Abstract
The Rhipicephalus (Boophilus) microplus complex currently consists of five taxa, namely R. australis, R. annulatus, R. (B.) microplus clade A sensu, R. microplus clade B sensu, and R. (B.) microplus clade C sensu. Mitochondrial DNA-based methods help taxonomists when they are facing the morpho-taxonomic problem of distinguishing members of the R. (B.) microplus complex. The purpose of this study was to perform molecular characterization of ticks in all five regions of Brazil and infer their phylogenetic relationships. Molecular analysis characterized 10 haplotypes of the COX-1 gene. Molecular network analysis revealed that haplotype H-2 was the most dispersed of the studied populations (n = 11). Haplotype H-3 (n = 2) had the greatest genetic differentiation when compared to other Brazilian populations. A Bayesian phylogenetic tree of the COX-1 gene obtained strong support. In addition, it was observed that the population of R. (B.) microplus haplotype H-3 exhibited diverging branches among the other Brazilian populations in the study. The study concludes that the different regions of Brazil have R. (B.) microplus tick populations with distinct haplotypes.
Keywords:
Rhipicephalus (Boophilus) microplus; cattle tick; COX-I gene; ITS-2 gene; Brazil
Resumo
Carrapatos do complexo R. (B.) microplus se distribuem em cinco taxa: R. australis, R. annulatus, R. (B.) microplus clado A sensu R. microplus clado B sensue e R. (B.) microplus clado C sensu. Métodos baseados no DNA mitocondrial podem auxiliar taxonomistas quando há dificuldades em estabelecer diferenças morfológicas para distinguir membros do complexo R. (B.) microplus. O objetivo deste estudo foi a caracterização molecular e a inferência de relações filogenéticas em carrapatos de todas as cinco regiões geográficas do Brasil. Para o gene COX-1, a análise molecular caracterizou 10 haplótipos. Na análise molecular em rede foi observado que o haplótipo H-2 é o mais disperso entre as populações (n=11). O haplótipo H-3 (n=2) foi o que obteve maior diferenciação genética ao ser comparado com outras populações brasileiras. A árvore filogenética Bayesiana de gene COX-1 gerou suporte robusto e foi observado que a população de R. (B.) microplus haplótipo H-3 apresentou ramificação com divergência entre as outras populações brasileiras apresentadas neste estudo. Conclui-se que as populações brasileiras possuem diversidade haplotípica com divergência entre as diversas populações de R. (B.) microplus no Brasil.
Palavras-chave:
Rhipicephalus (Boophilus) microplus; carrapato-do-boi; gene COX-I; gene ITS-2; Brasil
Introduction
The bovine tick Rhipicephalus (B.) microplus can be found in multiple tropical and subtropical regions worldwide (ESTRADA-PEÑA et al., 2006Estrada-Peña A, Bouattour A, Camicas JL, Guglielmone A, Horak I, Jongejan F, et al. The known distribution and ecological preferences of the tick subgenus (Acari: Ixodidae) in Africa and Latin America. BoophilusExp Appl Acarol 2006; 38(2-3): 219-235. http://dx.doi.org/10.1007/s10493-006-0003-5. PMid:16596355.
http://dx.doi.org/10.1007/s10493-006-000...
). This parasite may transmit pathogens that cause babesiosis (Babesia bovis and B. bigemina) and anaplasmosis (Anaplasma marginale) (PETER et al., 2005Peter RJ, Van den Bossche P, Penzhorn BL, Sharp B. Tick, fly, and mosquito Control-Lessons from the past, solutions for the future. Vet Parasitol 2005; 132(3-4): 205-215. http://dx.doi.org/10.1016/j.vetpar.2005.07.004. PMid:16099104.
http://dx.doi.org/10.1016/j.vetpar.2005....
; de la FUENTE et al., 2008de la Fuente J, Estrada-Peña A, Venzal JM, Kocan KM, Sonenshine DE. Overview: ticks as vectors of pathogens that cause disease in humans and animals. Front Biosci 2008; 13(13): 6938-6946. http://dx.doi.org/10.2741/3200. PMid:18508706.
http://dx.doi.org/10.2741/3200...
). Infestation of herds with R. (B.) microplus cause losses estimated at US$3.24 billion per year in Brazil (GRISI et al., 2014Grisi L, Leite RC, Martins JR, Barros AT, Andreotti R, Cançado PH, et al. Reassessment of the potential economic impact of cattle parasites in Brazil. Rev Bras Parasitol Vet 2014; 23(2): 150-156. http://dx.doi.org/10.1590/S1984-29612014042. PMid:25054492.
http://dx.doi.org/10.1590/S1984-29612014...
).
In most cattle-producing countries, control methods against this tick are costly and require the use of acaricides. However, acaricide-resistant R. (B.) microplus populations have become a worldwide problem, and molecular ecology studies and new tick control technologies are now required to preserve cattle production (RODRÍGUEZ-VIVAS et al., 2007Rodríguez-Vivas RI, Rivas AL, Chowell G, Fragoso SH, Rosario CR, García Z, et al. Spatial distribution of acaricide profiles ( strains susceptible or resistant to acaricides) in southeastern Mexico. Boophilus microplusVet Parasitol 2007; 146(1-2): 158-169. http://dx.doi.org/10.1016/j.vetpar.2007.01.016. PMid:17349747.
http://dx.doi.org/10.1016/j.vetpar.2007....
; ANDREOTTI et al., 2011Andreotti R, Guerrero FD, Soares MA, Barros JC, Miller RJ, Léon AP. Acaricide resistance of Rhipicephalus (Boophilus) microplus in State of Mato Grosso do Sul, Brazil. Rev Bras Parasitol Vet 2011; 20(2): 127-133. http://dx.doi.org/10.1590/S1984-29612011000200007. PMid:21722487.
http://dx.doi.org/10.1590/S1984-29612011...
