Resumos
O modelo de tight-binding é um dos pilares da Física da Matéria Condensada, permitindo descrever de forma bastante simples uma ampla gama de fenômenos de interesse, como por exemplo a estrutura de bandas dos materiais e o transporte eletrônico. Nesse trabalho, o Hamiltoniano de tight-binding, escrito em linguagem de segunda quantização, é utilizado na modelagem das densidades de estados de energia em polímeros orgânicos. O procedimento de segunda quantização permite obter as expressões para a determinação dos parâmetros de hopping de forma natural. No âmbito do modelo de tight-binding são analisadas a cadeia atômica infinita e as moléculas do benzeno, do ciclopentadieno e do tiofeno.
Palavras-chave:
Tight-binding; polímeros; hopping; segunda quantização
The tight-binding model is a cornerstone of Condensed Matter Physics, describing in a simple way a wide range of phenomena, such as the band structure of materials and the electronic transport. In this paper, the referred model, written in the language of second quantization, is used to describe the density of states in organic polymers. Using the second quantization method the expressions for the hopping parameters emerge quite naturally. The infinite atomic chain, the benzene, cyclopentadiene and tyophene molecules are analyzed within the tight-binding model.
Keywords
Tight-binding; polymers; hopping; second quantization
1. Introdução
Polímeros são macromoléculas formadas pela repetição de uma unidade básica denominada monômero. Tipicamente, os materiais poliméricos possuem em sua estrutura o carbono ou o silício. Aqueles que são originários da química do carbono são conhecidos como polímeros orgânicos [1[1] W. Callister Jr. e D.G. Rethwisch, Fundamentos da Ciência e Engenharia dos Materiais: Abordagem Integrada (LTC, Rio de Janeiro, 2014)., 2[2] J.D. Shackelford, Ciência dos Materiais (Pearson, São Paulo, 2008), 6a ed.]. Dentre os os polímeros inorgânicos os silicones, cuja unidade de repetição envolve o , são os mais comuns. Já como exemplos de polímeros à base do carbono podemos citar o DNA, a polianilina, o politiofeno e o polietileno. Materiais poliméricos orgânicos apresentam amplo espectro de propriedades físico-químicas, o que torna possível um vasto leque de aplicações tecnológicas. Por exemplo, do ponto de vista mecânico existem tanto polímeros rígidos quanto flexíveis e elásticos [1[1] W. Callister Jr. e D.G. Rethwisch, Fundamentos da Ciência e Engenharia dos Materiais: Abordagem Integrada (LTC, Rio de Janeiro, 2014).]. Do ponto de vista de condutividade elétrica, há desde os polímeros isolantes, atualmente empregados como isoladores nos sistemas de transmissão e distribuição de energia elétrica, passando pelos semicondutores e condutores [3[3] H. Shirakawa, E.J. Louis, A.G. MacDiarmid, C.K. Chiang e A.J. Heeger, Journal of the Chemical Society, Chemical Communications 16 , 578 (1977)., 4[4] C.K. Chiang, C.R. Finche, Y.W. Park, A.J. Heeger, H. Shirakawa, E.J. Lois, S.C. Gau e A.G. MacDiarmid, Phys. Rev. Lett. 39 , 1098 (1977)., 5[5] N.S. Sariciftci, L. Smilowitz, A.J. Heeger e F. Wuld, Science 258 , 1474 (1992)., 6[6] A.J. Heeger, S. Kivelson, J.R. Schrieffer e W.-P. Su, Rev. Mod. Phys. 60 , 781 (1988).], com aplicações na eletrônica orgânica e na fabricação de dispositivos fotovoltaicos [7[7] M. Pope, H.P. Kallmann e P. Magnante, J. Chem. Phys. 38 , 2042 (1963)., 8[8] J.J.M. Halls, Photoconductive properties of conjugated polymers . Doctoral Thesis, University of Cambridge, Cambridge (1997)., 9[9] B.R. Weinberger, M.Akhtar e S.C. Gau, Synthetic Metals 4 , 187 (1982)., 10[10] A.A. Lima, N.P.Menezes, S. Santos, B. Amorim, F. Thomazi, F. Zanella, A. Heilmann, E. Burkarter e C.A. Dartora, Revista Brasileira de Ensino de Física 42 , e20190191 (2020)., 11[11] F. Thomazi, M. Souza, C.K. Saul, G.A. Viana, F.C. Marques, R. Silvestre, M. Brehm, C.E.B. Marino, E. Burkarter e C.A. Dartora, Current Nanoscience 10 , 877 (2014)., 12[12] F. Thomazi, C.K. Saul, C. Marino, E. Burkarter e C.A. Dartora, Current Nanoscience 12 , 611 (2016)., 13[13] L.B. Schneider, F. Thomazi, E. Burkarter, C.E.B. Marino, C.K. Saul, A.S. Ito e C.A. Dartora, Current Nanoscience 14 , 403 (2018).].
No presente trabalho será dada maior ênfase aos polímeros conjugados, de maior relevância para a área da eletrônica. Seu estudo rendeu o Prêmio Nobel de Química no ano de 2000 para Alan MacDiarmid, Hideki Shirakawa e Alan J. Heeger [3[3] H. Shirakawa, E.J. Louis, A.G. MacDiarmid, C.K. Chiang e A.J. Heeger, Journal of the Chemical Society, Chemical Communications 16 , 578 (1977).]. Os polímeros conjugados são caracterizados pela alternância entre ligações simples e duplas, denominadas de sigma () e pi (), respectivamente. As ligações favorecem a condução de eletricidade, sendo desejáveis em aplicações como a optoeletrônica e a fotovoltaica [14[14] Z. Qiu, B.A.G. Hammer e K. Müllen, Progress in Polymer Science 100 , 101179 (2020).].
A compreensão mais detalhada do comportamento desses polímeros requer o entendimento mais aprofundado da estrutura eletrônica do carbono e suas hibridizações. Ressalta-se que os elementos sempre presentes nas cadeias conjugadas são o carbono e o hidrogênio. A configuração eletrônica do elemento carbono no seu estado fundamental é , havendo 4 elétrons na camada de valência, de número atômico principal . O processo de hibridização dos orbitais e do carbono favorece a formação das ligações químicas. No caso do carbono ocorrem três tipos de hibridização dos orbitais atômicos, a saber: i) , onde um orbital mistura-se ao orbital ; ii) , na qual dois dos orbitais misturam-se com o orbital ; e iii) , em que todos os orbitais do tipo participam da mistura com o orbital [15[15] C.A. Dartora, M.J. Saldaña Jimenez e F. Zanella, Revista Brasileira de Ensino de Física 37 , 3301 (2015)., 16[16] M. Alonso e E.J. Finn, Física (Editora Escolar, São Paulo, 2012).]. Enquanto na hibridização do ocorrem as ligações do tipo , bastante localizadas, na hibridização , em que o orbital não se superpõe ao orbital , ocorre a formação das ligações duplas ou do tipo .
Considerando-se as formas alotrópicas cristalinas do carbono, o diamante tem comportamento elétrico isolante devido ao fato de ocorrer a hibridização em sua estrutura, enquanto que o grafite tem comportamento condutor de eletricidade como consequência da hibridização . Os polímeros conjugados são caracterizados pela ocorrência da hibridização . Enquanto as ligações proporcionadas pelos orbitais hibridizados vão moldar a forma da estrutura formada, devido às ligações altamente direcionais, as ligações duplas provenientes dos orbitais irão concorrer de maneira decisiva para as propriedades de transporte de carga elétrica nesses polímeros [3[3] H. Shirakawa, E.J. Louis, A.G. MacDiarmid, C.K. Chiang e A.J. Heeger, Journal of the Chemical Society, Chemical Communications 16 , 578 (1977)., 14[14] Z. Qiu, B.A.G. Hammer e K. Müllen, Progress in Polymer Science 100 , 101179 (2020).].
