Resumos
Poliangeíte microscópica é uma forma de vasculite sistêmica de pequenos vasos, associada aos anticorpos anticitoplasma de neutrófilos, que preferencialmente acomete vênulas, capilares e arteríolas, e que pode, entretanto, envolver artérias e veias. Está entre as vasculites sistêmicas primárias de pequenos vasos mais freqüentes, e pode ter apresentação clínica indistinguível da granulomatose de Wegener e da síndrome de Churg-Strauss. Estas vasculites de pequenos vasos são histologicamente semelhantes e podem ser diferenciadas pela presença de granulomas na granulomatose de Wegener, ou de quadro clínico-funcional de asma na síndrome de Churg-Strauss. Relata-se o caso de um paciente do sexo masculino de 66 anos com poliangeíte microscópica com hemorragia alveolar difusa como forma de apresentação clínica, com ênfase no diagnóstico diferencial com outras vasculites pulmonares de pequenos vasos.
Poliangeíte microscópica; Hemorragia alveolar; Vasculites pulmonares
Microscopic Polyangiitis is a form of Anti-Neutrophil Cytoplasmic Antibody (ANCA)- associated small-vessel vasculitis that preferentially involves venules, capillaries and arterioles and may also involve arteries and veins. It is one of the most common primary systemic small-vessel vasculitis. Its clinical presentation is not distinguishable from the Wegener’s granulomatosis (WG) and the Churg-Strauss syndrome (CSS). These types of small-vessel vasculitis are histologically similar and can be differentiated by the presence of granulomatous inflammation in WG or asthma in CSS. The case of a 66-year-old man with microscopic polyangiitis presenting with alveolar hemorrhage is reported with a discussion of the differential diagnosis of other types of pulmonary small-vessel vasculitis.
Microscopic polyangiitis; Alveolar hemorrhage; Pulmonary vasculitis
RELATO DE CASO
Poliangeíte microscópica com hemorragia alveolar difusa* * Trabalho realizado: Serviço de Pneumologia, Hospital Universitário de Santa Maria, Universidade Federal de Santa Maria. Departamento de Patologia, Faculdade de Medicina, Universidade de São Paulo (USP).
José Wellington Alves dos Santos(TE SBPT); Gustavo Trindade Michel; Carlos Eurico da Luz Pereira(TE SBPT); Vera Luiza Capelozzi; Jader Nascimento Mileto; Cleber Antonio Fiorini
Endereço para correspondência Endereço para correspondência José Wellington Alves dos Santos Rua Venâncio Aires 2020 / 403 Centro CEP: 97010-004. Santa Maria, RS Tel: (55) 220 8585 e-mail: well@vant.com.br Recebido para publicação, em 5/5/3. Aprovado, após revisão, em 30/7/3.
RESUMO
Poliangeíte microscópica é uma forma de vasculite sistêmica de pequenos vasos, associada aos anticorpos anticitoplasma de neutrófilos, que preferencialmente acomete vênulas, capilares e arteríolas, e que pode, entretanto, envolver artérias e veias. Está entre as vasculites sistêmicas primárias de pequenos vasos mais freqüentes, e pode ter apresentação clínica indistinguível da granulomatose de Wegener e da síndrome de Churg-Strauss. Estas vasculites de pequenos vasos são histologicamente semelhantes e podem ser diferenciadas pela presença de granulomas na granulomatose de Wegener, ou de quadro clínico-funcional de asma na síndrome de Churg-Strauss. Relata-se o caso de um paciente do sexo masculino de 66 anos com poliangeíte microscópica com hemorragia alveolar difusa como forma de apresentação clínica, com ênfase no diagnóstico diferencial com outras vasculites pulmonares de pequenos vasos.