; GUERRERO et al., 2014Guerrero FD, Andreotti R, Bendele KG, Cunha RC, Miller RJ, Yeater K, et al. aquaporin as an effective vaccine antigen to protect against cattle tick infestations. Rhipicephalus (Boophilus) microplusParasit Vectors 2014; 7: 475. PMid:25306139.).
Six years ago, Labruna et al. (2009)Labruna MB, Naranjo V, Mangold AJ, Thompson C, Estrada-Peña A, Guglielmone AA, et al. Allopatric speciation in ticks: genetic and reproductive divergence between geographic strains of Rhipicephalus (Boophilus) microplus.BMC Evol Biol 2009; 9(1): 46. http://dx.doi.org/10.1186/1471-2148-9-46. PMid:19243585.
http://dx.doi.org/10.1186/1471-2148-9-46...
used 12S and 16S molecular markers to show a lack of divergence among populations in the Americas, including Brazil. Later, Burger et al. (2014)Burger TD, Shao R, Barker SC. Phylogenetic analysis of mitochondrial genome sequences indicates that the cattle tick, Rhipicephalus (Boophilus) microplus, contains a cryptic species. Mol Phylogenet Evol 2014; 76: 241-253. http://dx.doi.org/10.1016/j.ympev.2014.03.017. PMid:24685498.
http://dx.doi.org/10.1016/j.ympev.2014.0...
used the COX-1 molecular marker to show that the R. (B.) microplus populations of Brazil (GenBank: KC503261) are organized in clade A, along with populations of R. (B.) microplus from China (GenBank: HM193863) and Cambodia (GenBank: KC503260). Thus, molecular markers may resolve very close taxonomic relationships and provide new knowledge about population structure, allowing a deeper understanding of R. (B.) microplus populations (BURGER et al., 2014Burger TD, Shao R, Barker SC. Phylogenetic analysis of mitochondrial genome sequences indicates that the cattle tick, Rhipicephalus (Boophilus) microplus, contains a cryptic species. Mol Phylogenet Evol 2014; 76: 241-253. http://dx.doi.org/10.1016/j.ympev.2014.03.017. PMid:24685498.
http://dx.doi.org/10.1016/j.ympev.2014.0...
).
Although the history of the dissemination of R. (B.) microplus is not well documented, the species is known to have originated in India (HOOGSTRAL, 1986Hoogstral H. Theobald Smith: his scientific work and impact. Bull Entomol Soc Am 1986; 32(1): 22-35. http://dx.doi.org/10.1093/besa/32.1.22.
http://dx.doi.org/10.1093/besa/32.1.22...
). According to Barré & Uilenberg (2010)Barré N, Uilenberg G. Spread of parasites transported with their hosts: case study of two species of cattle tick. Rev Sci Tech 2010; 29(1): 149-160, 135-147. PMid:20617654., R. (B.) microplus originated in the southern and southeastern regions of Asia and was spread throughout the tropical and subtropical belts via cattle, arriving in Brazil between the 16th and 17th centuries.
Mitochondrial and nuclear genome markers are increasingly being used to better understand phylogenetic relationships in order to elucidate whether evolution has occurred among populations of a species (KANDUMA et al., 2012Kanduma EG, Mwacharo JM, Sunter JD, Nzuki I, Mwaura S, Kinyanjui PW, et al. Micro-and minisatellite-expressed sequence tag (EST) markers discriminate between populations of Rhipicephalus appendiculatus.Ticks Tick Borne Dis 2012; 3(3): 128-136. http://dx.doi.org/10.1016/j.ttbdis.2012.05.001. PMid:22789728.
http://dx.doi.org/10.1016/j.ttbdis.2012....
).
The DNA barcode has been proposed as a universal tool for identifying biological diversity, and can be used as a molecular marker (HEBERT et al., 2003Hebert PDN, Cywinska A, Ball SL, deWaard JR. Biological identifications through DNA barcodes. Proc Biol Sci 2003; 270(1512): 313-321.). DNA barcoding is based on information gathered from a fragment of approximately 688 base pairs of mitochondrial DNA base sequences (mtDNA) from the cytochrome oxidase I gene (COX-I) of different species. A number of studies have shown that the DNA barcode is a universal code highly effective for the identification of species (HEBERT et al., 2004Hebert PDN, Stoeckle MY, Zemlak TS, Francis CM. Identification of birds through DNA barcodes. PLoS Biol 2004; 2(10): e312. http://dx.doi.org/10.1371/journal.pbio.0020312. PMid:15455034.
http://dx.doi.org/10.1371/journal.pbio.0...
; BARRETT & HEBERT, 2005Barrett RDH, Hebert PDN. Identifying spiders through DNA barcodes. Can J Zool 2005; 83(3): 481-491. http://dx.doi.org/10.1139/z05-024.
http://dx.doi.org/10.1139/z05-024...
).
In addition to the DNA barcode, ribosomal DNA (rDNA) may be used as a nuclear molecular marker. The internal transcribed spacer 2 (ITS-2) is located between the 5.8S and 28S ribosomal subunits of rDNA (CRUICKSHANK, 2002Cruickshank RH. Molecular markers for the phylogenetics of mites and ticks. Syst Appl Acarol 2002; 7(1): 3-14. http://dx.doi.org/10.11158/saa.7.1.1.
http://dx.doi.org/10.11158/saa.7.1.1...