O objetivo principal deste trabalho é apresentar de forma didática os modelos de polímeros conjugados a partir do Hamiltoniano de tight-binding . Esse Hamiltoniano é um dos pilares da Física da Matéria Condensada, permitindo descrever de forma simples uma ampla gama de fenômenos de interesse, como por exemplo a estrutura de bandas dos materiais e o transporte eletrônico. Aqui o Hamiltoniano de tight-binding será escrito em linguagem de segunda quantização. Com o emprego dos métodos da segunda quantização as expressões matemáticas para a determinação dos parâmetros de hopping do modelo surgem de forma natural.
O presente trabalho está estruturado da seguinte maneira: Na próxima Seção será apresentado o Hamiltoniano de tight-binding em segunda quantização, para férmions não-relativísticos. Expressões gerais para o cálculo dos parâmetros de hopping no modelo de tight-binding serão apresentadas. Na Seção 3 será estudado o caso de interação entre primeiros vizinhos em uma cadeia de átomos infinitamente longa. Na Seção 4 serão abordadas alguns monômeros de interesse na obtenção de polímeros conjugados. O problema de hibridização e formação das ligações e será discutido em maiores detalhes. Finalmente, na Seção 5 serão apresentadas as considerações finais e conclusões.
2. O Modelo de Tight-Binding em Segunda Quantização
A literatura acerca da segunda quantização, ou teoria quântica dos campos, é vasta e com abordagens distintas, por isso aqui recomendamos ao leitor os livros-texto de Greiner/Reinhardt [17[17] W. Greiner e J. Reinhardt, Field Quantization (Springer-Verlag, Berlin, Heidelberg, 1996).] e de Kittel [18[18] C. Kittel, Quantum Theory of Solids (John Wiley & Sons, New York, 1987), 2a ed.], sendo que este último enfatiza as aplicações em Física do Estado Sólido. Como ponto de partida, considere o caso das partículas não-relativísticas em primeira quantização. Essa situação é de grande utilidade pois na matéria ordinária os elétrons, prótons e nêutrons podem ser tratados em boa aproximação como férmions não-relativísticos. A função de ondas descrevendo a amplitude de probabilidade de encontrar uma partícula em torno da posição no instante de tempo deve satisfazer a equação de Schrödinger:
Nessa situação, o Hamiltoniano de uma partícula é dado por , sendo é um termo de energia potencial independente do tempo.
A substituição de uma solução do tipo na equação de movimento (1) leva ao problema de autovalor . O conjunto das soluções do problema do autovalor, satisfazendo as condições de contorno impostas pelo Hamiltoniano e pela interpretação probabilística da Mecânica Quantica, formará uma base completa de funções, permitindo expandir o campo na forma que segue:
onde são coeficientes complexos, o índice corresponde a um conjunto de números quânticos associados à -ésima função . A condição de ortonormalidade é escrita da seguinte forma:
De acordo com o procedimento de segunda quantização, deve-se converter a função de ondas em um operador de campo fermiônico, alterando o status dos coeficientes complexos da expansão, na forma que segue:
onde é o operador de aniquilação de um férmion no estado quântico , e o transposto conjugado é o operador de criação de um férmion nesse mesmo estado. Esses operadores atuam em um espaço conhecido como espaço de Fock, cujos estados quânticos são auto-estados de número de partículas. Essa representação é também conhecida como representação de número. Os operadores de criação e aniquilação de férmions satisfazem uma álgebra anti-comutativa, introduzida originalmente por Pascual Jordan e conhecida como álgebra fermiônica, mostrada a seguir:
onde ou , é o anti-comutador entre os operadores e , é um estado com partículas no estado , partículas no estado e assim sucessivamente. O operador é o operador que conta o número de partículas no -ésimo estado quântico disponível, sendo denominado operador de número. Define-se o vácuo dos férmions, , como o estado no qual não há partículas, de tal forma que
pois não é possível aniquilar uma partícula do estado que já não possui nenhuma. Por outro lado, a equação (7) implica que , ou seja, não é possível adicionar mais do que uma partícula em um dado estado físico. Observe que:
onde corresponde ao estado de número com uma única partícula no estado quântico indexado por . Portanto, o princípio de exclusão de Pauli é uma consequência direta da própria álgebra fermiônica.
Indo mais adiante, em segunda quantização a média de um operador , obtida através da expressão , é convertida em um operador que age sobre os estados de número (ou superposições destes) no espaço de Fock, uma vez que o próprio campo é agora um operador.
Enquanto na primeira quantização a interpretação probabilística requer que , em segunda quantização essa integral corresponde ao operador de número total de partículas, , conforme segue:
Observe que é de fato o operador de número total, pois ao atuar sobre um estado de número produz como autovalor a somatória do número de partículas do sistema.
O próximo passo é converter a média do Hamiltoniano de uma partícula,
no operador Hamiltoniano em segunda quantização, que permite descrever o problema de muitas partículas, ao invés de uma única. Considere a expansão dos operadores de campo em termos de autofunções de energia, ou seja, . Desse modo:
O caso das partículas livres, quando a energia potencial é nula, , é de especial interesse, permitindo satisfazer a equação de Schrödinger através de ondas planas uniformes, de forma que, omitindo o spin, tem-se . Os autovalores de energia associados são dados por , e o Hamiltoniano em segunda quantização toma a forma seguinte:
A inclusão de interações entre partículas de um mesmo tipo, como por exemplo as interações de repulsão coulombiana entre elétrons, faz-se através do chamado ordenamento normal dos operadores de campo, no qual todos os operadores de criação devem aparecer à esquerda dos operadores de aniquilação associados àquele campo. Por exemplo, em uma interação entre pares de partículas idênticas situadas nas posições e , cuja energia potencial de interação tem a forma , a versão em segunda quantização será escrita da seguinte maneira:
Observe atentamente a ordem em que aparecem as variáveis e nos argumentos dos operadores de campo. De forma geral, para interações entre -tuplas de partículas tem-se:
Desse modo, o termo representando a interação de pares de partículas no Hamiltoniano em segunda quantização tem a forma forma geral . Por uma questão de simplicidade, todavia, no presente trabalho termos envolvendo essas interações serão desconsiderados.
É importante mencionar que nem sempre são conhecidos os auto-estados de energia do Hamiltoniano completo. Nesse cenário emprega-se na expansão dos campos uma base completa conhecida, proveniente da solução de um Hamiltoniano mais simples. A dependência temporal é incorporada aos operadores de criação e aniquilação e tem-se:
onde os coeficientes são definidos da seguinte maneira:
Os elementos diagonais são conhecidos como auto-energias associadas às funções . Já para , os coeficientes são denominados parâmetros de salto ou de hopping, e representam uma energia de transição entre os estados e .
Deixa-se como exercício para o leitor demonstrar, utilizando a equação de movimento de Heisenberg para um operador ,
que os operadores de aniquilação devem satisfazer a seguinte equação de movimento:
O modelo de tight-binding é uma aproximação em que as funções de base utilizadas na expansão dos operadores de campo são funções de onda localizadas em torno de cada sítio da rede atômica, e tipicamente envolvem orbitais atômicos, ou superposições de orbitais atômicos, sendo muito útil no cálculo da estrutura de bandas de energia de um sólido. Nas próximas duas seções o modelo de tight-binding será utilizado para descrever cadeias atômicas infinitamente longas e alguns monômeros de interesse para as cadeias poliméricas conjugadas.
Para finalizar a presente seção, cabe destacar que os orbitais atômicos provenientes de átomos vizinhos não são necessariamente ortogonais entre si, mas é possível encontrar um conjunto ortonormalizado, utilizando o método de Gram-Schmidt, por exemplo. Considerando um conjunto de orbitais atômicos em que é proveniente do -ésimo átomo da rede, não ortogonais entre si, é possível obter um novo conjunto:
Estão omitidos nas equações acima os fatores de renormalização dos estados a cada passo. Um método semelhante para obter novas funções ortogonalizadas é denominado de ortogonalização de Löwdin, e os orbitais assim obtidos possuem contribuições de átomos distantes [19[19] J.C. Slater e G.F. Koster, Physical Review 94 , 1498 (1954).]. De modo análogo, em cristais é possível utilizar um conjunto de funções ortogonais conhecido como funções de Wannier, que são localizadas em torno dos sítios atômicos, para construir o modelo de tight-binding .
