Abstract
Approximately 1% of all men in the general population suffer from azoospermia, and azoospermic men constitute approximately 10 to 15% of all infertile men. Thus, this group of patients represents a significant population in the field of male infertility. A thorough medical history, physical examination and hormonal profile are essential in the evaluation of azoospermic males. Imaging studies, a genetic workup and a testicular biopsy (with cryopreservation) may augment the workup and evaluation. Men with nonobstructive azoospermia should be offered genetic counseling before their spermatozoa are used for assisted reproductive techniques. This article provides a contemporary review of the evaluation of the azoospermic male.
Azoospermia; Male Infertility; Testis Biopsy; Testicular Biopsy
REVIEW
Update in the evaluation of the azoospermic male
Ahmet Gudeloglu; Sijo J. Parekattil
University of Florida, Winter Haven Hospital, Department of Urology, Winter Haven, FL, United States
ABSTRACT
Approximately 1% of all men in the general population suffer from azoospermia, and azoospermic men constitute approximately 10 to 15% of all infertile men. Thus, this group of patients represents a significant population in the field of male infertility. A thorough medical history, physical examination and hormonal profile are essential in the evaluation of azoospermic males. Imaging studies, a genetic workup and a testicular biopsy (with cryopreservation) may augment the workup and evaluation. Men with nonobstructive azoospermia should be offered genetic counseling before their spermatozoa are used for assisted reproductive techniques. This article provides a contemporary review of the evaluation of the azoospermic male.
Keywords: Azoospermia; Male Infertility; Testis Biopsy; Testicular Biopsy.
INTRODUCTION
Azoospermia is defined as the absence of sperm in at least two different ejaculate samples (including the centrifuged sediment) (1,2). In the general population, 10 to 15% of couples suffer from infertility issues (3,4). Approximately 50% of these cases can be attributable to a male issue. Of these infertile men, 10 to 20% (or 1% of all men in the general population) suffer from azoospermia (3). A detailed history, a physical examination, a hormone profile, imaging and genetic counseling are important to determine the specific clinical classification of the azoospermia. This distinction is important given that, for example, obstructive azoospermia (OA) and nonobstructive azoospermia (NOA) require different treatment approaches. In this chapter, we will provide a contemporary review of the evaluation process for males with azoospermia.
EVALUATION OF THE AZOOSPERMIC MALE
Definition and classification of azoospermia
Azoospermia is defined as the absence of spermatozoa in the semen. If no spermatozoa are observed in the wet preparation, the World Health Organization (WHO) recommends an examination of the centrifuged sample (3000 X g or greater for 15 minutes). If no sperm are observed in the centrifuged sample, the semen analysis should be repeated. The presence of a small number of spermatozoa in either of the centrifuged samples is defined as cryptozoospermia, and the complete absence of spermatozoa is defined as azoospermia (1).
The etiologic classification of azoospermia is divided into three primary categories: pretesticular, testicular, and post-testicular. Although the pretesticular and post-testicular causes of azoospermia are generally curable, the testicular causes of azoospermia are generally not.
Pretesticular azoospermia can be caused by endocrine abnormalities that are characterized by low levels of sex steroids and abnormal gonadotropin levels. These abnormalities can be congenital (e.g., Kallmann syndrome), acquired (e.g., hypothalamic or pituitary disorders) or secondary (e.g., an adverse effect from a medication).
Testicular causes include congenital, acquired or idio-pathic disorders that lead to spermatogenic failure. Congenital testicular causes consist of anorchia, testicular dysgenesis (cryptorchidism), genetic abnormalities (Y chromosome deletions), germ cell aplasia (Sertoli cell-only syndrome) and spermatogenic arrest (maturation arrest). Acquired testicular causes include trauma, torsion, infection (mumps orchitis), testicular tumors, medications, irradiation, surgery (compromising vascularization of testis), systemic diseases (cirrhosis, renal failure) and varicocele (5).
Post-testicular causes include ejaculatory disorders or obstructions, which impair the transport of spermatozoa from the testis. These obstructions can also be congenital, caused by a bilateral absence of the vas deferens (CBAVD), or acquired because of infection or surgery (vasectomy or an iatrogenic injury). Obstructive azoospermia (OA) is also classified according to the localization of the obstruction: epididymal (postinfection), vasal (vasectomy, CBAVD) or ductal (Mullerian cysts) (5).
Azoospermia may also be clinically classified as obstructive (post-testicular) and nonobstructive (pretesticular or testicular). Obstructive azoospermia (OA) is less common than nonobstructive azoospermia (NOA) and occurs in 15 to 20% of men with azoospermia (5). Although NOA indicates impaired sperm production of the entire testis by definition, it has been observed that focal normal spermatogenesis can be observed in 50 to 60% of men with NOA (6,7).