). These genes are arranged in repeated units known as ribosomal cistrons, which have repeated copies and have been used to study phylogenetic relationships (CAMPBELL et al., 1993Campbell BC, Steffen-Campbell JD, Werren JH. Phylogeny of the Nasonia species complex (Hymenoptera: Pteromalidae) inferred from an internal transcribed spacer (ITS2) and 28S rDNA sequences. Insect Mol Biol 1993; 2(4): 225-237. http://dx.doi.org/10.1111/j.1365-2583.1994.tb00142.x. PMid:9087560.
http://dx.doi.org/10.1111/j.1365-2583.19...
; SONG et al., 2011Song S, Shao R, Atwell R, Barker S, Vankan D. Phylogenetic and phylogeographic relationships in Ixodes holocyclus and (Acari: Ixodidae) inferred from COX1 and ITS2 sequences. Ixodes cornuatusInt J Parasitol 2011; 41(8): 871-880. http://dx.doi.org/10.1016/j.ijpara.2011.03.008. PMid:21540032.
http://dx.doi.org/10.1016/j.ijpara.2011....
; BURGER et al., 2014Burger TD, Shao R, Barker SC. Phylogenetic analysis of mitochondrial genome sequences indicates that the cattle tick, Rhipicephalus (Boophilus) microplus, contains a cryptic species. Mol Phylogenet Evol 2014; 76: 241-253. http://dx.doi.org/10.1016/j.ympev.2014.03.017. PMid:24685498.
http://dx.doi.org/10.1016/j.ympev.2014.0...
).
The objective of this study was to infer the phylogenetic and phylogeographic relationships among R. (B.) microplus tick populations in Brazil based on COX-I mitochondrial DNA gene sequences and ITS-2 nuclear DNA, and compare them with R. (B.) microplus tick populations from other countries.
Materials and Methods
Tick collection
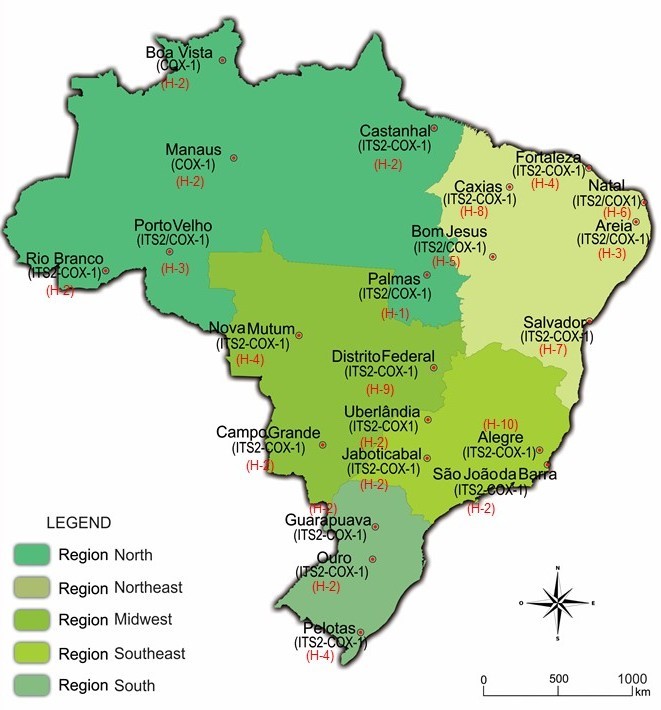
Ticks were obtained from 22 locations in different geographical regions of Brazil (Figure 1, Table 1). A pool of larvae and 22 engorged females were collected from natural environments. The morphology and identification key was based on Barros-Battesti et al. (2006)Barros-Battesti DM, Arzua M, Bechara GH. Carrapatos de importância médico-veterinária da região neotropical: Um guia ilustrado para identificação de espécies. São Paulo: Instituto Butantan, 2006. 223 p.. Samples were stored in an ultra-freezer at –80 °C for subsequent analysis.

Geographic locations where Rhipicephalus (Boophilus) microplus ticks were collected in five regions of Brazil.
Brazilian states, geographic coordinates related to the collection and GenBank accession number of this study.
DNA extraction and PCR amplification
DNA extraction was performed on a pool of larvae and individual samples of engorged females using DNAzol® Reagent (Invitrogen) according to the manufacturer’s recommendations. The DNA was quantified using a Nano Drop 1000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA), and an A260nm/A280nm ratio above 1.6 was established as acceptable. The sample concentration was corrected to 50 ng µL–1. Next, the DNA was used in a polymerase chain reaction (PCR) for COX-I and ITS-2 region amplification with new primers designed by our group. PCR generated products of 643bp and 580bp for COX-1 and ITS-2, respectively.
COX-I gene fragments were amplified using the COX-I.2F (5’-CTTCAGCCATTTTACCGCGA-3’) and COX-I.2R (5’-CTCCGCCTGAAGGGTCAAA-3’) starter oligonucleotides. ITS-2 fragments were amplified with the ITS-2F (5’-CGGATCACATATCAAGAGAG-3’) and ITS-2R (5’-CCCAACTGGAGTGGCCCAGTTT-3’) primers. The PCR was standardized for a final volume of 25 μL with 1X buffer, 1.5 mM MgCl, 25 mM dNTPs, 10 µM of each primer, 0.5 unit of Platinum® Taq DNA Polymerase High Fidelity (Invitrogen by Life TechnologiesTM, Massachusetts, USA), and 1µL of DNA at 50 ngµL-1. PCR conditions were optimized for each reaction, and the annealing temperature was adjusted to suit the primers used. General PCR conditions were 94 °C for 2 min, followed by 35 cycles of 94 °C for 1 min, 56 °C/60 °C (COX-I/ITS-2, respectively) for 30s, 68 °C for 1 min, and a final extension of 68 °C for 3 min. PCR products were visualized in a 2% agarose gel stained with ethidium bromide. The PCR-amplified products were purified using a Purelink®kit (Invitrogen by Life TechnologiesTM, Massachusetts, USA). Fragments were then cloned with pGEM®-T Easy Vector Systems cloning vector according to the manufacturer’s manual (Promega, Madison, WI-USA) and sequenced as described below.
