Abstracts
The relationship between cancer and coagulopathy was suggested by Trousseau nearly 150 years ago. Later, it became more evident that oncologic patients are at a higher risk of experiencing thromboembolic events. This can be due to activation of the coagulation system either by neoplastic cells or by prescribed therapies (chemotherapy or surgical procedures). In fact, these events can constitute the first manifestation of cancer, and their recurrence, despite efficient anticoagulation, has been described. The coagulation system is normally activated in order to provide healing. In the presence of neoplasms, this complex system is activated as a response to multiple stimuli and seems to contribute to cancer progression. Activation of the coagulation system has a greater effect on metastatic foci than on the primary tumor. However, most cancer victims die from complications caused by metastasis, which underscores the importance of this theme. In this area, various mechanisms have been described, creating promising perspectives for future treatments. The current success in using low-molecular-weight heparins against small cell lung cancer is encouraging. Although the knowledge of those mechanisms is relatively incipient, many basic research and clinical studies are underway.
Lung neoplasms; Thrombophilia
A relação entre câncer e alteração na coagulação já havia sido sugerida há quase 150 anos por Trousseau e, subseqüentemente, ficou claro o maior risco que os pacientes oncológicos têm de desenvolverem fenômenos tromboembólicos. Isto pode ser conseqüência da ativação do sistema de coagulação pelas células neoplásicas ou pelas terapias empregadas (quimioterapias e cirurgias). Tais fenômenos podem, ainda, ser a primeira manifestação do câncer e a sua recorrência, mesmo com anticoagulação adequada, foi descrita. O sistema de coagulação é ativado, normalmente, com finalidade reparativa. Na presença de neoplasias, este complexo sistema está atuante frente a variados estímulos e parece contribuir para a progressão tumoral. Este efeito é mais importante para os focos metastáticos que para o próprio tumor primário. Contudo, a maior parte das vítimas de neoplasias morre das complicações das metástases, revelando a importância deste tema. Nesta área, vários mecanismos já são conhecidos e geram interessantes perspectivas para tratamentos futuros. Atualmente, o sucesso obtido com as heparinas de baixo peso molecular no carcinoma de pequenas células de pulmão é animador. Embora o conhecimento sobre esses mecanismos sejam relativamente recentes, os campos de pesquisa e tratamento estão amplamente abertos.
Neoplasias pulmonares; Trombofilia
REVIEW ARTICLE
Hypercoagulability and lung cancer*
Felipe Costa de Andrade MarinhoI; Teresa Yae TakagakiII
IResident in the Department of Pulmonology of the University of São Paulo School of Medicine, São Paulo, Brazil
IISupervisor of the Department of Pulmonology of the University of São Paulo School of Medicine, São Paulo, Brazil
Correspondence to
ABSTRACT
The relationship between cancer and coagulopathy was suggested by Trousseau nearly 150 years ago. Later, it became more evident that oncologic patients are at a higher risk of experiencing thromboembolic events. This can be due to activation of the coagulation system either by neoplastic cells or by prescribed therapies (chemotherapy or surgical procedures). In fact, these events can constitute the first manifestation of cancer, and their recurrence, despite efficient anticoagulation, has been described. The coagulation system is normally activated in order to provide healing. In the presence of neoplasms, this complex system is activated as a response to multiple stimuli and seems to contribute to cancer progression. Activation of the coagulation system has a greater effect on metastatic foci than on the primary tumor. However, most cancer victims die from complications caused by metastasis, which underscores the importance of this theme. In this area, various mechanisms have been described, creating promising perspectives for future treatments. The current success in using low-molecular-weight heparins against small cell lung cancer is encouraging. Although the knowledge of those mechanisms is relatively incipient, many basic research and clinical studies are underway.
Keywords: Lung neoplasms; Thrombophilia.
Introduction
Among all malignancies, lung cancer has the greatest impact on mortality worldwide, and that impact is steadily increasing.(1,2) In Brazil, lung cancer was responsible for 12% of all deaths from cancer within the past 30 years.(1) Despite this growing problem, the treatment for lung cancer has hardly evolved since the ratification, in the 1980s, of the platin-based regimens. Survival curves do not seem to change significantly despite the new chemotherapeutic agents and target therapies, as well as conformal, adjuvant and neoadjuvant radiotherapy. Greater knowledge on the pathogenesis and pathophysiology of the disease can suggest new therapies or facilitate the understanding of the failure of other therapies. In this context, the relationship between these diseased cells and healthy tissues, as well as their systems, appears as a promising area. Although we might not be able to define the cure for a disease that depends on multiple variables, there is the perspective of improving local control or avoiding the dissemination of tumors.