Entretanto, a superposição dos orbitais provenientes de átomos vizinhos na rede é usualmente pequena, ou seja:
sendo da rede um vetor conectando o átomo na posição com um de seus primeiros vizinhos, e são orbitais atômicos, com conjuntos de números quânticos e , respectivamente. Nessa situação, a etapa de ortogonalização de Gram-Schmidt não se faz necessária na primeira aproximação, e considera-se que o conjunto de todos os orbitais atômicos localizados em torno de seus respectivos átomos forma uma base completa. Note ainda que orbitais atômicos do tipo hidrogenóide tem uma dependência na forma com relação à distância, significando que decaem exponencialmente com o aumento da distância em relação à sua origem.
3. A Cadeia Infinitamente Longa e Bandas de Energia
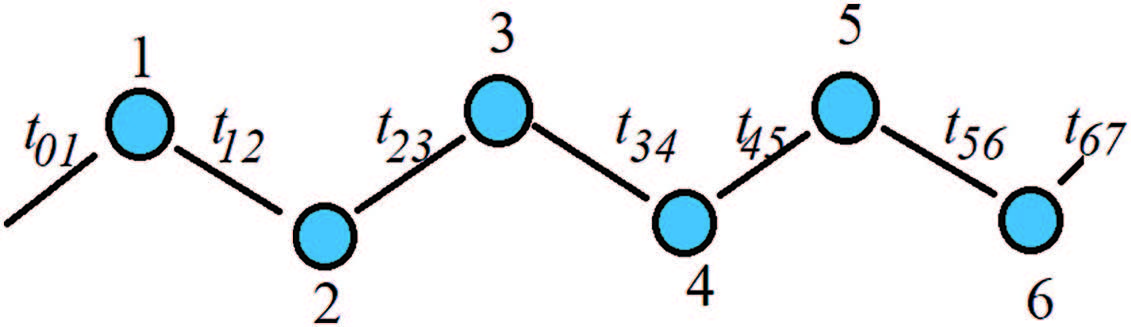
Uma cadeia monoatômica em uma dimensão espacial é ilustrada na Figura1, podendo representar uma macromolécula linear, como por exemplo o poliacetileno. O comprimento de uma macromolécula polimérica linear é dado aproximadamente por , sendo o número de átomos de carbono na cadeia e a distância entre eles, assumindo que esta seja constante. Nesse caso o valor de é conhecido como parâmetro de rede. Um modelo de brinquedo (toy model ) para uma macromolécula em que é obtido fazendo o limite .

Cadeia monoatômica em uma dimensão espacial, podendo representar um polímero linear, como por exemplo o poliacetileno.
Por uma questão de simplicidade, assuma uma base ortonormalizada formada considerando um único orbital relevante por átomo da rede unidimensional infinita. Em primeira aproximação, admite-se que os parâmetros são não negligenciáveis apenas entre primeiros vizinhos, ou seja, . Tanto a equação (21) quanto o requerimento de hermiticidade do Hamiltoniano implicam que . Sem perda de generalidade no que segue, assuma que , ou seja, o parâmetro de hopping é puramente real, dado por:
Note que a cadeia está orientada ao longo da direção e que os orbitais são considerados puramente reais. Claramente envolve a superposição entre orbitais provenientes de átomos vizinhos na cadeia.
Desse modo, o Hamiltoniano (20) pode ser escrito explicitamente da seguinte maneira:
A interpretação desse Hamiltoniano é bastante simples, sendo que o primeiro termo corresponde à energia de um elétron ocupando o orbital no sítio -ésimo da rede. Já o termo representa uma energia cinética proveniente de um elétron saltando do orbital inicialmente ocupado no -ésimo sítio para o orbital do sítio , que deve estar inicialmente desocupado. O termo corresponde ao movimento no sentido contrário ao do caso anterior.
Para diagonalizar o Hamiltoniano (27) é conveniente expressar os operadores de criação e aniquilação em termos de uma série de Fourier, na forma que segue:
onde é o número total de sítios na rede (podendo ser levado ao limite ), é um vetor de onda no espaço recíproco a , é o vetor de posição do -ésimo átomo na rede. A equação (30) é a representação da função delta de Kronecker. Para obter os resultados de uma rede unidimensional deve-se adotar e , de tal forma que . O leitor é desafiado a demonstrar que, fazendo as substituições de (28) e (29) em (27) e fazendo uso de (30), obtém-se como resultado:
Observe que na representação de posição o Hamiltoniano não é diagonal, mas fazendo uso da periodicidade da rede, torna-se diagonal no espaço recíproco, produzindo um espectro de energia . No limite em que , assume valores no continuum, sendo o intervalo (para o caso unidimenional) conhecido como primeira zona de Brillouin no espaço . Forma-se assim uma banda de energias permitidas, com ponto médio em e largura de banda . Portanto, quanto maior o valor do parâmetro de hopping mais larga é a banda de energia.
A generalização óbvia para esse modelo simples consiste em considerar mais de um orbital por sítio, dando origem a várias bandas de energia. No caso unidimensional o espectro de energia na -ésima banda, proveniente do -ésimo orbital atômico considerado, terá a forma . Quanto maior o valor de , correspondendo a um orbital mais externo, mais estendida é a função de onda no espaço real, ocasionando maior superposição entre os orbitais dos primeiros vizinhos na integral de hopping e consequentemente maior será o valor do parâmetro de hopping . Para o caso de uma cadeia linear, as bandas oriundas dos orbitais de maior energia tem maior largura de banda. Essas últimas afirmações podem ser entendidas adotando-se um cenário simplista, em que um átomo da rede corresponde a um poço de potencial simétrico e finito em uma dimensão espacial. Nesse caso, para obter os orbitais deve-se resolver a equação de Schrödinger 1D,
onde a energia potencial é dada por:
sendo . As soluções confinadas (ou ligadas) são aquelas para as quais as autoenergias satisfazem a condição , de tal modo que . Isso se deve ao fato de que nas regiões em que a solução tem a forma , sendo . Nesse caso, é fácil ver que quanto maior a energia associada à função , menor a diferença e menor o valor da constante , produzindo um orbital mais estendido no espaço.
A expansão de em séries de Taylor nas vizinhanças do ponto resulta em e tem-se uma relação de dispersão parabólica em torno de . Sabendo-se que a massa efetiva é calculada como , tem-se:
Se , a massa efetiva é negativa e o ponto corresponde ao máximo daquela banda. Nesse caso, ao invés de considerar um elétron de carga negativa e massa negativa, reinterpreta-se a ausência do elétron de massa efetiva negativa e carga negativa em torno desse ponto como uma partícula de massa positiva e carga elétrica positiva, conhecida como lacuna ou buraco. Por outro lado, para o ponto corresponderá ao mínimo da banda e presença de uma partícula de massa positiva e carga negativa é naturalmente interpretada como um elétron. Uma conclusão importante nesse contexto é que quanto maior o valor do módulo do parâmetro de hopping, menor será a massa efetiva da partícula associada, o que afeta a mobilidade eletrônica do portador de carga, dada por
sendo o módulo da carga eletrônica e o tempo associado ao livre caminho médio. Em semicondutores, tipicamente os elétrons na banda de condução têm maior mobilidade do que as lacunas na banda de valência, o que grosseiramente pode ser entendido considerando-se o fato de que o parâmetro de hopping associado às bandas de maior energia têm maior valor absoluto, levando a uma menor massa efetiva.
4. Monômeros de Interesse na Obtenção de Polímeros Conjugados
Como mencionado anteriormente, os polímeros conjugados são aqueles em que ocorre a alternância das ligações e entre os átomos de carbono da cadeia polimérica, sendo a ligação do tipo a principal responsável por conferir propriedades fotofísicas e de condutividade elétrica comparáveis à de semicondutores inorgânicos ou de alguns metais. Uma boa revisão pode ser encontrada nas notas da aula magna do Prêmio Nobel de Química do ano de 2000 [20[20] A. Heeger, Semiconducting and Metallic Polymers: The Fourth Generation of Polymeric Materials, disponível em: https://www.nobelprize.org/prizes/chemistry/2000/heeger/lecture/, acessado em 01/07/2021.
https://www.nobelprize.org/prizes/chemis...