Genetic analysis
All analyses generated in this study were performed in silico. Sanger sequencing was performed in a 48 well plate with robotic instrumentation using T7 Transcription Start (5’-CTAATACGACTCACTATAGGG-3’) universal sequencing primer (Promega, Madison, WI-USA). Samples were sequenced using an Applied BiosystemsTM ABI 3730 DNA Analyzer and the conditions described by Song et al. (2011)Song S, Shao R, Atwell R, Barker S, Vankan D. Phylogenetic and phylogeographic relationships in Ixodes holocyclus and (Acari: Ixodidae) inferred from COX1 and ITS2 sequences. Ixodes cornuatusInt J Parasitol 2011; 41(8): 871-880. http://dx.doi.org/10.1016/j.ijpara.2011.03.008. PMid:21540032.
http://dx.doi.org/10.1016/j.ijpara.2011....
. The sequencing reactions were done using the Big Dye® Terminator v3.1 cycle sequencing kit. The runs were performed in 36-cm capillaries using the POP7 polymer, and the sequences were generated by the Sequencing Analysis Software v5.3.1 through the Caller KB Phred program (EWING & GREEN, 1998Ewing B, Green P. Base-calling of automated sequencer traces using . II. Error probabilities. phredGenome Res 1998; 8(3): 186-194. http://dx.doi.org/10.1101/gr.8.3.186. PMid:9521922.
http://dx.doi.org/10.1101/gr.8.3.186...
; EWING et al., 1998Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using . I. Accuracy assessment. phredGenome Res 1998; 8(3): 175-185. http://dx.doi.org/10.1101/gr.8.3.175. PMid:9521921.
http://dx.doi.org/10.1101/gr.8.3.175...
).
Plasmid sequences were identified and removed using an NCBI (VecScreen) tool (ALTSCHUL et al., 1997Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997; 25(17): 3389-3402. http://dx.doi.org/10.1093/nar/25.17.3389. PMid:9254694.
http://dx.doi.org/10.1093/nar/25.17.3389...
), and a hash search algorithm was used to remove the contaminant sequences. Consensus sequences were aligned with the GenBank data using the BLASTN program (ALTSCHUL et al., 1997Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997; 25(17): 3389-3402. http://dx.doi.org/10.1093/nar/25.17.3389. PMid:9254694.
http://dx.doi.org/10.1093/nar/25.17.3389...
).
The partition homogeneity test was conducted using the ARLEQUIN program, version 3.5.1.2 (EXCOFFIER & LISCHER, 2010Excoffier L, Lischer HEL. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 2010; 10(3): 564-567. http://dx.doi.org/10.1111/j.1755-0998.2010.02847.x. PMid:21565059.
http://dx.doi.org/10.1111/j.1755-0998.20...
). The aligned COX-I and ITS-2 sequences comprised 681bpsand 649bps, respectively. Database sequence R. (B.) microplus MS- Brazil (GenBank: KC503261) clade A sensuBurger et al. (2014)Burger TD, Shao R, Barker SC. Phylogenetic analysis of mitochondrial genome sequences indicates that the cattle tick, Rhipicephalus (Boophilus) microplus, contains a cryptic species. Mol Phylogenet Evol 2014; 76: 241-253. http://dx.doi.org/10.1016/j.ympev.2014.03.017. PMid:24685498.
http://dx.doi.org/10.1016/j.ympev.2014.0...
, was used for alignment with samples from this study. This sequence alignment revealed 10 haplotypes (H1-H10) based on COX-I genes alone (Figure 2). Distinct sequences of COX-I (KP226159-KP226180) and ITS-2 (KP226139-KP226158) were deposited in NCBI GenBank.

(A) Bayesian phylogenetic tree recovered for cytochrome oxidase subunit 1 gene (COX-I) of 643 nucleotides. R. annulatus (GenBank: KC503256) was used as the outgroup (B) median joining network of R. (B.) microplus from five different regions populations in Brazil. A circle represents each haplotype. Relative areas of the circles indicate haplotype frequency of each sample.
A median-joining analysis implemented in the program Network Version 5.0 (BANDELT et al., 1999Bandelt HJ, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 1999; 16(1): 37-48. http://dx.doi.org/10.1093/oxfordjournals.molbev.a026036. PMid:10331250.
http://dx.doi.org/10.1093/oxfordjournals...
) was used for the intraspecific analysis of the evolutionary relationships among haplotypes. Uncorrected (p) pairwise genetic distances were calculated using ARLEQUIN program, version 3.5.1.2 (EXCOFFIER & LISCHER, 2010Excoffier L, Lischer HEL. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 2010; 10(3): 564-567. http://dx.doi.org/10.1111/j.1755-0998.2010.02847.x. PMid:21565059.
http://dx.doi.org/10.1111/j.1755-0998.20...
) to assess the genetic divergence of R. (B.) microplus in both COX-I and ITS-2 genes. To assess the level of genetic differentiation, we used the program DnaSP 5.0 (LIBRADO & ROZAS, 2009Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009; 25(11): 1451-1452. http://dx.doi.org/10.1093/bioinformatics/btp187. PMid:19346325.
http://dx.doi.org/10.1093/bioinformatics...
). Haplotype diversity (Hd), nucleotide diversity (pi), number of haplotypes (h), and genetic differentiation (FST) were determined.