Semen analysis of the azoospermic male
In 2010, the WHO updated the lower reference limits for semen characteristics, which are provided in Table 1 (1). In the absence of vasal agenesis or testicular atrophy, semen volume and serum FSH are key factors in determining the cause of azoospermia. Azoospermic men with a normal ejaculate volume may have either epididymal/vasal obstruction or an abnormality of spermatogenesis. In such cases, a hormonal evaluation would aid in the differential diagnosis. Azoospermia in men with a low semen volume and normal-sized testes can be caused by ejaculatory dysfunction; however, the most likely cause is ejaculatory duct obstruction (EDO) (8). Semen volume, pH and fructose levels are generally within normal ranges in NOA; however, these parameters can be lower in OA, depending on the location of the obstruction. Although there are no pathog-nomonic findings for the ejaculate evaluation of patients with EDO, the absence of spermatozoa in a centrifuged semen sample accompanied by a low ejaculate volume (<2.0 ml), a pH below 7.2, and the absence of fructose in the seminal fluid suggest EDO (9). The classic clinical presentation of EDO includes postejaculatory pain, hematospermia and infertility. This triad of clinical presentation warrants a transrectal ultrasound examination. The presence of dilated seminal vesicles and/or dilated ejaculatory ducts upon transrectal ultrasound examination, combined with a normal hormonal profile, support a diagnosis of EDO.
A congenital bilateral absence of the vas deferens (CBAVD) is responsible for 2 to 6% of cases of OA. CBAVD classically presents as a nonpalpable vas segment, low semen volume and low pH and fructose levels caused by an obstructed epididymis and the atrophia or congenital absence of the seminal vesicles (10,11). Nevertheless, ejaculatory disorders and retrograde ejaculation should be eliminated as a diagnosis for azoospermic patients with a low ejaculate volume. A postejaculation urine evaluation can identify patients with retrograde ejaculation.
Initial evaluation of the azoospermic male
A complete medical history, physical examination and hormonal investigation are the principal components of the initial evaluation of males with azoospermia (8).
Medical history
It is important to obtain a complete infertility medical history for both partners. The azoospermic male's history should include such conditions as trauma, torsion, cryp-torchidism; history of pelvic, inguinal or scrotal surgeries; any potentially compromising testicular vascularization; vasal patency; and ejaculatory function. Additionally, a late onset of puberty can indicate hypogonadotropic hypogo-nadism. Any prior fertility history can be beneficial in distinguishing between OA and NOA. If there is a history of vasectomy, the duration since the procedure and any reversal attempts may be helpful in developing a treatment plan. Genitourinary infections, such as urethritis and epididymitis can cause OA, while late pubertal mumps orchitis can cause NOA.
It is important to inquire about systemic diseases, given that diabetes mellitus, cirrhosis or chronic renal insufficiency can affect sperm production or transport. Previous malignancies, especially if treated with cytotoxic chemotherapy or radiotherapy, should be identified. Medications that include gonadotoxic agents, such as cimetidine, nitrofur-antoin and calcium channel blockers, should be documented.
Physical examination
The physical examination should begin with the inspection of the body habitus, given that hair distribution, gynecomastia and a eunuchoid stature can indicate a testosterone deficiency or hormonal imbalance. Possible disorders include low serum testosterone level, hyperpro-lactinemia, abnormalities in the estrogen-to-testosterone ratio, adrenal dysfunction, and genetic syndromes that are associated with subvirilization, such as Klinefelter's syndrome. Additionally, disproportionately long extremities (caused by testosterone deficiency at the time of puberty, leading to delayed closure of the epiphyseal plates) should be noted.
The genital examination should also include an inspection of the external genitalia. Penile curvature, hypospadias and surgical scars should be noted. Penile curvature and hypospadias may cause improper sperm placement into the vagina. Surgical scars may indicate possible injuries to the testicular blood supply and/or vas deferens.
The scrotal contents should be assessed via manual palpation to determine the testis size and consistency and the presence of testicular mass or asymmetry. An orchid-ometer, calipers or sonographic measurement techniques may be utilized to measure testicle size. The normal adult testicle should be 4x3 cm or approximately 20 ml (12). Although smaller and softer testes can signal inadequate sperm production, men with Klinefelter's syndrome have small (approximately 5 ml), firm testes that are devoid of germ cells (13). Epididymal enlargement, induration and cysts should be assessed and can be considered a cause of OA. The presence of the vas deferens and varicose veins (Figure 1) should be ascertained while palpating the spermatic cord. The Valsalva maneuver may aid in the identification of low-grade varicocele. Examiners should bear in mind that vasal agenesis may be accompanied by genetic or renal abnormalities. The physical examination findings for patients with CBAVD should be normal-sized and consistent testis; full and firm caput epididymis caused by obstructed efferent ducts (with the presence or absence of the distal two thirds of the epididymis); nonpalpable vas deferens; atrophic, dysfunctional or congenitally absent seminal vesicle; and normal male external genitalia (14).
A complete physical examination of males with azoos-permia must include a rectal examination to exclude ejaculatory duct obstruction. Midline prostatic cysts, such as Mullerian duct cysts and dilated seminal vesicles, can be easily palpated, and prostatic induration or tenderness supports a diagnosis of prostatitis.