Descritores: Poliangeíte microscópica. Hemorragia alveolar. Vasculites pulmonares
Siglas e abreviaturas utilizadas neste trabalho:
PM: Poliangeíte Microscópica
ANCA: Anticorpo anticitoplasma de neutrófilo
GW: Granulomatose de Wegener
SCS: Síndrome de Churg-Strauss
Introdução
Poliangeíte microscópica (PM) é uma vasculite necrosante sistêmica pauci-imune, geralmente associada ao anticorpo anticitoplasma de neutrófilos (ANCA), que afeta predominantemente pequenos vasos (capilares, vênulas e arteríolas)1-4. Glomerulonefrite necrosante com formação de crescentes e capilarite pulmonar hemorrágica são manifestações freqüentes e as mais importantes causas de morbidade e mortalidade da doença. A incidência de PM é de aproximadamente 1:100.000, com um leve predomínio no sexo masculino e uma média de idade de início dos sintomas de 50 anos, embora indivíduos de qualquer idade possam ser acometidos.1,3,4
Relato do caso
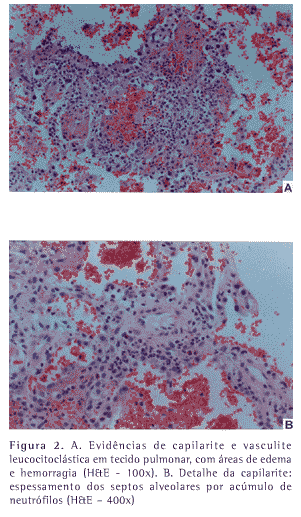
Um paciente do sexo masculino, branco, de 66 anos e ex-tabagista (60 anos/maço) procurou atendimento médico com história de dispnéia e tosse produtiva com escasso escarro mucóide havia quatro meses. Nos últimos 30 dias, havia apresentado vários episódios de hemoptise em pequena quantidade e emagrecimento de 3 kg. Na internação, o radiograma de tórax evidenciou infiltrado difuso com padrão intersticial e alveolar. O hemograma revelou anemia (hemoglobina: 9,5 g/dL). O exame qualitativo de urina demonstrou proteinúria, mais de 50 hemácias por campo, e cilindros hialinos com inclusão de hemácias. A depuração da creatinina endógena foi de 25 mL/min/1,73 m2 de superfície corporal e a proteinúria de 24 horas foi de 2,56 g. Fator anti-nuclear, pesquisa de células LE, fator reumatóide, VDRL e pesquisa de anticorpos anti-DNA e anti-SM foram negativos. O ecocardiograma e a ultra-sonografia renal não revelaram alterações. O radiograma e a tomografia computadorizada dos seios da face foram normais. Biópsia renal, realizada oito dias após a internação, evidenciou esclerose avançada, com formação de crescentes e a imunofluorescência do material mostrou escassos depósitos de C3 e ausência de depósitos lineares de IgG. A pesquisa de ANCA, realizada doze dias após a admissão, foi positiva e revelou padrão perinuclear (p-ANCA) através da imunofluorescência indireta. A tomografia computadorizada de tórax evidenciou áreas em vidro despolido, bilaterais e esparsas, com micronódulos densos, alguns deles confluentes (Figura 1). O paciente permanecia clinicamente estável e no décimo terceiro dia de internação foi submetido à biópsia pulmonar por videotoracoscopia. No dia seguinte à realização do procedimento o paciente evoluiu com hemorragia alveolar difusa e insuficiência respiratória. Apesar da instituição de terapia com corticosteróides sistêmicos em altas doses (500 mg/dia de metilprednisolona) e ciclofosfamida (150 mg/dia) o paciente faleceu 48 horas após a piora do quadro clínico. O estudo histopatológico do espécime cirúrgico obtido foi compatível com vasculite pauci-imune de pequenos vasos (Figura 2). Como o paciente não apresentava evidência histopatológica de doença granulomatosa, quadro clínico de asma, e possuía testes de função pulmonar normais, foi confirmado o diagnóstico de poliangeíte microscópica.