The levels of genetic differentiation were defined as FST > 0.25 (great differentiation), 0.15 to 0.25 (moderate differentiation) and FST< 0.05 (negligible differentiation) (WRIGHT, 1931Wright S. Evolution in mendelian populations. Genetics 1931; 16(2): 97-159. PMid:17246615.). The levels of gene flow were defined as Nm> 1 (high gene flow), 0.25 to 0.99 (intermediate gene flow) and Nm< 0.25 (low gene flow). The distinct COX-I and 16S rRNA haplotypes identified in the Brazilian R. (B.) microplus ticks were aligned with all representative sequences of Rhipicephalus taxa available in the NCBI GenBank. R. annulatus was used as an outgroup for the construction of Bayesian phylogenetic trees based on Brazilian R. (B.) microplus ticks COX-I only; Dermacentor nitens was used as an outgroup for the construction of Bayesian phylogenetic trees based on COX-I and ITS-2 sequences. The Bayesian phylogenetic analysis performed using MrBayes 3.1 (HUELSENBECK & RONQUIST, 2001Huelsenbeck JP, Ronquist F. MRBAYES: bayesian inference of phylogenetic trees. Bioinformatics 2001; 17(8): 754-755. http://dx.doi.org/10.1093/bioinformatics/17.8.754. PMid:11524383.
http://dx.doi.org/10.1093/bioinformatics...
; RONQUIST & HUELSENBECK, 2003Ronquist F, Huelsenbeck JP. MRBAYES 3: bayesian phylogenetic inference under mixed models. Bioinformatics 2003; 19(12): 1572-1574. http://dx.doi.org/10.1093/bioinformatics/btg180. PMid:12912839.
http://dx.doi.org/10.1093/bioinformatics...
) were plotted using FigTree software version 1.4.2 (TREE BIO, 2016Tree Bio. FigTree [online]. London; 2016 [cited 2016 Apr 11]. Available from: http://tree.bio.ed.ac.uk/
http://tree.bio.ed.ac.uk/...
).
The ARLEQUIN software revealed population structure by means of AMOVA, which uncovers the existence of population differentiation at both intra- and inter-population levels (EXCOFFIER & LISCHER, 2010Excoffier L, Lischer HEL. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 2010; 10(3): 564-567. http://dx.doi.org/10.1111/j.1755-0998.2010.02847.x. PMid:21565059.
http://dx.doi.org/10.1111/j.1755-0998.20...
).
Results and Discussion
Nucleotide analyses and Haplotype
The phylogenetic and phylogeographic relations of the Brazilian R. (B.) microplus were inferred from the COX-I sequences (22 samples), R. (B.) microplus Brazil (GenBank: KC503261) and the ITS2 sequences (20 samples) obtained from all five Brazilian geographic regions (Figure 1).
This is the only study that has compared a representative sample of R. (B.) microplus, which together represent the whole country and the authors were careful to represent each region by just having differentiated biogeography. Assays performed by Dantas-Torres (2015)Dantas-Torres F. Climate change, biodiversity, ticks and tick-borne diseases: the butterfly effect. Int J Parasitol Parasites Wildl 2015; 4(3): 452-461. http://dx.doi.org/10.1016/j.ijppaw.2015.07.001. PMid:26835253.
http://dx.doi.org/10.1016/j.ijppaw.2015....
, can demonstrate that certain factors such as climate change and biodiversity boost the expansion of tick populations.
The COX-I dataset showed haplotype diversity (0.767) greater than that revealed by the ITS-2 (0.567) gene. However, the ITS-2 showed greater nucleotide diversity (0.00277) than the COX-I (0.00432) dataset. Haplotype network analysis (COX-I) revealed ten distinct haplotype clusters among Brazilian populations, with no clear separation by tick populations/geographical areas, indicating that an overlap of the ten genetically divergent haplotypes of R. (B.) microplus (Figure 2A) exists.
Haplotype H-2 was the most widespread haplotype (n = 11) in Northern, Midwest, Southeast and Southern populations (Figure 2B). Haplotypes H-5, H-6 and H-8 (Northern region) originates from H-4 and H-2 haplotypes, as shown in Figure 2B. However, how these haplotypes are associated in other regions is not clear. ITS-2 genotypes indicate a lack of clear separation of tick populations (data no shown).
Genetic distance, genetic differentiation and gene flow
In the uncorrected “p” distance matrix, the COX-I gene indicated stronger resolving power than ITS-2 (1.36–11.00) (Table 2). The genetic distance of the COX-I H-2 haplotype of R. (B.) microplus indicated 0.73% of divergence within this population (red value, Table 2). Overall, a high level of significant genetic differentiation was observed among all populations (P < 0.001).
Percentage of uncorrected “p” distance matrix among the ten haplotypes of COX-1 of R. (B.) microplus in Brazil.
Great genetic differentiation (FST = 1.00), revealed by the COX-I gene, was found with in all haplotypes (Table 3). Additionally, genetic differentiation between H-2 haplotypes and others varied between 0.47-0.89 (Table 3). High levels (30.00) of gene flow (Nm) were observed among tick populations as shown by the COX-I (data no shown).
Genetic differentiation (FST) based on COX-1, in all the haplotypes found of R. (B.) microplus populations in Brazil.
Among the ten haplotypes studied, R. (B.) microplus haplotype H-3 from Rondônia (RO) and Paraíba (PB) were genetically diverse as supported by the highest haplotype and nucleotide diversity based on COX-I (Table 3). Higher inter-population levels (82.69%) and (FST = 0.8269) revealed the existence of population structure in all haplotypes analyzed by means of AMOVA (Table 4).
Livestock transition in Brazil is very intense. Whether these regions actually have different populations not found in other regions can only be answered by analyzing a larger number of samples from various municipalities in the States of Rondônia and Paraíba.