Hormonal analysis and hypogonadism
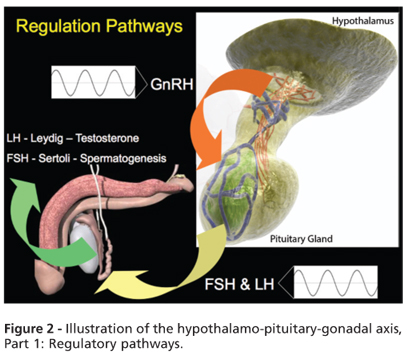
Sperm production is controlled by the hypothalamo-pituitary-gonadal axis (Figures 2 and 3). Gonadotropin-releasing hormone is secreted by the hypothalamus and stimulates the anterior pituitary gland to release luteinizing hormone (LH) and follicle stimulating hormone (FSH). LH and FSH stimulate Leydig cells and the germinal epithelium to produce testosterone and sperm, respectively. Testosterone is required for the completion of the meiotic division and spermatid development and thus plays an important role in the initiation and maintenance of spermatogenesis. FSH also stimulates Sertoli cells, which produce inhibin B. LH and FSH are under negative feedback control by testosterone and inhibin B, respectively (Figure 3) (15).
Normal levels of LH and FSH are expected in OA; however, LH and FSH can be low or elevated in NOA. Hypogonadism is defined by impaired testicular function, which potentially affects spermatogenesis and/or testosterone synthesis. Sussman et al. reported that the respective incidences of hypogonadism in males who visited an infertility clinic were 35.3%, 45% and 16.7% for those with normal sperm analysis, NOA and OA (15). Hypogonadism can be caused by primary testicular failure (hypergonado-tropic hypogonadism) or secondary testicular failure resulting from a hypothalmo-pituitary deficiency (hypogo-nadotropic hypogonadism). Rarely, hypogonadism can occur in complete (testicular feminization) or partial (Reifenstein's syndrome) androgen insensitivity syndrome. The end organs that are affected by hypogonadism are the external genitalia, the muscles, the bone, the skin and the brain. The symptoms include infertility, muscular hypotro-phy, low bone mineral density, anemia, decreased acne, alterations in body hair distributions and decreased libido (5,15).
In primary hypogonadism, normal or elevated LH and FSH levels accompany low serum testosterone levels. An FSH level greater than twice the normal upper limit is accepted as a clear elevation of the serum FSH level, and it is a reliable indicator of abnormal spermatogenesis (8). However, elevated FSH in men with azoospermia does not eliminate the possibility of obstruction and the capacity for fertility (16). Furthermore, a stronger correlation has been demonstrated between inhibin B and spermatogenesis
compared with FSH and spermatogenesis (17). Primary hypogonadism can be observed in patients who exhibit testicular failure caused by congenital (anorchia, undes-cended testes and genetic abnormalities, such as Klinefelter's syndrome or Y chromosome defects), acquired (trauma, tumor, torsion, orchitis or varicocele) or idiopathic causes. While evaluating testosterone levels, sex hormone-binding globulin should be considered, and an accurate calculation of free and bioavailable testosterone should be performed. Because of diurnal variation, blood samples used to measure testosterone should be taken prior to 10 o'clock in the morning. The Food and Drug Administration (FDA) defines the normal range of total testosterone levels as between 300 and 1000 ng/dL (5,15).
Hypogonadotropic hypogonadism is characterized by low serum FSH levels in association with low serum testosterone levels and, generally, low LH levels. According to a University of Illinois study, nearly half of all men who have NOA suffer from hypogonadotropic hypogonadism. This finding suggests that hypogonadotro-pic hypogonadism may be considerably more prevalent in the infertile male population than was previously believed. Genetic hypothalamic disorders, such as Kallmann syndrome, and congenital or acquired pituitary deficiencies, such as empty sella syndrome or pituitary tumors (functional or nonfunctional), can cause hypogonadotropic hypogonadism. In men who have azoospermia with anosmia, decreased libido, gynecomastia, headaches or visual field deficits, hypogonadotropic hypogonadism should be suspected, and a complete endocrine work-up, including cranial imaging, should be performed.
IMAGING TECHNIQUES
Scrotal ultrasonography (US), transrectal US (TRUS), TRUS-guided seminal vesiculography, seminal tract washout, vasography, endorectal magnetic resonance imaging, abdominal US and cranial imaging are all imaging studies that can be used to evaluate males with azoospermia.
Scrotal US is a first-line, basic imaging tool for all scrotal abnormalities, and it has also been demonstrated that testicular volume as measured by scrotal US is significantly correlated with testicular function. An increased resistive index and pulsatility index of the capsular branches of the testicular arteries on unenhanced color Doppler US examination may indicate impaired testicular microcirculation in patients with clinical varicocele. Doppler US is a promising method for assessing patients who are affected by azoos-permia. This technique allows the differentiation of obstructive azoospermia (normal vessel distribution) from nonobstructive azoospermia (reduced or absent testicular vessels) (18). The benefit of evaluating the intratesticular blood vessel distribution prior to the performance of any method to retrieve intra-testicular spermatozoa for intracy-toplasmic sperm injection has also been demonstrated (19).