The relationship between cancer and coagulation changes was suggested by Trousseau nearly 150 years ago,(3) It later became more evident that oncologic patients are at a higher risk of experiencing thromboembolic events. This can be due to activation of the coagulation system either by neoplastic cells or by prescribed therapies (chemotherapy or surgical procedures). In fact, these events can constitute the first manifestation of cancer, and their recurrence, despite efficient anticoagulation, has been described.(4)
However, these events are paraneoplastic effects and represent only the visible part of this binomial. Current knowledge indicates that tumor cells can utilize these mechanisms in order to promote the progression of the disease and guarantee the success of the clone.
The current success in using low-molecular-weight heparins (LMWHs) against small cell lung cancer is encouraging. Although the knowledge of those mechanisms is relatively incipient, many basic research and clinical studies are underway.
Pathogenesis
The coagulation system is fundamental to homeostasis. It avoids blood loss, initiates cicatrization and promotes tissue remodeling, as well as orchestrating cell proliferation, migration, synthesis and secretion. In normal cells, this complex system is activated as a response to various chemical or physical insults. Two main pathways are activated. The first pathway is activated by the action of procoagulant substances (such as tissue factor), and the second pathway is activated by the expression of urokinase-type plasminogen activator and tissue-type plasminogen activator (uPA and tPA, respectively), which, by activating plasminogen, coordinate cell proliferation and tissue remodeling.(5)
Although the pathogenesis of the interaction between neoplastic cells and the coagulation system is complex and not completely understood, it is known to be characterized by an imbalance between procoagulant and anticoagulant factors. In general, the host response to neoplasms involves inflammation, paraprotein production, necrosis and hemodynamic disorders. Factors external to this system, such as chemotherapies, surgical procedures and central venous catheters, also contribute to the genesis of the prothrombotic state. In addition, recent advances in biochemistry, molecular biology and cell biology have promoted better understanding of this subject, revealing the direct and the indirect activation of the coagulation cascade by substances released by cancer cells (Figure 1).
Coagulation abnormalities
Altered coagulation is detected in more than 50% of patients with cancer and in 90% of those who present metastases. Activation of the coagulation system generates thrombin, as well as promoting the formation and subsequent deposition in blood vessels of fibrin, thereby generating a process of disseminated intravascular coagulation (DIC), which is partially controlled by the fibrinolytic system.
High levels of fibrinopeptide A (a fibrinogen fragment created when fibrinogen is ruptured by thrombin), thrombin fragments and thrombin-antithrombin complexes can be detected in most patients with cancer. The most common biochemical alterations are slightly longer prothrombin time and partial thromboplastin time, as well as higher levels of fibrin degradation products, including D-dimers. This demonstrates increased activity of the fibrinolytic system. Fibrinogen levels are also high, revealing compensated DIC. Approximately 30% of the patients also present increases in platelet count and platelet activity.
Fibrinolysis inhibition, caused either by a decrease in fibrinolytic proteases (uPA and tPA) or by an increase in their inhibiting factors, such as plasminogen activator inhibitor 1 (PAI-1), has been reported in the presence of various tumor types.(6) However, some types of malignancy, such as promyelocytic leukemia and some cases of disseminated prostate cancer, over-activate the fibrinolytic system, via uPA and tPA pathways, causing severe bleeding.
Certain biochemical alterations can be quite sensitive for the detection of many such abnormalities. However, this does not help establish a clear relationship between the risk for thrombosis and the indication for long-term use of anticoagulants.(7)
Prothrombotic mechanisms of malignant cells
Procoagulant factors
Tumor cells produce procoagulant factors; the most widely studied and most often implicated factors are tissue factor and cancer procoagulant.(6-8)
Tissue factor, the main activator of the coagulation cascade, forms a complex with factor VII in order to activate factors X and XI. The final pathway of this process is the transformation of prothrombin into thrombin. Although tissue factor is not typically expressed by healthy endothelial cells, it can be induced by inflammatory stimuli, as well as by cytokines, such as tumor necrosis factor-alpha and interleukin-1 beta.(7) However, tissue factor is constitutively expressed by malignant cells, such as those of some types of lymphoma, leukemia and adenocarcinoma. Monocytes and macrophages can also be activated by the receptors in tumor cells and begin to express tissue factor.(6)
Cancer procoagulant is a vitamin-K-dependent protease found in extracts of some types of cancer, including sarcoma, neuroblastoma and melanomas, as well as lung, breast, kidney, colon, vagina and blood cancer. Cancer procoagulant can activate factor X independently of factor VII. Although cancer procoagulant cannot be found in normal cells, its use as a tumor marker or as an indicator of thrombosis is still unclear. Promyelocytic leukemia is an exception. The presence of cancer procoagulant in blasts is a predictor of malignant transformation and of the response to trans-retinoic acid.(6)