, 21[21] A.G. MacDiarmid, “Synthetic Metals”: A Novel Role for Organic Polymers, disponível: https://www.nobelprize.org/prizes/chemistry/2000/macdiarmid/lecture/, acessado em 02/07/2021.
https://www.nobelprize.org/prizes/chemis...
, 22[22] H. Shirakawa, The Discovery of Polyacetylene Film: The Dawning of an Era of Conducting Polymers, disponível em: https://www.nobelprize.org/prizes/chemistry/2000/shirakawa/lecture/, acessado em 01/07/2021.
https://www.nobelprize.org/prizes/chemis...
].
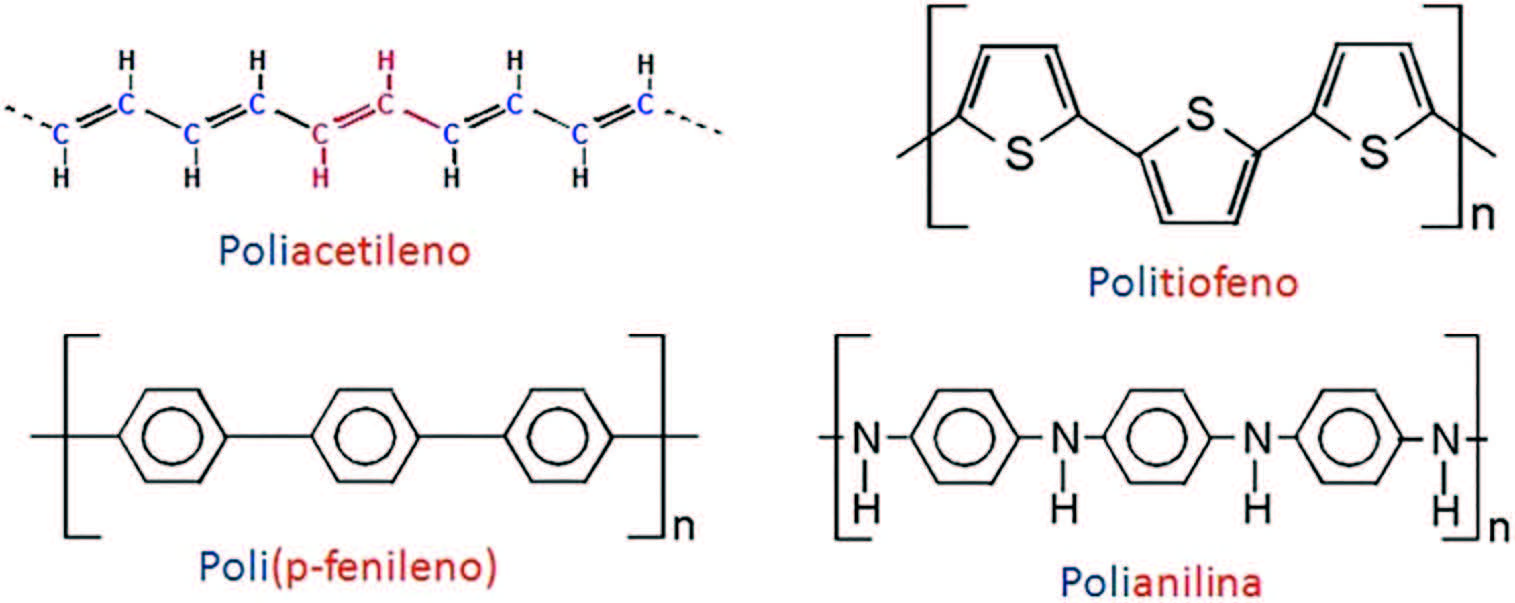
Na obtenção de polímeros conjugados, podem ser citados como monômeros de interesse o acetileno e algumas moléculas cíclicas, como o benzeno, que envolve o ciclo aromático de átomos de carbono apenas, bem como o tiofeno, o furano e o pirrol, em que o heteroátomo na cadeia cíclica é o enxofre, o oxigênio e o nitrogênio, respectivamente. A Figura2 ilustra os monômeros cíclicos aqui mencionando enquanto que algumas das cadeias poliméricas conjugadas que podem ser obtidas estão ilustradas na Figura3.


Com o intuito de melhor entender o problema dos polímeros conjugados, considere um orbital atômico, cuja expressão é dada por:
sendo o número quântico principal, o número quântico de momento angular orbital, o número quântico magnético e o número quântico de spin, refere-se às coordenadas espaciais no sistema esférico, é uma função com dependência radial, que controla a probabilidade de encontrar um elétron na distância a partir do núcleo atômico, são as funções harmônicas esféricas e é um espinor de Pauli. É sabido que a média do operador de posição de um elétron será tanto maior quanto maior o valor de , significando maior afastamento em relação ao núcleo atômico, e portanto menor energia de ligação. Na notação da Química o número quântico é representado pelas letras , respectivamente.
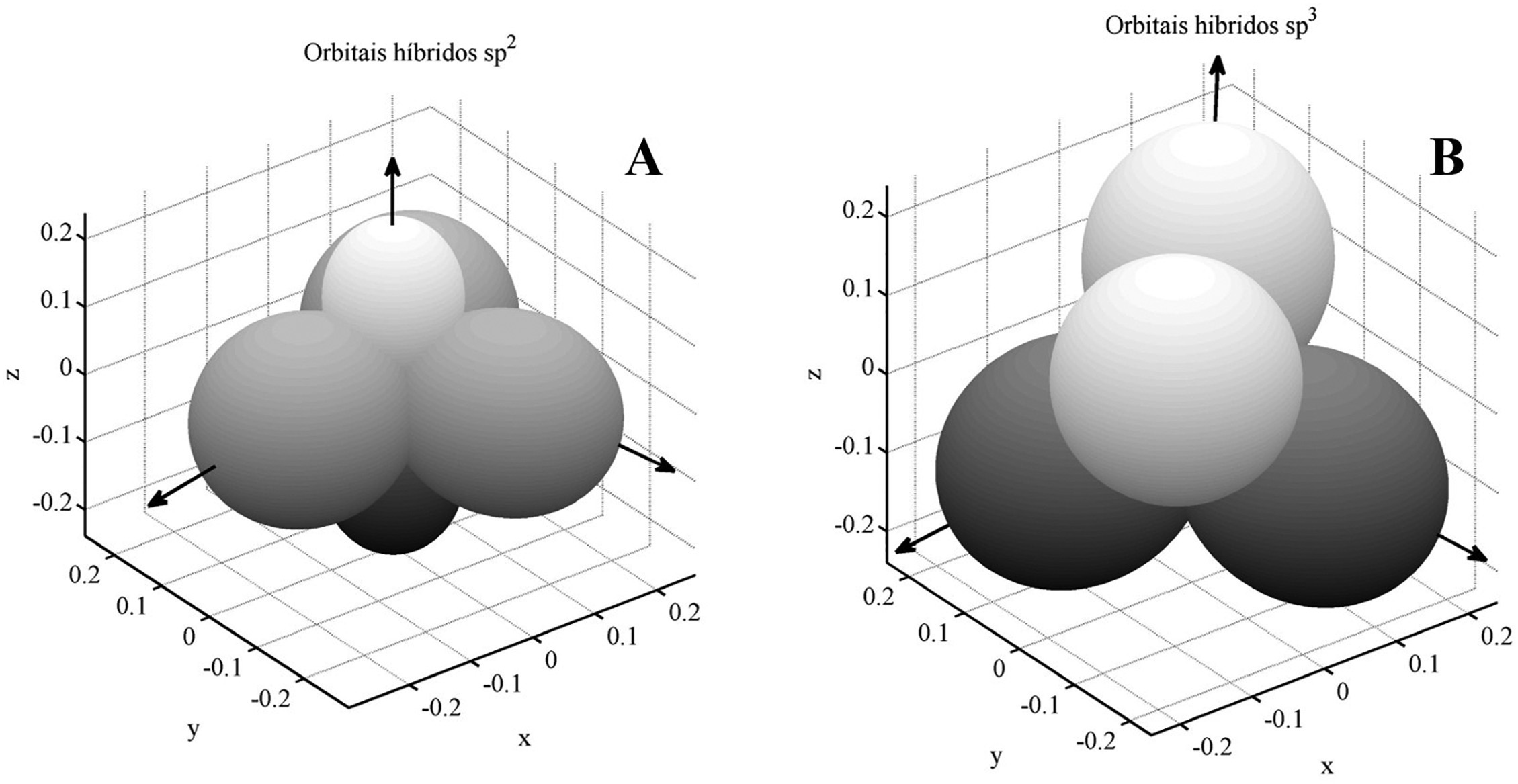
Sendo o carbono o principal elemento das cadeias poliméricas conjugadas, e considerando-se que os orbitais 2s e 2p desse elemento têm energias semelhantes, para a formação de ligações químicas mais estáveis ocorre a formação de orbitais atômicos híbridos, em que os orbitais , , e , ilustrados na Figura4 se misturam. As expressões matemáticas para a dependência angular dos orbitais e do carbono são dadas abaixo1 1 O número quântico de spin será omitido nas expressões para os orbitais 2s, 2px, 2py e 2pz e nas que se seguirão. Cada um deles é duplamente degenerado devido ao spin eletrônico, que pode assumir as projeções ↑ ou ↓, significando que caberia um total de 8 elétrons na camada n=2. :

No processo de hibridização, os novos orbitais atômicos originados da superposição serão necessariamente ortonormais no espaço de Hilbert. Das três possibilidades hibridização para o carbono, denominadas sp, e , as mais relevantes são as duas últimas. A hibridização se dá através da superposição entre o orbital e os orbitais , , formando assim três novos orbitais:
Estes são ortogonais no espaço de Hilbert, mas as direções de máxima propabilidade formam um ângulo de entre si para esses orbitais no espaço real, enquanto o orbital não se mistura aos demais, sendo orientado espacialmente num eixo ortogonal ao plano formado pelos orbitais . Essa situação é ilustrada na Figura5-A. Já a hibridização leva a uma superposição entre o orbital e todos os orbitais do tipo :

Dependência angular da densidade de probabilidade associada aos orbitais híbridos do tipo (A) e (B) .