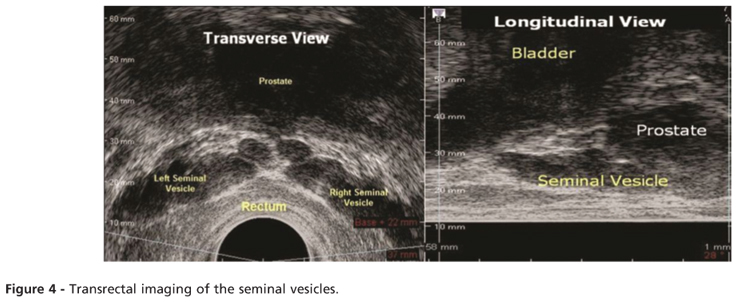
When evaluating the azoospermic male to diagnose EDO, TRUS is used for men with low ejaculate volume, but only rarely for those with normal ejaculates (8). A dilated seminal vesicle (anterior-posterior diameter >15 mm, Figure 4) and anechoic areas in the seminal vesicle are the TRUS abnormalities most frequently associated with EDO (20). TRUS is also able to identify other known and potentially correctable OA causes, such as Mullerian (utricular) or Wolffian (ejaculatory duct) cysts, ejaculatory duct calcifications, congenital unilateral or bilateral absence of the vas deferens, and obstructing seminal vesicle cysts (21).
TRUS-guided seminal vesiculography and seminal vesicle washout are other invasive imaging techniques used to investigate OA (22). In a prospective, comparative study, Purohit et al. observed that TRUS has a poor specificity for EDO evaluation when compared with vesiculography, seminal vesicle aspiration and duct chromotubation (23). Newly developed 3D-guided transrectal imaging devices enable easier visualization and needle guidance for vesicu-lography techniques (Figure 5).
The combination of azoospermia, a testis biopsy revealing complete spermatogenesis and at least one palpable vas deferens strongly warrant a vasography. If necessary, the biopsy should be performed at the same time as the scrotal exploration and the definitive repair of obstruction. Stricture or obstruction at the vasography site, vasal blood supply injury, hematoma and sperm granuloma are potential complications of vasography (24).
Endorectal coil magnetic resonance imaging assesses the relationships between the proximal prostatic urethra and the posterior wall of the ejaculatory ducts, which must be precisely known when endoscopic resection of the ejacula-tory ducts is planned (25).
An azoospermic male with hypogonadotropic hypogo-nadism may merit cranial imaging to identify hypothalamo-pituitary disorders, especially if prolactin levels are elevated.
Abdominal US imaging should be considered if there is a unilateral or bilateral congenital absence of the vas deferens. Because of the embryologic origins of the vas deferens and the kidney, anomalies in these organs tend to coexist. One study has demonstrated that 26% and 11% of men with unilateral or bilateral congenital absence of vas deferens, respectively also exhibit renal agenesis (26).
Testicular biopsy
In the evaluation of azoospermic males who have normal-sized testis and a normal hormone profile, a testicular biopsy is critical for distinguishing between OA and NOA. If feasible, it is best to plan for the cryopreservation of the sperm at the same time as this procedure. In NOA, the prognostic value of the testis biopsy is controversial. Although many groups have not demonstrated a relationship between testicular histology and sperm retrieval rate, certain groups argue that a prior testicular biopsy may help to determine the sperm retrieval success rate during a follow-up procedure, such as a microTESE (testicular sperm extraction) (27,28). It has also been demonstrated that sperm retrieval success rates are generally high in men with hypospermatogenesis, moderate in men with maturation arrest and limited in men with Sertoli cell-only syndrome. However, examiners should bear in mind that a single testicular biopsy is not representative of the entire testicle (29). A normal testicular biopsy implies obstruction at some level in the sperm transport system. Diagnostic testicular biopsies are of limited value in men with small testis size and elevated FSH levels (greater than twice the upper limit), which supports a diagnosis of NOA. Research recommends that the patient undergo a microTESE (combined with cryopreservation) in these cases and subsequent in vitro fertilization with intracytoplasmic sperm injection (ICSI); this procedure would enable several samples to be collected (5,8).
A diagnostic testicular biopsy may be considered for select azoospermic individuals who have risk factor(s) for a testicular germ cell tumor, such as cryptorchidism, atrophic testis and a testicular germ cell history accompanied by testicular microlithiasis on ultrasound imaging (30). Again, if feasible, any sperm that are detected should be cryopre-served at the time of this biopsy.
Genetic investigation
Genetic factors occupy an important place in the evaluation and management of the azoospermic male. Such factors can be pretesticular (e.g., Kallmann syndrome), testicular (e.g., Klinefelter's syndrome or Y chromosome microdele-tions) or post-testicular (CBAVD). Genetic counseling provides couples with information about the nature, inheritance pattern, and implications of genetic disorders to help them make informed medical and personal decisions. The common genetic disorders that are associated with azoospermia are reviewed below.
Kallmann syndrome
Kallmann syndrome is an X chromosome-linked disorder characterized by isolated gonadotropin releasing hormone (GnRH) deficiency accompanied by complete or partial anosmia. Six X chromosome-linked autosomal dominant and recessive genes have been identified; of these, KALI is the gene that is most commonly associated with Kallmann syndrome. This syndrome is essentially a hormonal disorder in which the lack of GnRH secretion leads to testicular insufficiency (i.e., hypogonadotropic hypogonadism) (31).