Mucin produced by carcinomas is another potent procoagulant agent and it can directly activate prothrombin to convert into thrombin when released in the circulatory system.(6)
Modulation of the fibrinolytic/anticoagulant system
Inhibiting the endogenous anticoagulant or fibrinolytic systems can also cause thrombosis. Plasminogen activators cause plasminogen to be transformed into plasmin, which lyses the fibrin formed during the coagulation process. Although the main inhibitor of this pathway, PAI-1, is typically produced by platelets and the endothelium, it is also produced by some tumor cells. In addition, angiostatin, which is a plasminogen product, inhibits angiogenesis, avoiding tumor progression. Therefore, the tumor production of inhibiting factors of the fibrinolytic system (PAI-1, for example) plays a role in hypercoagulability and can contribute to tumor angiogenesis.(6)
Proteins that regulate the fibrinolytic system, such as uPA and tPA, as well as their receptors, can also be expressed by tumor cells.(5,9) Modulation of this pathway interferes with tissue remodeling and tissue proliferation.(5)
Protein C (PC) and antithrombin III (ATIII) are the main factors in the anticoagulant system. The activation of PC is carried out by means of the binding between thrombin and thrombomodulin, which is a PC membrane cofactor; PC interacts with the coagulation cascade, inhibiting factors Va, VIIIa and PAI-1. In addition, ATIII inhibits coagulation proteases, including thrombin, and other factors, such as Xa. Serum levels of PC and ATIII are decreased in cancer-related DIC; their production in the liver is also decreased when there is metastasis. However, their levels do not correlate with the appearance of thrombotic episodes unfortunately.(6)
Cell activation
Platelets are particularly affected by cancer: up to 30% of patients present increases in platelet count, whereas 11% present decreases.(10) It has been demonstrated that in vitro platelets aggregate in the presence of cancer cells and there is thrombocytopenia when cancer cells are infused in vivo.(11) Platelet activation can be mediated by direct cell-to-cell contact and by the release of several factors, such as adenosine diphosphate, thrombin, other proteases and interleukins. Activated platelets have greater adhesive capacity and interact with other cells, such as leukocytes, endothelial cells and tumor cells.(12). Tumor emboli are usually enfolded within a fibrin-platelet net that contributes to endothelial adhesion. This tumor thrombus, adhered to the endothelium, causes a lesion, and consequently, there is the release of vascular permeability factor, as well as the local loss of inhibition of platelet aggregation, anticoagulation and vasodilation.(6)
Cytokines
The physiological response, mediated by macrophages and T cells activated by tumor antigens and procoagulant substances released in the circulation, causes the release of potent inflammatory mediators, such as interleukin-1 beta and tumor necrosis factor alpha.(6) These cytokines can induce the expression of tissue factor (even in the endothelium, macrophages and tumor cells), activate platelets and reduce thrombomodulin (and, consequently, the anticoagulant PC pathway).(6,7) Finally, the PAI-1 pathway is stimulated, and the PC pathway is inhibited. All of these changes contribute to promoting coagulation over the anticoagulant and fibrinolytic systems.(6)
Another cytokine derived from these cells, vascular endothelial growth factor (VEGF), can be produced by malignant cells. However, VEGF levels can increase due to the stimulus of tissue factor, and, feeding back to the system, VEGF also induces tissue factor in interposed endothelial cells. This suggests the participation of tissue factor in tumor neovascularization.(6,7)
These cytokines promote alterations in the thrombosis-anticoagulation binomial, as well as increasing the expression of adhesion molecules in endothelial cells, thereby facilitating the interaction and adhesion of platelets and leukocytes, which promotes local activation of coagulation and fibrin formation.
Cell interaction
Tumor cells, directly or indirectly, activate monocytes, which express tissue factor in their membranes. Polymorphonuclear leukocytes are also activated, releasing proteases and oxygen-derived free radicals. This is another mechanism acting on endothelial cell lesion and activation, promoting hypercoagulability.(7)
The induction of adhesion molecules into endothelial cells, leukocytes and platelets provides the substrate for the interaction of these cells with tumor cells. This factor takes on added importance in the hematogenous dissemination of the disease.
Tumor cells bound to the endothelium activate local coagulation, causing thrombus formation, as well as adhesion and activation of platelets and leukocytes (by means of the released cytokines). Therefore, these tumor cells can pass through the vessel walls. This entire process is made possible by thrombus formation, and leukocytes facilitate the invasion of vessel walls (Figure 2).(7)
Tumor progression
Various arms of this system contribute to the progression of the neoplastic disease, as described above. Cell interaction, induction of adhesion molecules, neovascularization, interleukins and cell activation (including defense cells, such as macrophages and polymorphonuclear leukocytes) are all potential aids for the progression of cancer.