Esses novos orbitais adquirem simetria tetraédrica, em que o ângulo formado pelas direções de máxima probabilidade desses orbitais é de aproximadamente , o que é mostrado na Figura5-B.
Deve-se à superposição de orbitais hibridizados de átomos de carbono vizinhos a formação dos orbitais moleculares do tipo , enquanto orbitais moleculares do tipo somente podem ocorrer pela superposição dos orbitais em átomos de carbono vizinhos, sendo que ambos devem estar hibridizados na forma . A hibridização do tipo somente permite a formação de ligações do tipo .
Tendo em vista que em polímeros conjugados devem ocorrer ligações do tipo , a hibridização se faz presente. Considere, por simplicidade, a molécula de etileno , em que a ligação química entre apenas dois átomos de carbono se dá através da hibridização , permitindo assim a ocorrência de uma ligação dupla entre os átomos de carbono, sendo uma dessas do tipo e a outra do tipo . Considerando apenas os efeitos da interação de Coulomb entre um elétron de valência e dois átomos de carbono situados nas posições e , o Hamiltoniano de uma partícula pode ser expressado da seguinte maneira:
onde e são os operadores de momento linear e posição do elétron, respectivamente, é o módulo da carga do elétron, a sua massa e é a permissividade dielétrica do vácuo, é o número atômico efetivo percebido pelos os elétrons de valência, já que a camada completa blinda parcialmente a carga do núcleo. Considerando que os orbitais atômicos hibridizados provenientes de cada átomo formam uma base completa, conforme discutido na Seção anterior, é possível calcular diretamente os elementos de matriz para os orbitais do tipo e , de tal modo que, utilizando as definições a seguir:
pode-se colocar o Hamiltoniano na forma matricial:
Observe que é um orbital do tipo e é um orbital do tipo , centrados no átomo de carbono , e ainda para quaisquer escolhas , tendo em vista que orbitais são ortogonais a orbitais . Um cálculo explícito nos mostra que , correspondendo às energias aproximadas do elétron em um orbital de átomo de carbono isolado, enquanto que as energias de interação satisfazem a condição , devido ao fato de que os orbitais que dão origem à ligação formarão um orbital molecular muito mais localizado do que o orbital molecular do tipo , originado pela superposição dos orbitais atômicos do tipo . É fácil diagonalizar (53), que tem a forma diagonal por blocos. Os autovalores de energia são:
onde o sinal corresponde a um orbital de menor(maior) energia e denominado de ligante(anti-ligante). Denotam-se convencionalmente os orbitais ligantes por e , com energias e , respectivamente, enquanto que os antiligantes são simbolizados por e , com energias e , respectivamente. Tendo em vista que , pode-se concluir que a separação energética entre os orbitais e será maior que aquela entre os orbitais e .

Os níveis de energia resultantes dos orbitais ligantes e anti-ligantes na molécula de etileno são ilustrados esquematicamente na Figura6. Observe que originalmente há 8 orbitais por átomo de carbono, sendo do tipo e do tipo , já considerada a duplicidade do spin, mas somente a metade desses níveis está ocupada em cada átomo, pelo fato de que o carbono possui apenas 4 elétrons de valência. Na formação da molécula, 4 orbitais do tipo orientados de modo a formar ângulos de com a linha que une os dois átomos de carbono estão envolvidos em ligações bastante localizadas com os átomos de hidrogênio da molécula. Sendo assim, do total de 4 elétrons de valência de um átomo de carbono, 2 estão envolvidos em ligações com átomos de hidrogênio, e outros 2 serão compartilhados com o outro átomo de carbono. Por conservação do número de graus de liberdade, ao formar a ligação química, 4 orbitais moleculares serão resultantes da superposição entre 2 orbitais do tipo de cada átomo, considerando-se a duplicidade do spin, esses sendo orientados ao longo da linha que une os átomos de carbono, e 4 orbitais moleculares resultantes da superposição entre 2 orbitais proveinentes de cada átomo. Como resultado final, orbitais moleculares corresponderão aos orbitais ligantes do tipo , aos orbitais ligantes , aos orbitais anti-ligantes do tipo e aos orbitais anti-ligantes , havendo um total de elétrons compartilhados entre os dois átomos de carbono. Desse modo, todos os orbitais ligantes e estarão preenchidos, enquanto que os orbitais anti-ligantes estarão desocupados. Na formação de uma cadeia polimérica de átomos os níveis discretos correspondentes aos orbitais , , e passam a se desdobrar em novos níveis, formando bandas de energia contínuas no limite de , denominadas bandas , , e . O nível de Fermi para esse sistema ficará localizado no ponto médio entre as energias e . Novamente, por conta da condição , os níveis de energia da banda estarão mais abaixo do nível de Fermi do que os níveis de energia da banda , e os níveis de energia da banda estarão mais acima do nível de Fermi do que os níveis de energia da banda . Portanto os processos de transporte e de fotocondutividade em baixas energias serão governados pelas bandas e .
No estudo de semicondutores orgânicos, denominam-se HOMO2 2 do inglês highest occupied molecular orbital e LUMO3 3 do inglês lowest unoccupied molecular orbital o estado de mais alta energia ocupado por um elétron e o estado de mais baixa energia que não esteja ocupado, respectivamente. De maneira usual, o bandgap do material é definido como a diferença de energia entre o LUMO e o HOMO.
Agora pretende-se analisar a molécula de benzeno, o ciclo conjugado mais estável, sendo constituído de 6 átomos de carbono e 6 átomos de hidrogênio, conforme ilustrado Figura7.

A molécula de benzeno: Note que as ligações , que se devem aos orbitais são direcionais, manténdo os elétrons fortemente localizados entre os átomos que formam a ligação. Já a ligação deve-se a orbitais perpendiculares ao plano mostrado. Nesse caso, os elétrons nesses orbitais podem saltar de um átomo para outro, fazendo as ligações duplas se moverem, com energia de transição proporcional ao parâmetro de hopping .