Kallmann syndrome is diagnosed clinically in the presence of anosmia, micropenis, cryptorchidism, diminished libido, erectile dysfunction and the absence of secondary sex characters. While serum testosterone level is low (<100ng/ml) in patients with Kallmann syndrome, pituitary and hypothalamus imaging studies are normal. Adult males with Kallmann syndrome tend to exhibit prepubertal testicular volume (<4 ml) and have eunuchoid body habitus caused by delayed skeletal maturation (31). One study recently demonstrated that testicular morphology in patients with Kallmann syndrome can vary (32). However, spermatogenesis can be easily induced by hormonal stimulation (5). Depending on the type of gene mutation, nonreproductive phenotypes in men with Kallmann syndrome can include unilateral renal agenesis, dyskinesia and/or skeletal abnormalities, cleft lip/palate, ear/hearing defects, coloboma (eye defect) and hyperlaxity of the joints.
Klinefelter's syndrome
Klinefelter's syndrome has a wide spectrum of clinical presentations. It is a chromosomal disorder in which at least one additional X chromosome is observed in the male karyotype. Although there are several mosaic forms of this syndrome, most cases are of the nonmosaic form, 47, XXY. Klinefelter's syndrome is the most common chromosome aneuploidy in human beings and the most common form of male hypogonadism, with a prevalence of 0.1 to 0.2% in the general population and up to 3.1% in the infertile population. The presence of the extra X chromosome sets in motion several undefined events that lead to spermatogenic and androgenic failure, gynecomastia, and learning difficulties (13,14,33). The extra X chromosome may originate from either the maternal or paternal side.
The clinical presentation of Klinefelter's syndrome varies according to the age at diagnosis and the severity of the mosaicism. It is difficult to differentiate prepubertal boys with Klinefelter's syndrome from normal boys based solely on their phenotype. Small, firm testes and varying symptoms of androgen deficiency characterize Klinefelter's syndrome in adolescence and after puberty (13). On one end of the spectrum are boys who are identified as having Klinefelter's syndrome because they have failed to undergo puberty and virilization as a result of nearly complete androgenic malfunction. These boys exhibit a eunuchoid appearance. On the opposite end of the spectrum are phenotypically normal boys who are diagnosed with Klinefelter's syndrome during an evaluation for azoosper-mia (14).
Although the exact mechanism of androgen deficiency is unknown, most patients with Klinefelter's syndrome exhibit low serum testosterone concentrations and elevated FSH levels. This reflects spermatogenic compromise and the compensatory elevation of LH levels that results because the Leydig cells are being maximally stimulated and have a small reserve capacity (14). The majority of these patients also suffer from decreased libido and erectile dysfunction. Generally, the patients' ejaculate presents with azoosper-mia. A testis biopsy reveals extensive fibrosis, hyalinization of seminiferous tubules and hyperplasia of the interstitium. However, the tubules may exhibit residual foci of sperma-togenesis (34).
Congenital bilateral absence of vas deference
Congenital bilateral absence of vas deference (CBAVD) is observed in 2 to 6% of men with OA and is responsible for infertility in approximately 1% of infertile men (10,35). A strong association between CBAVD and the cystic fibrosis transmembrane conductance regulator (CFTR) gene has been demonstrated (36). This gene is located on the short arm of chromosome 7 and encodes the CFTR protein, which is crucial for maintaining proper sodium/chloride balance in epithelial secretions. This balance is necessary to optimize the viscosity and fluidity of these secretions. Approximately 1,500 different mutations of the CFTR gene have been described. Nearly all male patients with clinically diagnosed cystic fibrosis have CBAVD, and approximately 80% of patients with CBAVD have mutations in at least one CFTR allele. The inability to identify a second mutation is presumed to result from the fact that these mutations are located elsewhere in the noncoding regions of the CFTR gene (5,8,37).
Cystic fibrosis (CF) is characterized by elevated concentrations of electrolytes in the sweat, chronic pulmonary disease resulting from thickened respiratory epithelial secretions and pancreatic exocrine insufficiency secondary to thickened and occlusive ductal secretions. Both maternal and paternal mutant alleles must be present to cause clinical CF. However, the clinical presentation of CF depends on the severity of the mutations in each CFTR allele and/or in the noncoding regions of the genes (e.g., the 5T alleles). Thus, whereas a subset of patients with CFTR mutations suffer from severe pulmonary disease and pancreatic dysfunctions, the bilateral absence of the vas deferens may be the only observable effect in other patients (14,37).
The physical examination of the patients with CBAVD reveals normal-sized and full testes, a full and firm caput epididymis caused by efferent ducts that are distended with sperm, a present or absent distal two-thirds of the epididymis and a bilateral absence of the vas deferens. Semen analysis reveals a low-volume (0.5 ml), acidic ejaculate that is devoid of fructose and seminal vesicle fluid because of atrophic, dysfunctional or absent seminal vesicles. The seminal vesicle anomalies can be confirmed with TRUS imaging (14). Most men with CBAVD exhibit normal spermatogenesis, but it has been observed that a large proportion exhibit impaired spermatogenesis. Prior to sperm harvesting, other potential coexisting causes of impaired spermatogenesis should be investigated (38). A careful abdominal US should be performed because as many as 10% of patients with CBAVD may also exhibit renal agenesis (26).
Y chromosome microdeletions
The relationship between deletions on the Y chromosome and azoospermia was first recognized in 1976. With the elucidation of the molecular anatomy of the Y chromosome, specific microdeletions that are associated with azoospermia or severe oligospermia were discovered in the 1990s (39). Since this time, several case series have been published. A study with a large number of participants demonstrated that the prevalence of microdeletions was approximately 3% in unselected infertile men, 8% in men with NOA and 5.5% in men with severe OA (40). Although rare, these micro-deletions have also been reported to occur in fertile men (41).