All haplotypes are derived from haplotype H-2. However, it is not possible to assert where haplotype H-2 has originated. Additionally, there is no haplotype H-2 in northeastern Brazil.
The fact that the country has cattle production for imported and local breeds and that animals are transported between different regions do not allow us to infer the exact origin of haplotypes. The natural dispersal ability of ticks might occur in parallel with human-mediated dispersal, causing genetic admixture and high gene flow in Brazilian populations.
Phylogenetic analyses
Bayesian analyses produced phylogenetic trees with high valued probabilities (Figure 2 and Figure 3). The 10 haplotypes (H1-H10) generated from the COX-I sequences of Brazilian R. (B.) microplus were subjected to phylogenetic analyses. By the topology of the phylogenetic trees generated, the R. (B.) microplus Brazilian population comprises two clades: clade A (H3) and clade B (H1, H2 and H4 to H10) (Figure 2A). Different than results revealed by the haplotype network analysis (Figure 2B), according to Bayesian phylogenetic tree, haplotypes H5, H6 and H8 were associated with ticks from the North region of the country and are derived from haplotype H-4. The tree also indicates that all haplotypes derive from H-2.

Bayesian phylogenetic tree recovered for cytochrome oxidase subunit 1 gene (COX-I) of 643 nucleotides, for Rhipicephalus taxa samples. In blue and green are the smallest nodes formed the Brazilian populations of R. (B.) microplus. Dermacentor nitens (GenBank: KC503258) was used as the outgroup.
The ITS-2 Bayesian phylogenetic tree revealed one R. (B.) microplus genetic clade (Figure 4). The COX-I Bayesian phylogenetic tree revealed two genetic clades of R. (B.) microplus, Clade A (Brazil, Texas, Malaysia, R. australis) and Clade B (R. (B.) microplus China and R. annulatus). Rhipicephalus (B.) microplus clade A showed a sister relationship with R. australis. By contrast, R. (B.) microplus clade B is more closely related to R. annulatus, with which it forms a sister group relationship, corroborating the data by Burger et al. (2014)Burger TD, Shao R, Barker SC. Phylogenetic analysis of mitochondrial genome sequences indicates that the cattle tick, Rhipicephalus (Boophilus) microplus, contains a cryptic species. Mol Phylogenet Evol 2014; 76: 241-253. http://dx.doi.org/10.1016/j.ympev.2014.03.017. PMid:24685498.
http://dx.doi.org/10.1016/j.ympev.2014.0...
(Figure 3).

Bayesian Cladogram recovered for internal transcript spacer 2 (ITS-2) of 580 nucleotides, for sample Rhipicephalus taxa. Dermacentor nitens KC503275 was used as the outgroup.
The R. (B.) microplus populations in Brazil have internal nodes, indicating the occurrence of subpopulations (GenBank: KP226160, KP226167, KP226169, KP226172, KP226174, KP226177). The haplotype H-3 formed another smaller node (GenBank: KP226176, KP226178) (Figure 3).
The intraspecific genetic distance or haplotype frequency, like that of haplotype H2 reported here, are notably higher than previously described for R. (B.) microplus sensuLow et al. (2015)Low VL, Tay ST, Kho KL, Koh FX, Tan TK, Lim YAL, et al. Molecular characterisation of the tick Rhipicephalus microplus in Malaysia: new insights into the cryptic diversity and distinct genetic assemblages throughout the world. Parasit Vectors 2015; 8(1): 341. http://dx.doi.org/10.1186/s13071-015-0956-5. PMid:26104478.
http://dx.doi.org/10.1186/s13071-015-095...
. Furthermore, the two, and perhaps three, genetic assemblages inferred from Brazilian R. (B.) microplus are more genetically diverse than those reported from other regions (Figure 3).
This study sought to obtain high support for the topology of tree and the COX-I molecular marker was found to have the advantage of presenting greater mutation rates than the nuclear marker, corroborating the data presented by Lv et al. (2014)Lv J, Wu S, Zhang Y, Chen Y, Feng C, Yuan X, et al. Assessment of four DNA fragments (COI, 16S rDNA, ITS2, 12S rDNA) for species identification of the Ixodida (Acari: Ixodida). Parasit Vectors 2014; 7(1): 93. http://dx.doi.org/10.1186/1756-3305-7-93. PMid:24589289.
http://dx.doi.org/10.1186/1756-3305-7-93...
. However, the ITS-2 molecular marker failed to resolve discrepancies between the populations of R. (B.) microplus and this study corroborates the findings of Burger et al. (2014)Burger TD, Shao R, Barker SC. Phylogenetic analysis of mitochondrial genome sequences indicates that the cattle tick, Rhipicephalus (Boophilus) microplus, contains a cryptic species. Mol Phylogenet Evol 2014; 76: 241-253. http://dx.doi.org/10.1016/j.ympev.2014.03.017. PMid:24685498.
http://dx.doi.org/10.1016/j.ympev.2014.0...
in which they showed that the ITS-2 molecular marker has insufficient power to distinguish between very close species.
The comparative analysis in this study shows that R. (B.) microplus ticks represent at least two different populations. Moreover, this study also attempted to investigate whether haplotypes are distributed according to the phylogeographical areas. It is unknown whether this indicates a cause-effect association of haplotypes and geographical areas.
However, the prevalence of ticks has been typically associated with cattle breeds, the highly invasive and widespread movement of R. (B.) microplus may facilitate its occurrence (IBELLI et al., 2012Ibelli AM, Ribeiro AR, Giglioti R, Regitano LC, Alencar MM, Chagas ACS, et al. Resistance of cattle of various genetic groups to the tick and the relationship with coat traits. Rhipicephalus microplusVet Parasitol 2012; 186(3-4): 425-430. http://dx.doi.org/10.1016/j.vetpar.2011.11.019. PMid:22115946.
http://dx.doi.org/10.1016/j.vetpar.2011....