Discussão
A PM é uma forma de vasculite sistêmica de pequenos vasos que se caracteriza pela ausência ou escassez de depósitos imunes vasculares e é associada com ANCA em aproximadamente 90% dos casos.3
As manifestações clínicas são variadas e inespecíficas. Hematúria, hemoptise, púrpura, neuropatia periférica, dor abdominal, hemorragia gastrintestinal, sinusite, febre, perda ponderal, mialgias e artralgias são manifestações freqüentes.5,6,7 Hemorragia alveolar ocorre em até 30% dos casos e é normalmente acompanhada de manifestações extratorácicas, principalmente renais (97%). Queixas articulares e musculares podem preceder por anos o surgimento de alterações renais e/ou pulmonares. Contudo, hemorragia alveolar difusa tem sido descrita como a única manifestação em pacientes com PM.6 Quando se apresenta com hemorragia alveolar difusa, os principais sintomas são dispnéia (90%), a qual é geralmente severa, tosse (90%) e hemoptise (79%).6
Achados laboratoriais freqüentemente incluem anemia, hematúria, proteinúria, cilindrúria e perda de função renal. Inicialmente, o radiograma de tórax pode mostrar opacidades alveolares bilaterais sugestivas de hemorragia alveolar. Com a progressão da doença, pode apresentar infiltrado reticulonodular difuso.6
Em um estudo envolvendo 29 pacientes com PM e hemorragia alveolar, Lauque et al. encontraram, no radiograma de tórax, padrão alveolar em 28 (97%) casos, com acometimento bilateral em 26 (90%) deles. Nesse estudo, a tomografia computadorizada de tórax, realizada em 18 casos, revelou, como no presente estudo, envolvimento bilateral em 100% dos pacientes, com predomínio de consolidações (67%) e padrão em vidro despolido (61%).6 Como o achado de infiltrado alveolar bilateral é inespecífico, o lavado bronco-alveolar progressivamente hemorrágico e uma captação aumentada de monóxido de carbono no teste de difusão podem auxiliar o diagnóstico.
A PM tem sido relatada como a principal causa de síndrome reno-pulmonar8, devendo ser considerada em pacientes com hemoptise que apresentem alterações da função renal ou do sedimento urinário. Entretanto, outras formas de vasculite que cursam com síndrome reno-pulmonar devem ser excluídas para que se confirme o diagnóstico de PM com acometimento pulmonar.2,3,7
O estudo histopatológico evidencia vasculite de pequenos vasos, embora artérias maiores possam ser envolvidas em alguns casos. A lesão vascular básica da PM consiste em necrose vascular segmentar com infiltração de neutrófilos e monócitos, e com escassez ou ausência de depósitos imunes (pauci-imune). O rim é o órgão-alvo mais freqüentemente acometido pela PM,1-4,9 sendo afetado em aproximadamente 90% dos casos. A mais freqüente lesão renal é a glomerulonefrite necrosante com formação de crescentes. A lesão glomerular detectada pelo estudo histopatológico é idêntica na PM, na Granulomatose de Wegener (GW) e na Síndrome de Churg-Strauss (SCS). No caso aqui relatado o paciente apresentava manifestações clínicas inicialmente pulmonares, com quatro meses de evolução, o que torna pouco provável a presença de doenças infecciosas como causa da hemorragia alveolar , como por exemplo leptospirose e hantavirose. Além disso, apresentava comprometimento da função renal já na admissão.
Os ANCA são auto-anticorpos que possuem especificidade para constituintes citoplasmáticos de monócitos e neutrófilos. Permanece incerto se estes anticorpos estão diretamente envolvidos na patogênese das vasculites ou são apenas um fenômeno associado.9
Os ANCA traduzem-se em marcadores diagnósticos úteis na suspeita clínica de vasculite de pequenos vasos.3,5 Nos pacientes com suspeita clínica de vasculite pauci-imune de pequenos vasos, a positividade na pesquisa de ANCA tem valor preditivo positivo de 90%, o que, no entanto, não prescinde da avaliação histopatológica. Entretanto, um teste negativo não exclui completamente este grupo de doenças.10 Em pacientes com fracas evidências clínicas, o teste possui um valor preditivo negativo de 99%.8 A PM deve ser diferenciada principalmente da GW e da SCS, pois também são vasculites pauci-imunes de pequenos vasos associadas com ANCA e com aparência patológica idêntica. Qualquer tipo de ANCA pode ser observado nas vasculites associadas a eles, contudo, o padrão c-ANCA tem predominância na GW e o p-ANCA na SCS.3 Na PM, há um predomínio do padrão p-ANCA, porém o c-ANCA também pode ser encontrado.5
Uma vez estabelecido o diagnóstico de vasculite pauci-imune de pequenos vasos associada aos ANCA, a seqüência da investigação diagnóstica deve ser a determinação de um diagnóstico específico.2,3,11 Por definição, pacientes com PM não têm inflamação granulomatosa nos pulmões ou em outros órgãos. A presença de inflamação granulomatosa de pequenos vasos deve ser indicativo de GW ou SCS.2,3 Quando o paciente tem vasculite acompanhada de evidências de inflamação necrosante granulomatosa não infecciosa, especialmente nos tratos respiratórios superior e inferior, sem história de asma, o diagnóstico apropriado é GW. Na presença de lesões granulomatosas, eosinofilia periférica, história clínica e testes funcionais compatíveis com asma, o diagnóstico apropriado é SCS. Se o paciente não apresenta evidência de inflamação granulomatosa ou asma, o diagnóstico apropriado é PM. Achados clínicos tais como nódulos ou cavidades pulmonares, ou lesões ósseas destrutivas no trato respiratório superior, podem ser usados como evidência de inflamação granulomatosa, o que dispensa a confirmação em tecido.9 Portanto, o diagnóstico da PM é por exclusão.