Three microdeletions have been mapped to three different regions on the long arm of the Y chromosome. These regions are referred to as azoospermia factors (AZF)a, AZFb and AZFc and are observed proximally, centrally and distally on Yq11, respectively (42). Multiple genes are distributed throughout these regions; most are involved in spermato-genesis but are still poorly characterized. For example, the deleted-in-azoospermia (DAZ) gene is located in the AZFc region. This gene encodes a transcription factor that is generally present in men with normal fertility (43). The most frequently deleted region is AZFc (65-70%), and the least frequently deleted region is AZFa (5%). AZFb, AZFb+c and AZFa+b+c deletions are responsible for approximately 25 to 30% of Y microdeletions. It has been reported that Y microdeletions are observed nearly exclusively in patients with severe oligospermia (<1 million spermatozoa/ml) and are extremely rare in patients with sperm concentrations >5 million spermatozoa/ml (44).
Genetic testing for Y microdeletions may predict the outcome of sperm retrieval techniques. One study has reported that sperm retrieval is possible in approximately 50% of patients with AZFc and partial AZFb deletions. The same study reported that the possibility of detecting mature spermatozoa in patients with complete AZFb deletions is virtually zero (45). In another study, Kamp et al. demonstrated a strong association between AZFa deletions and Sertoli cell-only syndrome (46).
It is also important to know whether AZFc microdeletions are present in patients with oligospermia given the evidence of a progressive decrease in sperm count over time in such men. The cryopreservation of spermatozoa in these cases may avoid invasive sperm retrieval procedures in the future (45).
Hormonal analysis (FSH and inhibin B levels) studies have not been reliable in discriminating between patients with idiopathic and microdeletion-associated oligospermia and azoospermia (44).
The male offspring of men with Y chromosome micro-deletions are likely to inherit the same abnormality and may also be infertile. It is unclear whether Y chromosome microdeletions can cause additional health risks or affect the results of assisted reproductive techniques.
Genetic counseling may be offered whenever a genetic abnormality is suspected in either the male or the female partner. Men with NOA should receive genetic counseling and should be offered karyotyping and Y chromosome microdeletion analysis before their sperm is used in assisted reproductive techniques (8).
EXPERT COMMENTARY
Azoospermic men constitute a significant portion of the infertile male population. A detailed and complete medical history, physical examination and hormonal profile are essential in the evaluation of the azoospermic male. Imaging studies, testicular biopsies (with cryopreservation) and genetic testing are also important. Men with NOA should be offered genetic counseling before their sperm is used in assisted reproductive techniques. The accurate classification and evaluation of the azoospermic male is important when determining the therapeutic approach that will be chosen for such patients.
KEY ISSUES
a) Azoospermia is defined as the absence of sperm in at least two different ejaculate samples (including the centri-fuged sediment) (1,2). Ten to fifteen percent of couples in the general population suffer from infertility issues (3,4). Approximately 50% of these cases can be attributable to a male issue. Ten to twenty percent of these men (or 1% of men in the general population) suffer from azoospermia-induced infertility (3).
b) A complete medical history, physical examination and hormonal investigation are the principal components of the initial evaluation of the azoospermic male (8).
c) Sperm production is controlled by the hypothalamo-pituitary-gonadal axis (Figures 2 and 3).
d) Scrotal ultrasonography (US), transrectal US (TRUS), TRUS-guided seminal vesiculography, seminal tract washout, vasography, endorectal magnetic resonance imaging, abdominal US and cranial imaging studies can be performed when evaluating the azoospermic male.
e) In the evaluation of azoospermic patients who have normal-sized testes and a normal hormone profile, testicular biopsy has a critical role in distinguishing between OA and NOA. It is best to plan for the cryopreservation of sperm at the time of the biopsy, if feasible.
f) Diagnostic testicular biopsies are of limited value in men with small testes and elevated FSH levels (greater than twice the upper limit), which support a diagnosis of NOA. It is recommended that such patients undergo a microTESE (combined with cryopreservation) and subsequent in vitro fertilization via intracytoplasmic sperm injection (ICSI); this procedure would enable several samples to be taken (5,8).
g) Genetic factors are important in the evaluation and management of azoospermic males. These factors can be pretesticular (Kallmann syndrome), testicular (Klinefelter's syndrome or Y chromosome microdeletions) or post-testicular (CBAVD). Genetic counseling provides couples with information about the nature, inheritance patterns, and implications of genetic disorders to help them make informed medical and personal decisions.
ACKNOWLEDGMENTS
We would like to thank Dr. Ashok Agarwal, Dr. Johannes Vieweg and Tom Crawford for their continued support of our program.
AUTHOR CONTRIBUTIONS
Gudeloglu A performed the research review, collected the data and wrote the preliminary draft of the manuscript. Parekattil S performed an additional research review and edited and revised the manuscript.
Received for publication on June 25, 2012
Accepted for publication on July 10, 2012
No potential conflict of interest was reported.
E-mail: sijo.parekattil@winterhavenhospital.org
Tel.: 1-863-292-4652
- 1. World Health Organization. WHO laboratory manual for the examination and processing of human semen. 5th ed. Geneva: World Health Organization; 2010.
