the northern portion of their ranges. Conserv Genet . 2014; 15(6):1281-98.
- Miller RR. First Fossil record (Plio-Pleistocene) of Threadfin Shad, Dorosoma petenense, from the Gatuna Formation of southeastern New Mexico. J Paleontol. 1982; 56(2):423-25.
- Miller MA, Pfeiffer W, Schwartz T. Creating the CIPRES science gateway for inference of large phylogenetic trees. In: Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, 14 November 2010, pp. 1-8.
- Moen D, Morlon H. Why does diversification slow down? Trends Ecol Evol [serial on the Internet]. 2014; 29(4):190-97. Available from: https://doi.org/10.1016/j.tree.2014.01.010
» https://doi.org/10.1016/j.tree.2014.01.010 - Montes C, Cardona A, Jaramillo C, Pardo A, Silva JC, Valencia V, Ayala C, Pérez-Angel LC, Rodriguez-Parra LA, Ramirez V, Niño H. Middle Miocene closure of the Central American Seaway. Science [serial on the Internet]. 2015; 348(6231):226-29. Available from: http://dx.doi.org/10.1126/science.aaa2815
» http://dx.doi.org/10.1126/science.aaa2815 - Mulcahy DG, Noonan BP, Moss T, Townsend TM, Reeder TW, Sites JW et al Estimating divergence dates and evaluating dating methods using phylogenomic and mitochondrial data in squamate reptiles. Mol Phylogenet Evol [serial on the Internet]. 2012; 65(3):974-91. Available from: http://dx.doi.org/10.1016/j.ympev.2012.08.018
» http://dx.doi.org/10.1016/j.ympev.2012.08.018 - Nee S, Mooers AO, Harvey PH. Tempo and mode of evolution revealed from molecular phylogenies. Proc Natl Acad Sci. 1992. 89(17):8322-26.
- Pamilo P, Nei M. Relationships between gene trees and species trees. Mol Biol Evol . 1988; 5(5):568-83.
- Platt RN, Faircloth BC, Sullivan KAM, Kieran TJ, Glenn TC, Vandewege MW et al Conflicting evolutionary histories of the mitochondrial and nuclear genomes in New World myotis bats. Syst Biol [serial on the Internet]. 2018; 67(2):236-49. Available from: http://dx.doi.org/10.1093/sysbio/syx070
» http://dx.doi.org/10.1093/sysbio/syx070 - Pybus OG, Harvey PH. Testing macro-evolutionary models using incomplete molecular phylogenies. Proc R Soc B [serial on the Internet]. 2000; 267(1459):2267-72. Available from: http://dx.doi.org/10.1098/rspb.2000.1278
» http://dx.doi.org/10.1098/rspb.2000.1278 - Rabosky DL. LASER: a maximum likelihood toolkit for detecting temporal shifts in diversification rates from molecular phylogenies. Evol Bioinform [serial on the Internet]. 2006; 2(Januray 1):247-250. Available from: https://doi.org/10.1177%2F117693430600200024
» https://doi.org/10.1177%2F117693430600200024 - Rabosky DL. Ecological limits and diversification rate: alternative paradigms to explain the variation in species richness among clades and regions. Ecol Lett [serial on the Internet]. 2009a; 12(8):735-43. Available from: http://dx.doi.org/10.1111/j.1461-0248.2009.01333.x
» http://dx.doi.org/10.1111/j.1461-0248.2009.01333.x - Rabosky DL. Ecological limits on clade diversification in higher taxa. Am Nat . 2009b; 173(5):662-74.
- Rabosky DL. Diversity-dependence, ecological speciation, and the role of competition in macroevolution. Annu Rev Ecol Evol Syst [serial on the Internet]. 2013; 44:481-502. Available from: https://doi.org/10.1146/annurev-ecolsys-110512-135800
» https://doi.org/10.1146/annurev-ecolsys-110512-135800 - Rabosky DL, Glor RE. Equilibrium speciation dynamics in a model adaptive radiation of island lizards. Proc Natl Acad Sci [serial on the Internet]. 2010. 107(51):22178-83. Available from: https://doi.org/10.1073/pnas.1007606107
» https://doi.org/10.1073/pnas.1007606107 - Rabosky DL, Hurlbert AH. Species richness at continental scales is dominated by ecological limits. Am Nat [serial on the Internet]. 2015; 185(5):572-83. Available from: http://dx.doi.org/10.1086/680850
» http://dx.doi.org/10.1086/680850 - Rabosky DL, Lovette IJ. Density-dependent diversification in North American wood warblers. Proc R Soc B [serial on the Internet]. 2008; 275(1649):2363-71. Available from: http://dx.doi.org/10.1098/rspb.2008.0630
» http://dx.doi.org/10.1098/rspb.2008.0630 - Rambaut A, Suchard MA, Xie D, Drummond AJ. Tracer v1.6 [Inter net]. 2014. Available from: http://tree.bio.ed.ac.uk/softwar/tracer
» http://tree.bio.ed.ac.uk/softwar/tracer - Ricklefs RE. A biogeographical perspective on ecological systems: some personal reflections. J Biogeogr [serial on the Internet]. 2011; 38(11):2045-56. Available from: https://doi.org/10.1111/j.1365-2699.2011.02520.x
» https://doi.org/10.1111/j.1365-2699.2011.02520.x - Roure B, Baurain D, Philippe H. Impact of missing data on phylogenies inferred from empirical phylogenomic data sets. Mol Biol Evol [serial on the Internet]. 2013; 30(1):197-214. Available from: http://dx.doi.org/10.1093/molbev/mss208
» http://dx.doi.org/10.1093/molbev/mss208 - Schluter D. Ecological causes of adaptive radiation. Am Nat . 1996; 148:40-64.