; BUSCH et al., 2014Busch JD, Stone NE, Nottingham R, Araya-Anchetta A, Lewis J, Hochhalter C, et al. Widespread movement of invasive cattle fever ticks (Rhipicephalus microplus) in southern Texas leads to shared local infestations on cattle and deer. Parasit Vectors 2014; 7(1): 188. http://dx.doi.org/10.1186/1756-3305-7-188. PMid:24742041.
http://dx.doi.org/10.1186/1756-3305-7-18...
).
Conclusion
This study provides new insights into the distinct genetic assemblages of the tick R. (B.) microplus in Brazil and reveals that the Brazilian R. (B.) microplus population consists of at least two different populations. In addition, future studies using a large number of samples from the Americas using COX-1 may be able to show how these populations differ in their inter- and intra- relationships.
Acknowledgements
We thank FUNDECT - MS, the State of Mato Grosso do Sul, CAPES and CNPq for financial support.
-
§ Bolsista DCR Fundect/MS.
References
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997; 25(17): 3389-3402. http://dx.doi.org/10.1093/nar/25.17.3389 PMid:9254694.
» http://dx.doi.org/10.1093/nar/25.17.3389 - Andreotti R, Guerrero FD, Soares MA, Barros JC, Miller RJ, Léon AP. Acaricide resistance of Rhipicephalus (Boophilus) microplus in State of Mato Grosso do Sul, Brazil. Rev Bras Parasitol Vet 2011; 20(2): 127-133. http://dx.doi.org/10.1590/S1984-29612011000200007 PMid:21722487.
» http://dx.doi.org/10.1590/S1984-29612011000200007 - Bandelt HJ, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 1999; 16(1): 37-48. http://dx.doi.org/10.1093/oxfordjournals.molbev.a026036 PMid:10331250.
» http://dx.doi.org/10.1093/oxfordjournals.molbev.a026036 - Barré N, Uilenberg G. Spread of parasites transported with their hosts: case study of two species of cattle tick. Rev Sci Tech 2010; 29(1): 149-160, 135-147. PMid:20617654.
- Barrett RDH, Hebert PDN. Identifying spiders through DNA barcodes. Can J Zool 2005; 83(3): 481-491. http://dx.doi.org/10.1139/z05-024
» http://dx.doi.org/10.1139/z05-024 - Barros-Battesti DM, Arzua M, Bechara GH. Carrapatos de importância médico-veterinária da região neotropical: Um guia ilustrado para identificação de espécies. São Paulo: Instituto Butantan, 2006. 223 p.
- Burger TD, Shao R, Barker SC. Phylogenetic analysis of mitochondrial genome sequences indicates that the cattle tick, Rhipicephalus (Boophilus) microplus, contains a cryptic species. Mol Phylogenet Evol 2014; 76: 241-253. http://dx.doi.org/10.1016/j.ympev.2014.03.017 PMid:24685498.
» http://dx.doi.org/10.1016/j.ympev.2014.03.017 - Busch JD, Stone NE, Nottingham R, Araya-Anchetta A, Lewis J, Hochhalter C, et al. Widespread movement of invasive cattle fever ticks (Rhipicephalus microplus) in southern Texas leads to shared local infestations on cattle and deer. Parasit Vectors 2014; 7(1): 188. http://dx.doi.org/10.1186/1756-3305-7-188 PMid:24742041.
» http://dx.doi.org/10.1186/1756-3305-7-188 - Campbell BC, Steffen-Campbell JD, Werren JH. Phylogeny of the Nasonia species complex (Hymenoptera: Pteromalidae) inferred from an internal transcribed spacer (ITS2) and 28S rDNA sequences. Insect Mol Biol 1993; 2(4): 225-237. http://dx.doi.org/10.1111/j.1365-2583.1994.tb00142.x PMid:9087560.
» http://dx.doi.org/10.1111/j.1365-2583.1994.tb00142.x - Cruickshank RH. Molecular markers for the phylogenetics of mites and ticks. Syst Appl Acarol 2002; 7(1): 3-14. http://dx.doi.org/10.11158/saa.7.1.1
» http://dx.doi.org/10.11158/saa.7.1.1 - Dantas-Torres F. Climate change, biodiversity, ticks and tick-borne diseases: the butterfly effect. Int J Parasitol Parasites Wildl 2015; 4(3): 452-461. http://dx.doi.org/10.1016/j.ijppaw.2015.07.001 PMid:26835253.
» http://dx.doi.org/10.1016/j.ijppaw.2015.07.001 - de la Fuente J, Estrada-Peña A, Venzal JM, Kocan KM, Sonenshine DE. Overview: ticks as vectors of pathogens that cause disease in humans and animals. Front Biosci 2008; 13(13): 6938-6946. http://dx.doi.org/10.2741/3200 PMid:18508706.
» http://dx.doi.org/10.2741/3200 - Estrada-Peña A, Bouattour A, Camicas JL, Guglielmone A, Horak I, Jongejan F, et al. The known distribution and ecological preferences of the tick subgenus (Acari: Ixodidae) in Africa and Latin America. BoophilusExp Appl Acarol 2006; 38(2-3): 219-235. http://dx.doi.org/10.1007/s10493-006-0003-5 PMid:16596355.
» http://dx.doi.org/10.1007/s10493-006-0003-5 - Ewing B, Green P. Base-calling of automated sequencer traces using . II. Error probabilities. phredGenome Res 1998; 8(3): 186-194. http://dx.doi.org/10.1101/gr.8.3.186 PMid:9521922.