A ligação entre os átomos de carbono que dá a forma a essa molécula ocorre por meio de interações no plano que contém todos os átomos de carbono e de hidrogênio. Essas ligações devem-se aos orbitais hibridizados do carbono. Como esses formam um ângulo de entre si, os átomos serão arranjados na forma de um hexágono. Os orbitais dos átomos de hidrogênio se misturam a orbitais do tipo do carbono. Ao todo nas ligações estarão compartilhados 3 elétrons por átomo de carbono e 1 elétrons por átomo do hidrogênio, totalizando assim elétrons. Restam ainda elétrons provenientes dos átomos de carbono, que ocupam orbitais do tipo e que serão misturados para formar ligações . Uma vez que os orbitais são perpendiculares ao plano da molécula, é equiprovável, por exemplo, para um elétron do átomo 1 na rede ser compartilhado com o átomo 2 ou com o átomo 6 (vide Figura7), e essa ligação flutua, originando assim uma nuvem. As propriedades de condutividade elétrica e absorção óptica são fortemente influenciadas por essas ligações. Para fins do cálculo que segue, serão levados em conta somente esses orbitais, e o parâmetro de hopping pode ser calculado a partir da expressão (52). Em segunda quantização, o Hamiltoniano da molécula para os orbitais tem a seguinte forma:
com a identificação . Note que nesse modelo simplificado, considera-se nula a possibilidade de mudança do número quântico de spin quando ocorre o salto de um átomo para outro, e também está sendo negligenciada a interação repulsiva de Coulomb, quando dois desses elétrons estão no mesmo átomo. A forma mais simples de incluir um termo repulsivo se dá adicionando ao Hamiltoniano a expressão , sendo a energia de Coulomb associada, levando ao conhecido modelo de Hubbard. Nesse caso, note que há um custo energético para manter dois elétrons de spins contrários no mesmo sítio.
A solução desse problema levando em conta os 6 elétrons simultaneamente tem como base estados da forma , com as condições de que indicando que o estado no -ésimo átomo com spin está vazio ou ocupado, e que o número total de elétrons satisfaz . Esse problema torna-se exponencialmente complexo com o aumento do número de graus de liberdade, por exemplo, número total de elétrons e de orbitais atômicos disponíveis.
Para um melhor entendimento da natureza do problema, considere como exemplo apenas 2 elétrons, mas que podem ser colocados em orbitais distintos, indexados por , já considerando o spin. Suponha que um estado na base de número seja escrito na forma , com e . Nesse caso, a lista de possibilidades é a seguinte: , totalizando estados. Isso significa que a representação matricial do Hamiltoniano a ser diagonalizado terá dimensão . De forma geral, a dimensão do problema será dada pela combinação de estados disponíveis, que podem ser ocupados por elétrons. Na molécula de benzeno, para os orbitais teríamos 12 estados disponíveis, já incluindo o spin, para preencher com elétrons, o que resulta em uma dimensão do Hamiltoniano de 924 na base de número.
Portanto, uma simplificação se faz necessária na solução desse tipo de problema, consistindo em considerar o cenário com apenas um elétron, que pode ser colocado em qualquer um dos estados disponíveis, e uma vez diagonalizado o Hamiltoniano resultante nessa base de um único elétron, faz-se o preenchimento dos níveis de acordo com o princípio de exclusão de Pauli, sempre indo do menor para o maior nível de energia, até que se esgote o número total de elétrons. No caso da molécula de benzeno esse número é .
Para colocar o Hamiltoniano de tight-binding (58) na forma matricial, tendo em vista que o spin nesse problema representa apenas um fator de multiplicidade, pode-se omitir esse grau de liberdade e então calcular os elementos de matriz de na base , onde significa que o elétron está localizado no orbital do -ésimo átomo. Considerando-se que e que , pode-se obter:
Explicitamente em forma matricial o Hamiltoniano toma a seguinte forma:
Lembre que devem ser varridos de 1 a 6, e que 7 deve ser identificado com 1, por conta da ciclicidade da molécula. Os autovalores de (60) correspondendo às energias dos orbitais moleculares , são dados por e , enquanto que para os níveis têm-se e . Lembre que todos esses níveis são duplamente degenerados devido ao spin, produzindo orbitais moleculares distintos. Os 6 orbitais ligantes vão contribuir para formação de uma densidade de estados semelhante à banda de valência de um semicondutor, enquanto que os 6 orbitais antiligantes vão contribuir para a formação de uma densidade de estados similar a uma banda de condução. Uma vez que existem 6 elétrons disponíveis, todos os orbitais estarão preenchidos e todos os orbitais estarão vazios. O bandgap no presente caso, correspondendo à distância entre o HOMO e o LUMO tem valor de .
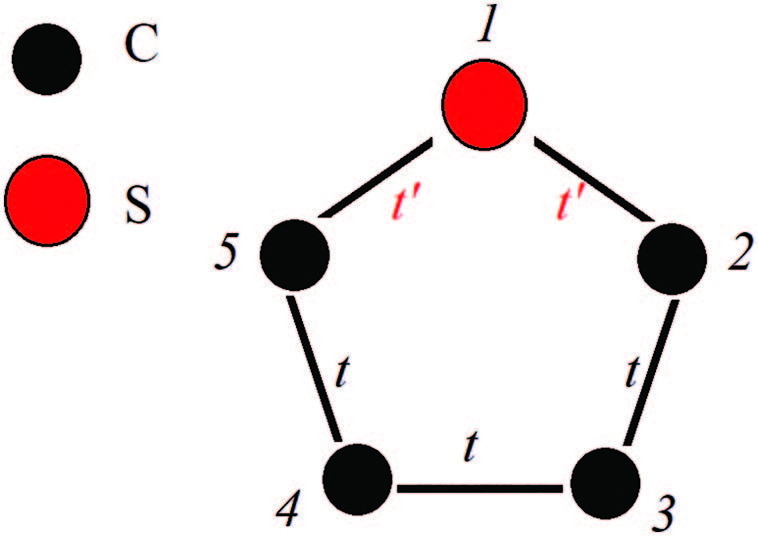
De maneira fenomenológica, é possível montar diretamente a matriz que representa o Hamiltoniano de tight-binding para um elétron, supondo conhecidos os vínculos entre os átomos primeiros vizinhos, e se for o caso, também segundos vizinhos. Como exercício, considere uma outra molécula cíclica de fundamental importância para o ramo dos polímeros conjugados, o tiofeno (), ilustrado na Figura8. Este monômero é essencial na síntese do P3HT, que é utilizado em estudos com células solares orgânicas [11[11] F. Thomazi, M. Souza, C.K. Saul, G.A. Viana, F.C. Marques, R. Silvestre, M. Brehm, C.E.B. Marino, E. Burkarter e C.A. Dartora, Current Nanoscience 10 , 877 (2014).].

A molécula de tiofeno: um dos átomos de carbono no ciclo é trocado pelo enxofre, cujo índice de sítio é . Esse átomo tem um nível diferente de energia no orbital que participa das ligações com os orbitais do carbono, bem como o parâmetro de salto para um elétron entre os átomos de enxofre e carbono tem valor .