The activation of the coagulation cascade results in thrombin generation and subsequent fibrin formation. These two substances have effects that further cancer growth and dissemination.(7) Fibrin, in particular, can modulate VEGF expression in cancer cells and in normal endothelial cells. This effect is particularly relevant in the formation of new blood vessels (Figure 3).(13)
Different tumors also have different responses regarding the activation mechanism of the coagulation system; some use the tissue factor pathway and some use the fibrinolytic system pathway (uPA). However, tumors can also share common pathways, such as interactions with platelets, dependence on growth factors and neovascularization. This enables various patients to benefit from the effects of anticoagulant medications.(5) In this component of the fibrinolytic system, PAI-1 stimulates angiogenesis, regulating plasmin-mediated proteolysis.(14) In addition, various proteolytic enzymes, such as collagenases, metalloproteinases and proteases, including plasmin, are involved in tumor invasion,(14) since they are fundamental for the cleavage of the basal membrane and in the extracellular matrix. In this aspect, matrix metalloproteinase 2 (MMP-2) and matrix metalloproteinase 9 (MMP-9) play an important role, promoting the release of local growth factors and modulating surface molecules.(15,16) The expression of MMP-9 in adenocarcinomas in the initial stages has prognostic value.(17)
However, determining the real importance of this system in situ is not as simple as determining serum levels or enzymatic reactions in plasma. Using immunohistochemical probes against factor Xa and thrombin, as well as using monoclonal antibodies against either the thrombin-antithrombin complex or fibrin (active fibrinogen by thrombin cleavage), are feasible methodologies. Similar techniques can detect activation of the uPA pathway.(5,14) These experimental techniques can define, in the various tumor types, which coagulation pathways are initially implicated.(18)
Associated conditions
Immobilization
Immobilization is one of the greatest risk factors in this group of patients, especially during prolonged hospitalizations. In a series of autopsies, pulmonary embolism was found in up to 14% of hospitalized patients with cancer and in only 8% of patients without cancer.(19)
Chemotherapy
The use of cytotoxic agents can have a thrombogenic effect due to various mechanisms, principally direct endothelial lesion, as well as the lysis-induced release of procoagulant products and cytokines. Patients who received 5-fluorouracil, with or without cisplatin, presented a decrease in their levels of protein S and PC, as well as an increase in PAI-1.(6,20) In a retrospective study involving patients submitted to chemotherapy, the rate of thromboembolic complications during the first 3 months of treatment was unexpectedly high, resulting in an annual rate of 11%.(21)
Surgery and radiotherapy
The incidence of deep vein thrombosis in surgical patients not submitted to thromboprophylaxis is two-times higher in patients with cancer than in those without cancer.(22) This incidence remains higher even when prophylactic anticoagulant medications are given, since other factors, such as a more severe clinical profile, lower performance status, immobilization, etc., work in conjunction. The risk persists if patients are discharged without proper prophylactic medication.(23)
Although it is believed that radiotherapy is implicated in the higher risk for thrombotic events, no studies have proven this relationship.(7)
Central venous catheters
Although long-term catheterization is valuable in managing the treatment of patients with cancer, it is associated with a higher risk of venous thrombosis in the upper limbs. Without prophylaxis, the risk of this complication can reach an incredible 37%.(24) However, recent data have shown the current incidence to be lower than that, probably due to the use of more modern catheters and less invasive procedures.(7)
Effects of anticoagulants on cancer
Studies using anticoagulant medications have shown an increase in the survival rate of patients with cancer, especially in those with lung cancer. This strengthens the hypothesis that coagulation plays a role in the progression of this disease.(5) In 2000, a meta-analysis comparing the use of low-molecular-weight heparin (LMWH) and unfractionated heparin (UFH) for the treatment of deep venous thrombosis revealed a tendency toward better survival among patients treated with LMWH. The subgroup analyses in 5 of the 9 studies revealed that this tendency resulted from the inclusion of patients with cancer.(25)
Anticoagulant medications have direct effects on the products of system activation (thrombin and fibrin). The levels of these two substances increase as a direct or indirect consequence of the tumor; in return, they stimulate tumor growth. The mere interruption of this circle explains the potential benefit of these medications. Thrombin is a protease that has high angiogenic capacity in endothelial cells and acts as a growth factor for tumor cells. Fibrin induces the endothelial expression of tissue factor and interleukin-8, which have angiogenic properties. Fibrin also works as a matrix for the migration of cancer cells out of the circulatory system, and the fibrin matrix serves as a substrate for neovascularization, which is necessary for tumor nutrition. In addition to these properties, fibrin forms a protective layer on tumor cells, protecting them from attack by the immune system, creating resistance to chemotherapy and strengthening their anchorage to the vascular wall.(7)