- 2. Corea M, Campagnone J, Sigman M. The diagnosis of azoospermia depends on the force of centrifugation. Fertil Steril. 2005;83(4):920-2, http://dx.doi.org/10.1016/jiertnstert.2004.09.028
- 3. Jarow JP, Espeland MA, Lipshultz LI. Evaluation of the azoospermic patient. J Urol. 1989;142(1):62-5.
- 4. Stephen EH, Chandra A. Declining estimates of infertility in the United States: 1982-2002. Fertil Steril. 2006;86(3):516-23, http://dx.doi.org/10.1016/j.fertnstert.2006.02.129
- 5. Jungwirth A, Diemer T, Dohle GR, Giwercman A, Kopa Z, Krausz C, et al. European Association of Urology Guidelines on Male Infertility. 2012.
- 6. Colpi GM, Piediferro G, Nerva F, Giacchetta D, Colpi EM, Piatti E. Sperm retrieval for intra-cytoplasmic sperm injection in non-obstructive azoospermia. Minerva Urol Nefrol. 2005;57(2):99-107.
- 7. Esteves SC, Miyaoka R, Agarwal A. Sperm retrieval techniques for assisted reproduction. Int Braz J Urol. 2011;37(5):570-83.
- 8. Evaluation of the azoospermic male. Fertil Steril. 2008;90(5 Suppl):S74-7.
- 9. Smith JF, Walsh TJ, Turek PJ. Ejaculatory duct obstruction. Urol Clin North Am. 2000;35(2):221, viii-7.
- 10. Donat R, McNeill AS, Fitzpatrick DR, Hargreave TB. The incidence of cystic fibrosis gene mutations in patients with congenital bilateral absence of the vas deferens in Scotland. Br J Urol. 1997;79(1):74-7.
- 11. Grangeia A, Niel F, Carvalho F, Fernandes S, Ardalan A, Girodon E, et al. Characterization of cystic fibrosis conductance transmembrane regulator gene mutations and IVS8 poly(T) variants in Portuguese patients with congenital absence of the vas deferens. Hum Reprod. 2004;19(11):2502-8, http://dx.doi.org/10.1093/humrep/deh462
- 12. Sabanegh E, Agarwal A. Male Infertility. In: Kavoussi LR, Novick AC, Partin AW, Peters CA, editors. Campbell-Walsh Urology. Tenth ed. United States of America: ELSEVIER SAUNDERS; 2012. p. 616-47.
- 13. Lanfranco F, Kamischke A, Zitzmann M, Nieschlag E. Klinefelter's syndrome. Lancet. 2004;364(9430):273-83, http://dx.doi.org/10.1016/S0140-6736(04)16678-6
- 14. Oates RD. The genetic basis of male reproductive failure. Urol Clin North Am. 2008;35(2):257, ix-70.
- 15. Sussman EM, Chudnovsky A, Niederberger CS. Hormonal evaluation of the infertile male: has it evolved? Urol Clin North Am. 2008;35(2):147, vii-55.
- 16. Hauser R, Temple-Smith PD, Southwick GJ, de Kretser D. Fertility in cases of hypergonadotropic azoospermia. Fertil Steril. 1995;63(3):631-6.
- 17. Pierik FH, Vreeburg JT, Stijnen T, De Jong FH, Weber RF. Serum inhibin B as a marker of spermatogenesis. J Clin Endocrinol Metab. 1998;83(9):3110-4, http://dx.doi.org/10.1210/jc.83.9.3110
- 18. Schurich M, Aigner F, Frauscher F, Pallwein L. The role of ultrasound in assessment of male fertility. Eur J Obstet Gynecol Reprod Biol. 2009;144 Suppl 1:S192-8, http://dx.doi.org/10.1016/j.ejogrb.2009.02.034
- 19. Foresta C, Garolla A, Bettella A, Ferlin A, Rossato M, Candiani F. Doppler ultrasound of the testis in azoospermic subjects as a parameter of testicular function. Hum Reprod. 1998;13(11):3090-3, http://dx.doi.org/10.1093/humrep/13.11.3090
- 20. Colpi GM, Negri L, Nappi RE, Chinea B. Is transrectal ultrasonography a reliable diagnostic approach in ejaculatory duct sub-obstruction? Hum Reprod. 1997;12(10):2186-91, http://dx.doi.org/10.1093/humrep/12.10. 2186
- 21. Kuligowska E, Fenlon HM. Transrectal US in male infertility: spectrum of findings and role in patient care. Radiology. 1998;207(1):173-81.
- 22. Donkol RH. Imaging in male-factor obstructive infertility. World J Radiol. 2010;2(5):172-9.
- 23. Purohit RS, Wu DS, Shinohara K, Turek PJ. A prospective comparison of 3 diagnostic methods to evaluate ejaculatory duct obstruction. J Urol. 2004;171(1):232-5; discussion 5-6.
- 24. Goldstein M. Vasography. In: Goldstein M, editor. Surgery of Male Infertility. 1st ed., United States of America: SAUNDERS; 1995. p. 26-31.