Para construir o Hamiltoniano de tight-binding dessa molécula, considerando apenas interações com primeiros vizinhos e orbitais do tipo , pode-se observar na Figura8, que o heteroátomo na estrutura, aqui o enxofre, está indexado por , enquanto o carbono ocupa os sítios . Átomos de hidrogênio presentes, que participam em ligações do tipo e não são relevantes para o que segue e foram omitidos na Figura8. Naturalmente, considerando a energia dos orbitais do carbono como referência, a energia associada a um elétron ocupando o orbital do enxofre tem valor . A probabilidade de salto do elétron no enxofre, para um dos carbonos vizinhos, nas posições e é mensurada pelo parâmetro de hopping . Nesse caso, o elemento de matriz que conecta um elétron no sítio ao sítio , denotado , terá valor . O salto no sentido inverso é dado por . Aqui vamos admitir por simplicidade que e são reais. Como outro exemplo, os elementos e da matriz Hamiltoniana devem representar a energia de salto entre os sítios e , ocupados por átomos de carbono no presente caso. Realizando a análise de todos os vínculos, obtém-se o Hamiltoniano do tiofeno para um elétron:
Sabe-se ainda que o parâmetro de hopping da ligação satisfaz a condição , sendo o hopping nas ligações . Para fins de comparação, considere uma molécula cíclica muito similar ao tiofeno, composta apenas de átomos de carbono no ciclo, denominada ciclopentadieno (), cujo Hamiltoniano é dado abaixo:
A análise desse caso relativamente simples permite compreender, dentre outras coisas, o efeito de uma impureza substitucional, nesse caso o enxofre, que é colocado no lugar de um átomo de carbono. O cálculo da densidade de estados, , pode ser feito utilizando-se o método da função de Green retardada , através das equações abaixo [18[18] C. Kittel, Quantum Theory of Solids (John Wiley & Sons, New York, 1987), 2a ed., 23[23] H.N. Nazareno, Mecânica Estatística e Funções de Green (UnB, Brasília, 2010), 2a ed.]:
onde é a matriz identidade com a dimensão de , Im denota a parte imaginária e Tr denota o traço de matriz. Idealmente, deve-se tomar o limite . Todavia, para fins de cálculo numérico é necessário considerar um valor finito para o parâmetro . E isso não é necessariamente uma limitação muito importante, já que na prática, esse parâmetro pode ser usado para descrever efeitos de relaxação dos níveis de energia.
Para fins de comparação, as densidades de estados das moléculas ciclopentadieno e tiofeno são ilustradas na Figura9. Os valores utilizados, normalizados em relação ao parâmetro de hopping da ligação , foram os seguintes: , e . Naturalmente, a molécula de ciclopentadieno tem uma simetria maior do que o tiofeno. A quebra de simetria é realizada pela substituição de um átomo de carbono por um de enxofre. É sabido que situações mais simétricas favorecem a degenerescência dos níveis de energia. Na Figura9-A pode-se perceber que para o ciclopendadieno há dois níveis de energia duplamente degenerados, sem considerar ainda o spin, em e , admitidos aqui como os orbitais LUMO e HOMO, respectivamente, produzindo um bandgap. Há ainda um nível de energia superior localizado em , não degenerado, a menos daquela devida ao spin. O grau de degenerescência de cada nível pode ser inferido a partir das amplitudes das densidades de estados. Para a molécula de tiofeno a densidade de estados está mostrada na Figura9-B. Note que aqueles níveis de energia degenerados no ciclopentadieno se separam, e além da remoção da degenerescência ocorre uma redução do bandgap para . A lição que se pode tirar desse exemplo é que a introdução de impurezas substitucionais remove pelo menos algumas degenerescências no espectro de e ainda altera o bandgap de energia.

Densidades de estados para as moléculas de (A) ciclopentadieno e (B) tiofeno. Note que, dada a similaridade estrutural das duas moléculas, os níveis de energia estão localizados em valores próximos. No entanto, a substituição de um átomo de carbono pelo enxofre remove degenerescências e reduz o valor do bandgap .
Finalizando esta presente Seção, considere cadeias poliméricas lineares, como o poliacetileno, cujo estudo pode ser realizado utilizando um Hamiltoniano de tight-binding com possibilidade de salto entre primeiros vizinhos, cuja forma matricial para o problema de um elétron é a seguinte:
onde é o número de átomos da cadeia, é a energia do orbital atômico relevante no -ésimo átomo da cadeia e é o parâmetro de hopping entre os átomos localizados nos sítios e , . No caso em que todos os átomos da cadeia são idênticos e não há defeitos deve-se assumir que e para qualquer . A inclusão de defeitos e impurezas se dá através da modificação de alguns valores de energia e de parâmetro de hopping no Hamiltoniano, correspondentes aos sítios onde ocorrem. No caso mais simples da uma cadeia polimérica linear de átomos de carbono, o Hamiltoniano (65) pode ser escrito explicitamente:
sendo um caso particular de matriz de Toeplitz [24[24] R.M. Gray, Foundations and Trends in Communications and Information Theory 2 , 155 (2006).], cujos autovalores são dados por:
Tomando-se o limite o espectro de energias tende ao continuum e a largura total da banda de energia terá valor , inexistindo um gap, reproduzindo o resultado obtido na Seção 3 do presente trabalho. Todavia, para finito e com o preenchimento de níveis até a metade, haverá uma diferença de energia entre o LUMO e o HOMO que dependerá do comprimento da cadeia, aqui representado pelo número total de átomos.
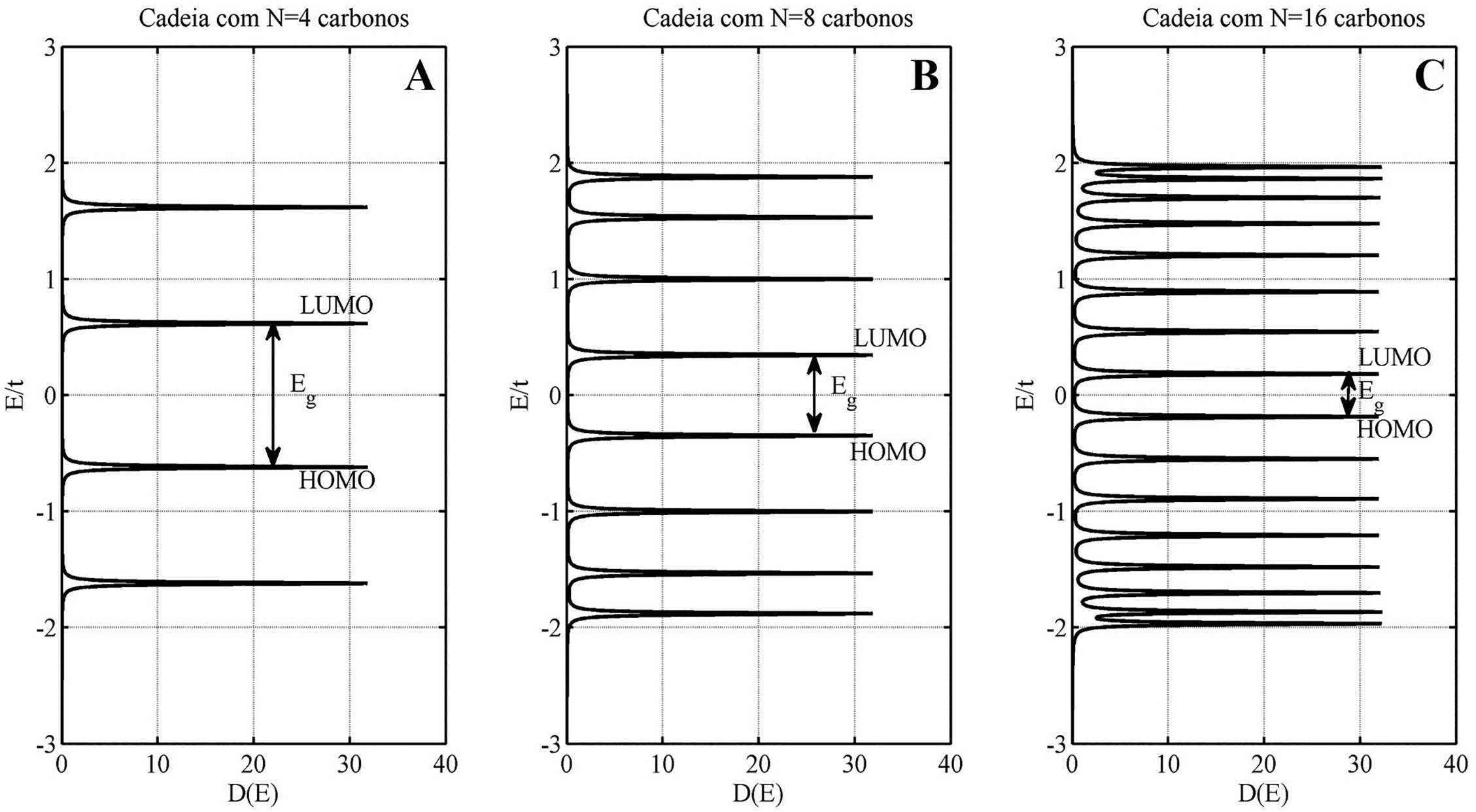
As densidades de estados para cadeias lineares contendo e 16 átomos de carbono, calculadas através das equações (63) e (64), são mostradas na Figura10. O que fica evidente é a diminuição do gap entre o LUMO e o HOMO com o aumento de . Quando o valor do número de átomos na cadeia polimérica conjugada é grande, formam-se estruturas de bandas similares aos dos semicondutores inorgânicos (Si,Ge, etc). Os valores do bandgap, medidos pela separação entre o HOMO e o LUMO, assumem valores tipicamente entre 1.5 a 3.5 eV, sendo determinantes nas propriedades eletro-ópticas dessas cadeias.