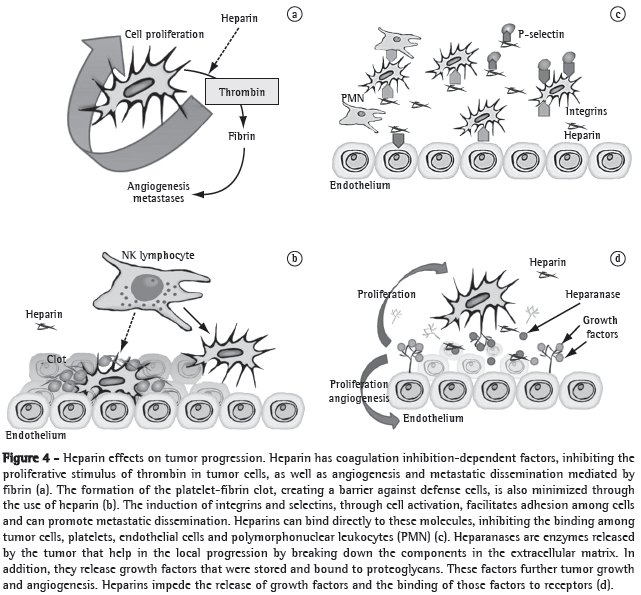
Heparins form a special group of this class because they have specific antitumor properties. Firstly, heparins impede the binding of malignant cells with platelets, leukocytes and the endothelium, since heparins bind to adhesion molecules, such as P-selectin or integrins. These medications also inhibit heparanase, a tumor enzyme, which facilitates the malignant penetration into the vascular basement membrane because it breaks down the polysaccharide barrier. Heparanase also facilitates the release of vascular growth factors that are bound to heparan sulphate in the extracellular matrix into the tumor microenvironment. Heparins can also directly bind to endothelial receptors and interfere with the effects of growth factors. Finally, heparins play a regulatory role in the immune system, modulating various cytokines involved in leukocyte migration and cancer destruction (Figure 4).(7) In addition, LMWHs bind more strongly to some growth factor receptors than do UFHs,(26) as well as inhibiting other receptors, such as those of VEGF and beta-fibroblast growth factor.(27) Due to these advantages, as well as to the fact that LMWHs have better pharmacokinetic properties and lower toxicity than do UFHs, LMWHs have become the medications of choice in clinical studies.(5)
As previously mentioned, different activation pathways of the coagulation system can be triggered by various tumor types. This might be partially explained by the heterogeneous tumor response to anticoagulant treatments. A better understanding in this area might lead to the development of better approaches depending on the histological type. For example, although breast, prostate, colon and non-small cell lung cancer express uPA and uPA receptors, they cannot express a thrombin-generating pathway.(5,28) Aprotinin, an antifibrinolytic agent, by inhibiting plasmin formation (uPA pathway), seems to have a beneficial effect on tumor progression in patients submitted to resection of hepatic metastasis of colon cancer.(29) This type of cancer does not respond to vitamin-K antagonists.(30) However, small cell lung cancer responds to medications with a coagulation-inhibiting effect, such as warfarin or heparins.(30,31)
Finally, clinical studies have shown that treatment with relatively short courses of these medications, such as aprotinin or LMWH, improve cancer outcome for months and even years, maybe because these medications induce a reprogramming of malignant cells.(5,29)
Use of anticoagulant medications in lung cancer
The first study showing a real beneficial effect of anticoagulant medications (warfarin) on small cell lung cancer was published by Zacharski in 1981.(32) A randomized study involving 328 patients confirmed that result.(33) In a recent meta-analysis on the efficacy and safety of anticoagulant use (warfarin or ximelagatran) in patients with cancer, 5 studies were analyzed, and the conclusion was that the only patients that actually benefit from this strategy are those with small cell lung cancer. This finding was attributed to the inclusion of patients with greater disease severity, and the principal impact was on six-month survival. There was an increased risk of severe bleeding in all groups.(34) Previous meta-analyses on anticoagulant use against deep venous thrombosis and pulmonary thromboembolism revealed that patients with cancer benefit from anticoagulant use and that LMWH is more beneficial than is UFH.(25) Based on such results, heparins have been more frequently studied than have been oral anticoagulants and apparently provide better results.
In 2004, 385 patients with advanced solid tumors with no evidence of thromboembolism were randomized to receive prophylactic doses of an LMWH (dalteparin) or a placebo for 12 months. Overall survival was similar in the two groups. However, in the subgroup of patients with better prognosis (survival for more than 17 months after randomization), there were statistically significant differences, favoring the patients who received heparin, in two-year and three-year survival rates.(35) In another study,(36) the use of therapeutic doses of an LMWH (nadroparin) for 2 weeks, followed by half doses for 4 weeks, was evaluated in a group of 302 patients with solid tumors but without evidence of thromboembolism. There was a decrease in 12-month and 24-month mortality (12% and 10%, respectively), and an increase in overall survival (from 6.6 months to 8 months). Again, the benefit was found to be greater in the subgroup of patients with better prognosis at the beginning of the study (life expectancy longer than 6 months), for whom the mean survival was 15.4 months, compared with 9.4 months in the control (placebo) group.