- 25. Cornud F, Belin X, Delafontaine D, Amar T, Helenon O, Moreau JF. Imaging of obstructive azoospermia. Eur Radiol. 1997;7(7):1079-85, http://dx.doi.org/10.1007/s003300050258
- 26. Schlegel PN, Shin D, Goldstein M. Urogenital anomalies in men with congenital absence of the vas deferens. J Urol. 1996;155(5):1644-8.
- 27. Hauser R, Yogev L, Paz G, Yavetz H, Azem F, Lessing JB, et al. Comparison of efficacy of two techniques for testicular sperm retrieval in nonobstructive azoospermia: multifocal testicular sperm extraction versus multifocal testicular sperm aspiration. J Androl. 2006;27(1):28-33.
- 28. Ramasamy R, Schlegel PN. Microdissection testicular sperm extraction: effect of prior biopsy on success of sperm retrieval. J Urol. 2007;177(4):1447-9.
- 29. Dohle GR, Elzanaty S, van Casteren NJ. Testicular biopsy: clinical practice and interpretation. Asian J Androl. 2012;14(1):88-93.
- 30. van Casteren NJ, Looijenga LH, Dohle GR. Testicular microlithiasis and carcinoma in situ overview and proposed clinical guideline. Int J Androl. 2009;32(4):279-87.
- 31. Pallais JC, Au M, Pitteloud N, Seminara S, Crowley WF. Kallmann Syndrome. In: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, editors. GeneReviews. Seattle (WA). 1993.
- 32. Nishio H, Mizuno K, Moritoki Y, Kamisawa H, Kojima Y, Mizuno H, et al. Clinical features and testicular morphology in patients with Kallmann syndrome. Urology. 2012;79(3):684-6, http://dx.doi.org/10.1016/j.urology.2011.10.032
- 33. Visootsak J, Graham JM, Jr. Klinefelter syndrome and other sex chromosomal aneuploidies. Orphanet J Rare Dis. 2006;1:42.
- 34. Wikstrom AM, Dunkel L. Klinefelter syndrome. Best Pract Res Clin Endocrinol Metab. 2011;25(2):239-50, http://dx.doi.org/10.1016/j.beem.2010.09.006
- 35. Jequier AM, Ansell ID, Bullimore NJ. Congenital absence of the vasa deferentia presenting with infertility. J Androl. 1985;6(1):15-9.
- 36. Anguiano A, Oates RD, Amos JA, Dean M, Gerrard B, Stewart C, et al. Congenital bilateral absence of the vas deferens.A primarily genital form of cystic fibrosis. JAMA. 1992;267(13):1794-7, http://dx.doi.org/10.1001/jama.1992.03480130110034
- 37. Chillon M, Casals T, Mercier B, Bassas L, Lissens W, Silber S, et al. Mutations in the cystic fibrosis gene in patients with congenital absence of the vas deferens. N Engl J Med. 1995;332(22):1475-80.
- 38. Meng MV, Black LD, Cha I, Ljung BM, Pera RA, Turek PJ. Impaired spermatogenesis in men with congenital absence of the vas deferens. Hum Reprod. 2001;16(3):529-33, http://dx.doi.org/10.1093/humrep/16.3.529
- 39. Tiepolo L, Zuffardi O. Localization of factors controlling spermatogen-esis in the nonfluorescent portion of the human Y chromosome long arm. Hum Genet. 1976;34(2):119-24, http://dx.doi.org/10.1007/BF00278879
- 40. Ferlin A, Arredi B, Speltra E, Cazzadore C, Selice R, Garolla A, et al. Molecular and clinical characterization of Y chromosome microdeletions in infertile men: a 10-year experience in Italy. J Clin Endocrinol Metab. 2007;92(3):762-70.
- 41. Pryor JL, Kent-First M, Muallem A, Van Bergen AH, Nolten WE, Meisner L, et al. Microdeletions in the Y chromosome of infertile men. N Engl J Med. 1997;336(8):534-9.
- 42. Vogt PH, Edelmann A, Kirsch S, Henegariu O, Hirschmann P, Kiesewetter F, et al. Human Y chromosome azoospermia factors (AZF) mapped to different subregions in Yq11. Hum Mol Genet. 1996;5(7):933-43, http://dx.doi.org/10.1093/hmg/5.7.933
- 43. Jobling MA, Tyler-Smith C. The human Y chromosome: an evolutionary marker comes of age. Nat Rev Genet. 2003;4(8):598-612, http://dx.doi. org/10.1038/nrg1124
- 44. Krausz C, Forti G, McElreavey K. The Y chromosome and male fertility and infertility. Int J Androl. 2003;26(2):70-5.
- 45. Krausz C, Quintana-Murci L, McElreavey K. Prognostic value of Y deletion analysis: what is the clinical prognostic value of Y chromosome microdeletion analysis? Hum Reprod. 2000;15(7):1431-4, http://dx.doi.org/10.1093/humrep/15.7.1431
- 46. Kamp C, Huellen K, Fernandes S, Sousa M, Schlegel PN, Mielnik A, et al. High deletion frequency of the complete AZFa sequence in men with Sertoli-cell-only syndrome. Molecular Hum Reprod. 2001;7(10):987-94, http://dx.doi.org/10.1093/molehr/7.10.987
Publication Dates
-
Publication in this collection
11 Mar 2013 -
Date of issue
2013
History
-
Received
25 June 2012 -
Accepted
10 July 2012