- Siebert, DJ. Notes on the anatomy and relationships of Sundasalanx Roberts (Teleostei, Clupeidae), with descriptions of four new species from Borneo. Bull Br Mus Nat His Zool 1997; 63: 13-26.
- Stamatakis A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics [serial on the Internet]. 2014; 30(9):1312-13. Available from: http://dx.doi.org/10.1093/bioinformatics/btu033
» http://dx.doi.org/10.1093/bioinformatics/btu033 - Thomson RC, Shaffer HB. Sparse supermatrices for phylogenetic inference: taxonomy, alignment, rogue taxa, and the phylogeny of living turtles. Syst Biol [serial on the Internet]. 2010; 59(1):42-58. Available from: http://dx.doi.org/10.1093/sysbio/syp075
» http://dx.doi.org/10.1093/sysbio/syp075 - Toews DPL, Brelsford A. The biogeography of mitochondrial and nuclear discordance in animals. Mol Ecol [serial on the Internet]. 2012; 21(16):3907-30. Available from: http://dx.doi.org/10.1111/j.1365-294X.2012.05664.x
» http://dx.doi.org/10.1111/j.1365-294X.2012.05664.x - Vermeij GJ, Dudley R. Why are there so few evolutionary transitions between aquatic and terrestrial ecosystems? Biol J Linn Soc [serial on the Internet]. 2000; 70(4):541-54. Available from: https://doi.org/10.1111/j.1095-8312.2000.tb00216.x
» https://doi.org/10.1111/j.1095-8312.2000.tb00216.x - Wallis GP, Cameron-Christie SR, Kennedy HL, Palmer G, Sanders TR, Winter DJ. Interspecific hybridization causes long-term phylogenetic discordance between nuclear and mitochondrial genomes in freshwater fishes. Mol Ecol [serial on the Internet]. 2017; 26(12):3116-27. Available from: http://dx.doi.org/10.1111/mec.14096
» http://dx.doi.org/10.1111/mec.14096 - Waters JM, Rowe DL, Burridge CP, Wallis GP. Gene trees versus species trees: Reassessing life-history evolution in a freshwater fish radiation. Syst Biol [serial on the Internet]. 2010; 59(5):504-17. Available from: https://doi.org/10.1093/sysbio/syq031
» https://doi.org/10.1093/sysbio/syq031 - Weir JT. Divergent timing and patterns of species accumulation in lowland and highland neotropical birds. Evolution. 2006; 60(4):842-55.
- Weir JT, Mursleen S. Diversity-dependent cladogenesis and trait evolution in the adaptive radiation of Auks (Aves: Alcidae). Evolution [serial on the Internet]. 2013; 67(2):403-16. Available from: https://doi.org/10.1111/j.1558-5646.2012.01786.x
» https://doi.org/10.1111/j.1558-5646.2012.01786.x - Whitehead P. King Herring: His place amongst the clupeoids. Can J Fish Aquat Sci. 1985a; 42(1):3-20.
- Whitehead PJ. FAO species catalogue. Clupeoid fishes of the world (suborder Clupeioidei). An annotated and illustrated catalogue of the herrings, sardines, pilchards, sprats, shads, anchovies and wolf-herrings. FAO Fish Synop. 1985b; 125(7):1-303.
- Wiens JJ, Kuczynski CA, Stephens PR. Discordant mitochondrial and nuclear gene phylogenies in emydid turtles: Implications for speciation and conservation. Biol J Linn Soc [serial on the Internet]. 2010; 99(2):445-61. Available from: https://doi.org/10.1111/j.1095-8312.2009.01342.x
» https://doi.org/10.1111/j.1095-8312.2009.01342.x - Winemiller KO, Leslie MA. Fish assemblages across a complex, tropical freshwater/marine ecotone. Environ Biol Fishes. 1992; 34(1):29-50.
- Yang Z. Computational Molecular Evolution. Oxford: Oxford University Press; 2006.
- Yoder JB, Clancey E, Roches SD, Eastman JM, Gentry L, Godsoe W et al Ecological opportunity and the origin of adaptive radiations. J Evol Biol [serial on the Internet]. 2010; 23(8):1581-96. Available from: https://doi.org/10.1111/j.1420-9101.2010.02029.x
» https://doi.org/10.1111/j.1420-9101.2010.02029.x - Zheng Y, Peng R, Kuro-O M, Zeng X. Exploring patterns and extent of bias in estimating divergence time from mitochondrial DNA sequence data in a particular lineage: A case study of salamanders (Order Caudata). Mol Biol Evol [serial on the Internet]. 2011; 28(9):2521-35. Available from: http://dx.doi.org/10.1093/molbev/msr072
» http://dx.doi.org/10.1093/molbev/msr072
Edited by
Publication Dates
-
Publication in this collection
11 Oct 2018 -
Date of issue
2018
History
-
Received
13 Mar 2018 -
Accepted
09 Aug 2018