A randomized, prospective, double-blind clinical lung cancer study was carried out in 2004.(31) In that study, 84 patients with small cell lung cancer were treated with an 18-week course of the LMWH dalteparin in combination with the chemotherapy regimen. Mean survival increased by 5 months, mean survival being 13 months in the LMWH+chemotherapy group, compared with 8 months in the chemotherapy-only group. In that study, as well as in others using heparins, the risk of severe bleeding was not significantly higher.(31,35,36)
Perspectives
Heparins form a heterogeneous group of polysaccharide molecules of various sizes. Their ability to block some points in hypercoagulability, as well as other mechanisms in tumor progression, such as growth factors or angiogenesis, seem to be due to some specific molecules in their composition. In a study,(37) it has been demonstrated that molecules with 8 or 10 saccharide units (octasaccharides and decasaccharides, respectively), both of which induced by growth factors (fibroblast growth factor 2), impede angiogenesis in an animal model, as well as in more complex, in vitro models involving lung and endometrial cancer cells. These molecules do not present the same anticoagulant or thrombocytopenic potential as do heparin fractions with higher molecular weights, which makes them promising targets for future clinical trials.
Genetic therapy can provide great improvement in the control of some tumor cell mechanisms that cause hypercoagulability. One group of authors showed that the ex vivo treatment of lung cancer cells with adenovirus, which impedes the expression of uPA receptors, decreased tumor invasion and metastasis potential in this group of cells.(38) The same authors more recently reported better results after using a genetically modified adenovirus that infects and impedes the genetic expression of tumor cells in order to block two points in that system: the uPA receptor and the MMP-9 receptor. In another animal model of lung cancer, that same strategy also reduced in vitro tumor invasion, cell motility and tumor-dependent angiogenesis, as well as inhibiting tumor growth and in vivo metastasis potential; no metastases were detected in the animals receiving the virus.(15) In another study employing similar methodology, MMP-2 inhibition also showed the therapeutic potential of this specific blockade in slowing the progression of lung cancer.(39)
The benefits from the blockade of this aspect of coagulation and tumor progression have spawned various studies, although none of those have yet produced any significant results. Some of the metalloproteinase inhibitors that have been studied in non-small cell lung cancer are batimastat (BB-94), marimastat (BB-2516), prinomastat (AG-3340) and BMS-275291.(40-42) The results have been discouraging, probably because some metalloproteinases can appear in a defense mechanism against tumor genesis.(15,41,43)
In conclusion, other points, such as tissue factor (chemical inhibitors, monoclonal antibodies), factor Xa, thrombin (hirudin, ximelagatran), some platelet membrane receptors (anti-glycoprotein IIb-IIIa), inhibitors of the tissue factor pathway, etc., can be blocked in a more specific fashion.(5) New knowledge will allow the use of more focused treatments so that we can obtain more concrete, solid answers.
Conclusion
The relationship between our tissue repair (coagulation) system and malignant cells is complex. In addition to various external factors (therapies, immobilization, catheters, etc.), tumor cells activate that system, either directly (tissue factor or cancer procoagulant) or indirectly (cytokines, inflammation, increased cell adhesion, cell activation and cell interaction). New data also implicate other important factors, such as variables in the tumor microenvironment (extracellular matrix, metalloproteinases, proteases, growth factor, vascular neoformation) and modulation of the fibrinolytic system. This relationship goes beyond thromboembolic events, showing a notable involvement with cancer progression. To a certain extent, cancer can be seen as a solid phase of this coagulopathy,(5) which causes a physiological response; however, there is neither a decrease in that stimulus nor a correction of the initial damage/lesion.
In this context, anticoagulant medications promise to play a role by minimizing the tumor-promoting effects of this relationship, not only by inhibiting direct products, such as thrombin and fibrin, but also by inhibiting tissue penetration, angiogenesis and local stimulation of trophic factors. Until then, the effects and safety profiles of heparins, LMWHs in particular, will apparently continue to improve.
Recent studies have revealed promising benefits of LMWHs in the evolution of solid tumors,(35,36) especially in small cell lung cancer.(31) For this type of cancer, warfarin has been used successfully since the 1980s.(32-34) Since there have been only a few reliable studies on the use of LMWH, none of which have involved large patient samples, the indiscriminate prescription of this medication is not presently viable.
Experimental studies have focused on the tumor microenvironment, metalloproteinases and modulation of the fibrinolytic system. Based on new pathophysiological discoveries, interventions in these areas can produce benefits, impeding tumor progression and distant metastases.
Although metastasis depends more on these complicated relationships than on tumor development per se, most deaths from cancer are due to metastatic complications rather than to the primary tumor. This encourages further research in this area, which might lead to the development of medications with less toxic potential than that of the current chemotherapeutic agents. Genetic therapy can promote specific treatment, with the objective of impeding various mechanisms of coagulation activation by malignant cells.
References
- 1. Boing AF, Rossi TF. Temporal trend in and spatial distribution of lung cancer mortality in Brazil between 1979 and 2004: magnitude, regional patterns, and gender-related differences. J Bras Pneumol. 2007;33(5):544-51.
- 2. Malta DC, de Moura L, de Souza Mde F, Curado MP, Alencar AP, Alencar GP. Lung cancer, cancer of the trachea, and bronchial cancer: mortality trends in Brazil, 1980-2003. J Bras Pneumol. 2007;33(5):536-43.