Densidades de estados obtidas através da função de Green retardada para cadeias lineares contendo A) , B) e C) átomos de carbono, evidenciando a diminuição do gap com o aumento de .
Tendo em vista que um material polimérico orgânico é tipicamente obtido da condensação de um conjunto de cadeias poliméricas lineares cujos comprimentos são variáveis, existirá uma estatística associada ao níveis de energia dos orbitais HOMO e LUMO. Uma boa aproximação é considerar distribuições gaussianas em torno dos níveis médios do HOMO e do LUMO. Além disso, podem ocorrer níveis de energia dentro do gap, que são evidências da presença de defeitos, como quebras, impurezas substitucionais, enovelamentos e isomerizações nas cadeias. A dispersão estatística em relação ao comprimento das cadeias poliméricas afeta propriedades de transporte de carga elétrica,como a mobilidade dos portadores de carga, por exemplo.
5. Conclusão
No presente trabalho foi apresentado o procedimento geral de segunda quantização, especializado para a obtenção dos modelos de tight-binding, que constituem um dos pilares da Física da Matéria Condensada. A conversão das funções de ondas descrevendo a amplitude de probabilidade dos elétrons na primera quantização em operadores de campo é realizada através de uma álgebra fermiônica, que foi discutida brevemente. Primeiramente, o método geral foi empregado no estudo de um modelo de brinquedo para uma macromolécula polimérica linear muito longa, através da aproximação de cadeia atômica unidimensional infinita. A diagonalização do Hamiltoniano de tight-binding nesse caso leva à formação de uma banda de energia, cuja largura depende do diretamente do parâmetro de salto. Adicionalmente, foi possível prever, com base no comportamento do parâmetro de hopping, que a mobilidade de elétrons na banda de condução tende a ser maior do que a mobilidade das lacunas na banda de valência. Na sequência, alguns monômeros de relevância na área de polímeros orgânicos conjugados foram analisados, dentre eles o benzeno e o tiofeno. O método de tight-binding permitiu determinar os níveis de energia HOMO e LUMO do benzeno e, indo um pouco adiante, as densidades de estados do tiofeno e do ciclopentadieno, bem como discutir aspectos gerais associados à estrutura dos níveis de energia de cadeias poliméricas.
Agradecimentos
O presente trabalho foi realizado com apoio da Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) – Código de Financiamento 001.
Referênces
-
[1]W. Callister Jr. e D.G. Rethwisch, Fundamentos da Ciência e Engenharia dos Materiais: Abordagem Integrada (LTC, Rio de Janeiro, 2014).
-
[2]J.D. Shackelford, Ciência dos Materiais (Pearson, São Paulo, 2008), 6a ed.
-
[3]H. Shirakawa, E.J. Louis, A.G. MacDiarmid, C.K. Chiang e A.J. Heeger, Journal of the Chemical Society, Chemical Communications 16 , 578 (1977).
-
[4]C.K. Chiang, C.R. Finche, Y.W. Park, A.J. Heeger, H. Shirakawa, E.J. Lois, S.C. Gau e A.G. MacDiarmid, Phys. Rev. Lett. 39 , 1098 (1977).
-
[5]N.S. Sariciftci, L. Smilowitz, A.J. Heeger e F. Wuld, Science 258 , 1474 (1992).
-
[6]A.J. Heeger, S. Kivelson, J.R. Schrieffer e W.-P. Su, Rev. Mod. Phys. 60 , 781 (1988).
-
[7]M. Pope, H.P. Kallmann e P. Magnante, J. Chem. Phys. 38 , 2042 (1963).
-
[8]J.J.M. Halls, Photoconductive properties of conjugated polymers . Doctoral Thesis, University of Cambridge, Cambridge (1997).
-
[9]B.R. Weinberger, M.Akhtar e S.C. Gau, Synthetic Metals 4 , 187 (1982).
-
[10]A.A. Lima, N.P.Menezes, S. Santos, B. Amorim, F. Thomazi, F. Zanella, A. Heilmann, E. Burkarter e C.A. Dartora, Revista Brasileira de Ensino de Física 42 , e20190191 (2020).
-
[11]F. Thomazi, M. Souza, C.K. Saul, G.A. Viana, F.C. Marques, R. Silvestre, M. Brehm, C.E.B. Marino, E. Burkarter e C.A. Dartora, Current Nanoscience 10 , 877 (2014).
-
[12]F. Thomazi, C.K. Saul, C. Marino, E. Burkarter e C.A. Dartora, Current Nanoscience 12 , 611 (2016).
-
[13]L.B. Schneider, F. Thomazi, E. Burkarter, C.E.B. Marino, C.K. Saul, A.S. Ito e C.A. Dartora, Current Nanoscience 14 , 403 (2018).
-
[14]Z. Qiu, B.A.G. Hammer e K. Müllen, Progress in Polymer Science 100 , 101179 (2020).
-
[15]C.A. Dartora, M.J. Saldaña Jimenez e F. Zanella, Revista Brasileira de Ensino de Física 37 , 3301 (2015).
-
[16]M. Alonso e E.J. Finn, Física (Editora Escolar, São Paulo, 2012).
-
[17]W. Greiner e J. Reinhardt, Field Quantization (Springer-Verlag, Berlin, Heidelberg, 1996).
-
[18]C. Kittel, Quantum Theory of Solids (John Wiley & Sons, New York, 1987), 2a ed.
-
[19]J.C. Slater e G.F. Koster, Physical Review 94 , 1498 (1954).
-
[20]A. Heeger, Semiconducting and Metallic Polymers: The Fourth Generation of Polymeric Materials, disponível em: https://www.nobelprize.org/prizes/chemistry/2000/heeger/lecture/, acessado em 01/07/2021.
» https://www.nobelprize.org/prizes/chemistry/2000/heeger/lecture/ -
[21]A.G. MacDiarmid, “Synthetic Metals”: A Novel Role for Organic Polymers, disponível: https://www.nobelprize.org/prizes/chemistry/2000/macdiarmid/lecture/, acessado em 02/07/2021.
» https://www.nobelprize.org/prizes/chemistry/2000/macdiarmid/lecture/ -
[22]H. Shirakawa, The Discovery of Polyacetylene Film: The Dawning of an Era of Conducting Polymers, disponível em: https://www.nobelprize.org/prizes/chemistry/2000/shirakawa/lecture/, acessado em 01/07/2021.
» https://www.nobelprize.org/prizes/chemistry/2000/shirakawa/lecture/ -
[23]H.N. Nazareno, Mecânica Estatística e Funções de Green (UnB, Brasília, 2010), 2a ed.
-
[24]R.M. Gray, Foundations and Trends in Communications and Information Theory 2 , 155 (2006).
-
1
O número quântico de spin será omitido nas expressões para os orbitais , , e e nas que se seguirão. Cada um deles é duplamente degenerado devido ao spin eletrônico, que pode assumir as projeções ou , significando que caberia um total de 8 elétrons na camada .
-
2
do inglês highest occupied molecular orbital
-
3
do inglês lowest unoccupied molecular orbital
Datas de Publicação
-
Publicação nesta coleção
05 Jan 2022 -
Data do Fascículo
2022
Histórico
-
Recebido
21 Set 2021 -
Revisado
05 Nov 2021 -
Aceito
26 Nov 2021