Em casos de suspeita clínica de vasculite, principalmente quando o paciente apresenta hemorragia alveolar e disfunção renal, o início do tratamento não deve ser retardado. Corticosteróides em altas doses (metilprednisolona 7 mg/kg/dia) e ciclofosfamida (2 mg/kg/dia) são o tratamento de escolha para a indução da remissão da PM.3,4,5 Após a remissão da doença reduz-se a dose dos corticosteróides até a retirada, o que geralmente ocorre em três a cinco meses. A ciclofosfamida é mantida por seis a doze meses, podendo ser substituída por azatioprina após a remissão. A morbidade e mortalidade dependem diretamente da instituição precoce da terapia5. A presença de vasculite de pequenos vasos envolvendo os pulmões, que neste caso foi evidenciada apenas após a piora do quadro clínico, eleva a mortalidade precoce na PM. Mais de 25% dos pacientes falecem durante o primeiro episódio de hemorragia alveolar difusa, como ocorreu no caso relatado, apesar do tratamento adequado. Naqueles que sobrevivem, a resposta ao tratamento é boa, mas episódios recorrentes são esperados. Fibrose pulmonar e doença pulmonar obstrutiva têm sido descritas como complicações crônicas de episódios recorrentes de hemorragia alveolar difusa.7
- 1. Savage CO, Winearls CG, Evans DJ, Reis AJ, Lockwood CM. Microscopic polyarteritis: presentation, pathology and prognosis. Q J Med 1985; 56:467-83.
- 2. Jennette JC, Falk RJ, Andrassy K, Bacon PA, Churg J, Gross WL, et al. Nomenclature of systemic vasculitis: the proposal of an international consensus conference. Arthritis Rheum 1994;37:187-92.
- 3. Jennette JC, Falk RJ. Small vessel vasculitis. N Engl J Med 1997;337:1512-23.
- 4. Guillevin L, Durand-Gasselin B, Cevallos R, Grayraud M, Lhote F, Callard P, et al. Microscopic polyangiitis: clinical and laboratory findings in eighty-five patients. Arthritis Rheum 1999;42:421-30.
- 5. Jennette JC, Thomas DB, Falk RJ. Microscopic polyangiitis (microscopic polyarteritis). Semin Diag Pathol 2001;18:3-13.
- 6. Lauque D, Cadranel J, Lazor R, Pourrat J, Ronco P, Guillevin L, et al. Microscopic polyangiitis with alveolar hemorrhage. A study of 29 cases and review of the literature. Medicine 2000;79:222-33.
- 7. Marvin IS, Kevin KB. Small vessel vasculitis of the lung. Thorax 2000;55:502-10.
- 8. Niles JL, Bottinger EP, Saurina GR, Kelley KJ, Pan G, Collins AB, et al. The syndrome of lung hemorrhage and nephritis is usually an ANCA-associated condition. Arch Intern Med 1996;156:440-5.
- 9. Jennette JC, Falk RJ. The pathology of vasculitis involving the kidney. Am J Kidney Dis 1994;24:130-41.
- 10. Lim LCL, Taylor JG, Schmitz JL, Folds JD, Wilkman AS, Falk RJ, et al. Diagnostic usefulness of antineutrophil cytoplasmic autoantibody serology: Comparative evaluation of commercial indirect fluorescent antibody kits and enzyme immunoassays kits. Am J Clin Pathol 1999;111:363-9.
- 11. Sorensen SF, Slot O, Tvede N, Petersen J. A prospective study of vasculitis patients collected in a five year period: evaluation of the Chapel Hill nomenclature. Ann Rheum Dis 2000;59:478-82.
Datas de Publicação
-
Publicação nesta coleção
08 Jun 2004 -
Data do Fascículo
Abr 2004
Histórico
-
Recebido
05 Maio 2003 -
Aceito
07 Jul 2003