- 3. Trousseau A. Phlegmasia Alba dolens. In: Trousseau, A, editor. Clinique médicale de l'Hôtel-Dieu de Paris. 2nd ed. Paris: Baillière; 1865. p. 654-712.
- 4. Prandoni P, Lensing AW, Piccioli A, Bernardi E, Simioni P, Girolami B, et al. Recurrent venous thromboembolism and bleeding complications during anticoagulant treatment in patients with cancer and venous thrombosis. Blood. 2002;100(10):3484-8.
- 5. Zacharski LR. Anticoagulants in cancer treatment: malignancy as a solid phase coagulopathy. Cancer Lett. 2002;186(1):1-9.
- 6. De Cicco M. The prothrombotic state in cancer: pathogenic mechanisms. Crit Rev Oncol Hematol. 2004;50(3):187-96.
- 7. Prandoni P, Falanga A, Piccioli A. Cancer and venous thromboembolism. Lancet Oncol. 2005;6(6):401-10.
- 8. Gale AJ, Gordon SG. Update on tumor cell procoagulant factors. Acta Haematol. 2001;106(1-2):25-32.
- 9. Kwaan HC, Keer HN. Fibrinolysis and cancer. Semin Thromb Hemost. 1990;16(3):230-5.
- 10. Sun NC, McAfee WM, Hum GJ, Weiner JM. Hemostatic abnormalities in malignancy, a prospective study of one hundred eight patients. Part I. Coagulation studies. Am J Clin Pathol. 1979;71(1):10-6.
- 11. Gasic GJ, Gasic TB, Galanti N, Johnson T, Murphy S. Platelet-tumor-cell interactions in mice. The role of platelets in the spread of malignant disease. Int J Cancer. 1973;11(3):704-18.
- 12. Varon D, Brill A. Platelets cross-talk with tumor cells. Haemostasis. 2001;31(Suppl 1):64-6.
- 13. Shoji M, Hancock WW, Abe K, Micko C, Casper KA, Baine RM, et al. Activation of coagulation and angiogenesis in cancer: immunohistochemical localization in situ of clotting proteins and vascular endothelial growth factor in human cancer. Am J Pathol. 1998;152(2):399-411.
- 14. Offersen BV, Pfeiffer P, Andreasen P, Overgaard J. Urokinase plasminogen activator and plasminogen activator inhibitor type-1 in nonsmall-cell lung cancer: relation to prognosis and angiogenesis. Lung Cancer. 2007;56(1):43-50.
- 15. Rao JS, Gondi C, Chetty C, Chittivelu S, Joseph PA, Lakka SS. Inhibition of invasion, angiogenesis, tumor growth, and metastasis by adenovirus-mediated transfer of antisense uPAR and MMP-9 in non-small cell lung cancer cells. Mol Cancer Ther. 2005;4(9):1399-408.
- 16. Bredin CG, Liu Z, Hauzenberger D, Klominek J. Growth-factor-dependent migration of human lung-cancer cells. Int J Cancer. 1999;82(3):338-45.
- 17. Kodate M, Kasai T, Hashimoto H, Yasumoto K, Iwata Y, Manabe H. Expression of matrix metalloproteinase (gelatinase) in T1 adenocarcinoma of the lung. Pathol Int. 1997;47(7):461-9.
- 18. Zacharski LR, Wojtukiewicz MZ, Costantini V, Ornstein DL, Memoli VA. Pathways of coagulation/fibrinolysis activation in malignancy. Semin Thromb Hemost. 1992;18(1):104-16.
- 19. Shen VS, Pollak EW. Fatal pulmonary embolism in cancer patients: is heparin prophylaxis justified? South Med J. 1980;73(7):841-3.
- 20. Kuzel T, Esparaz B, Green D, Kies M. Thrombogenicity of intravenous 5-fluorouracil alone or in combination with cisplatin. Cancer. 1990;65(4):885-9.
- 21. Otten HM, Mathijssen J, ten Cate H, Soesan M, Inghels M, Richel DJ, et al. Symptomatic venous thromboembolism in cancer patients treated with chemotherapy: an underestimated phenomenon. Arch Intern Med. 2004;164(2):190-4.
- 22. Bern MM, Bothe A Jr, Bistrian B, Champagne CD, Keane MS, Blackburn GL. Prophylaxis against central vein thrombosis with low-dose warfarin. Surgery. 1986;99(2):216-21.
- 23. Bergqvist D, Agnelli G, Cohen AT, Eldor A, Nilsson PE, Le Moigne-Amrani A, et al. Duration of prophylaxis against venous thromboembolism with enoxaparin after surgery for cancer. N Engl J Med. 2002;346(13):975-80.
- 24. Bern MM, Lokich JJ, Wallach SR, Bothe A Jr, Benotti PN, Arkin CF, et al. Very low doses of warfarin can prevent thrombosis in central venous catheters. A randomized prospective trial. Ann Intern Med. 1990;112(6):423-8.
- 25. Dolovich LR, Ginsberg JS, Douketis JD, Holbrook AM, Cheah G. A meta-analysis comparing low-molecular-weight heparins with unfractionated heparin in the treatment of venous thromboembolism: examining some unanswered questions regarding location of treatment, product type, and dosing frequency. Arch Intern Med. 2000;160(2):181-8.
- 26. Jayson GC, Gallagher JT. Heparin oligosaccharides: inhibitors of the biological activity of bFGF on Caco-2 cells. Br J Cancer. 1997;75(1):9-16.
- 27. Niers TM, Klerk CP, DiNisio M, Van Noorden CJ, Büller HR, Reitsma PH, et al. Mechanisms of heparin induced anti-cancer activity in experimental cancer models. Crit Rev Oncol Hematol. 2007;61(3):195-207.
- 28. Dunbar SD, Ornstein DL, Zacharski LR. Cancer treatment with inhibitors of urokinase-type plasminogen activator and plasmin. Expert Opin Investig Drugs. 2000;9(9):2085-92.
- 29. Lentschener C, Li H, Franco D, Mercier FJ, Lu H, Soria J, et al. Intraoperatively-administered aprotinin and survival after elective liver resection for colorectal cancer metastasis A preliminary study. Fibrinol Proteol. 1999;13(1):39-45.
- 30. Zacharski LR, Henderson WG, Rickles FR, Forman WB, Cornell CJ Jr, Forcier RJ, et al. Effect of warfarin anticoagulation on survival in carcinoma of the lung, colon, head and neck, and prostate. Final report of VA Cooperative Study #75. Cancer. 1984;53(10):2046-52.
- 31. Altinbas M, Coskun HS, Er O, Ozkan M, Eser B, Unal A, et al. A randomized clinical trial of combination chemotherapy with and without low-molecular-weight heparin in small cell lung cancer. J Thromb Haemost. 2004;2(8):1266-71.
- 32. Zacharski LR, Henderson WG, Rickles FR, Forman WB, Cornell CJ Jr, Forcier RJ, et al. Effect of warfarin on survival in small cell carcinoma of the lung. Veterans Administration Study No. 75. JAMA. 1981;245(8):831-5.
- 33. Chahinian AP, Propert KJ, Ware JH, Zimmer B, Perry MC, Hirsh V, et al. A randomized trial of anticoagulation with warfarin and of alternating chemotherapy in extensive small-cell lung cancer by the Cancer and Leukemia Group B. J Clin Oncol. 1989;7(8):993-1002.
- 34. Ak EA, Kamath G, Kim SY, Yosuico V, Barba M, Terrenato I, et al. Oral anticoagulation may prolong survival of a subgroup of patients with cancer: a Cochrane systematic review. J Exp Clin Cancer Res. 2007;26(2):175-84.
- 35. Kakkar AK, Levine MN, Kadziola Z, Lemoine NR, Low V, Patel HK, et al. Low molecular weight heparin, therapy with dalteparin, and survival in advanced cancer: the fragmin advanced malignancy outcome study (FAMOUS). J Clin Oncol. 2004;22(10):1944-8.
- 36. Klerk CP, Smorenburg SM, Otten HM, Lensing AW, Prins MH, Piovella F, et al. The effect of low molecular weight heparin on survival in patients with advanced malignancy. J Clin Oncol. 2005;23(10):2130-5.
- 37. Hasan J, Shnyder SD, Clamp AR, McGown AT, Bicknell R, Presta M, et al. Heparin octasaccharides inhibit angiogenesis in vivo Clin Cancer Res. 2005;11(22):8172-9.
- 38. Lakka SS, Rajagopal R, Rajan MK, Mohan PM, Adachi Y, Dinh DH, et al. Adenovirus-mediated antisense urokinase-type plasminogen activator receptor gene transfer reduces tumor cell invasion and metastasis in non-small cell lung cancer cell lines. Clin Cancer Res. 2001;7(4):1087-93.
- 39. Chetty C, Bhoopathi P, Joseph P, Chittivelu S, Rao JS, Lakka S. Adenovirus-mediated small interfering RNA against matrix metalloproteinase-2 suppresses tumor growth and lung metastasis in mice. Mol Cancer Ther. 2006;5(9):2289-99.
- 40. Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295(5564):2387-92.
- 41. Leslie WT, Bonomi PD. Novel treatments in non-small cell lung cancer. Hematol Oncol Clin North Am. 2004;18(1):245-67.
- 42. Bonomi P. Matrix metalloproteinases and matrix metalloproteinase inhibitors in lung cancer. Semin Oncol. 2002;29(1 Suppl 4):78-86.
- 43. McCawley LJ, Crawford HC, King LE Jr, Mudgett J, Matrisian LM. A protective role for matrix metalloproteinase-3 in squamous cell carcinoma. Cancer Res. 2004;64(19): 6965-72.
Publication Dates
-
Publication in this collection
02 June 2008 -
Date of issue
May 2008
History
-
Accepted
07 Jan 2008 -
Received
29 Nov 2007