» http://dx.doi.org/10.1101/gr.8.3.186 - Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using . I. Accuracy assessment. phredGenome Res 1998; 8(3): 175-185. http://dx.doi.org/10.1101/gr.8.3.175 PMid:9521921.
» http://dx.doi.org/10.1101/gr.8.3.175 - Excoffier L, Lischer HEL. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 2010; 10(3): 564-567. http://dx.doi.org/10.1111/j.1755-0998.2010.02847.x PMid:21565059.
» http://dx.doi.org/10.1111/j.1755-0998.2010.02847.x - Grisi L, Leite RC, Martins JR, Barros AT, Andreotti R, Cançado PH, et al. Reassessment of the potential economic impact of cattle parasites in Brazil. Rev Bras Parasitol Vet 2014; 23(2): 150-156. http://dx.doi.org/10.1590/S1984-29612014042 PMid:25054492.
» http://dx.doi.org/10.1590/S1984-29612014042 - Guerrero FD, Andreotti R, Bendele KG, Cunha RC, Miller RJ, Yeater K, et al. aquaporin as an effective vaccine antigen to protect against cattle tick infestations. Rhipicephalus (Boophilus) microplusParasit Vectors 2014; 7: 475. PMid:25306139.
- Hebert PDN, Cywinska A, Ball SL, deWaard JR. Biological identifications through DNA barcodes. Proc Biol Sci 2003; 270(1512): 313-321.
- Hebert PDN, Stoeckle MY, Zemlak TS, Francis CM. Identification of birds through DNA barcodes. PLoS Biol 2004; 2(10): e312. http://dx.doi.org/10.1371/journal.pbio.0020312 PMid:15455034.
» http://dx.doi.org/10.1371/journal.pbio.0020312 - Hoogstral H. Theobald Smith: his scientific work and impact. Bull Entomol Soc Am 1986; 32(1): 22-35. http://dx.doi.org/10.1093/besa/32.1.22
» http://dx.doi.org/10.1093/besa/32.1.22 - Huelsenbeck JP, Ronquist F. MRBAYES: bayesian inference of phylogenetic trees. Bioinformatics 2001; 17(8): 754-755. http://dx.doi.org/10.1093/bioinformatics/17.8.754 PMid:11524383.
» http://dx.doi.org/10.1093/bioinformatics/17.8.754 - Ibelli AM, Ribeiro AR, Giglioti R, Regitano LC, Alencar MM, Chagas ACS, et al. Resistance of cattle of various genetic groups to the tick and the relationship with coat traits. Rhipicephalus microplusVet Parasitol 2012; 186(3-4): 425-430. http://dx.doi.org/10.1016/j.vetpar.2011.11.019 PMid:22115946.
» http://dx.doi.org/10.1016/j.vetpar.2011.11.019 - Kanduma EG, Mwacharo JM, Sunter JD, Nzuki I, Mwaura S, Kinyanjui PW, et al. Micro-and minisatellite-expressed sequence tag (EST) markers discriminate between populations of Rhipicephalus appendiculatus.Ticks Tick Borne Dis 2012; 3(3): 128-136. http://dx.doi.org/10.1016/j.ttbdis.2012.05.001 PMid:22789728.
» http://dx.doi.org/10.1016/j.ttbdis.2012.05.001 - Labruna MB, Naranjo V, Mangold AJ, Thompson C, Estrada-Peña A, Guglielmone AA, et al. Allopatric speciation in ticks: genetic and reproductive divergence between geographic strains of Rhipicephalus (Boophilus) microplus.BMC Evol Biol 2009; 9(1): 46. http://dx.doi.org/10.1186/1471-2148-9-46 PMid:19243585.
» http://dx.doi.org/10.1186/1471-2148-9-46 - Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009; 25(11): 1451-1452. http://dx.doi.org/10.1093/bioinformatics/btp187 PMid:19346325.
» http://dx.doi.org/10.1093/bioinformatics/btp187 - Low VL, Tay ST, Kho KL, Koh FX, Tan TK, Lim YAL, et al. Molecular characterisation of the tick Rhipicephalus microplus in Malaysia: new insights into the cryptic diversity and distinct genetic assemblages throughout the world. Parasit Vectors 2015; 8(1): 341. http://dx.doi.org/10.1186/s13071-015-0956-5 PMid:26104478.
» http://dx.doi.org/10.1186/s13071-015-0956-5 - Lv J, Wu S, Zhang Y, Chen Y, Feng C, Yuan X, et al. Assessment of four DNA fragments (COI, 16S rDNA, ITS2, 12S rDNA) for species identification of the Ixodida (Acari: Ixodida). Parasit Vectors 2014; 7(1): 93. http://dx.doi.org/10.1186/1756-3305-7-93 PMid:24589289.
» http://dx.doi.org/10.1186/1756-3305-7-93 - Peter RJ, Van den Bossche P, Penzhorn BL, Sharp B. Tick, fly, and mosquito Control-Lessons from the past, solutions for the future. Vet Parasitol 2005; 132(3-4): 205-215. http://dx.doi.org/10.1016/j.vetpar.2005.07.004 PMid:16099104.
» http://dx.doi.org/10.1016/j.vetpar.2005.07.004 - Rodríguez-Vivas RI, Rivas AL, Chowell G, Fragoso SH, Rosario CR, García Z, et al. Spatial distribution of acaricide profiles ( strains susceptible or resistant to acaricides) in southeastern Mexico. Boophilus microplusVet Parasitol 2007; 146(1-2): 158-169. http://dx.doi.org/10.1016/j.vetpar.2007.01.016 PMid:17349747.
» http://dx.doi.org/10.1016/j.vetpar.2007.01.016 - Ronquist F, Huelsenbeck JP. MRBAYES 3: bayesian phylogenetic inference under mixed models. Bioinformatics 2003; 19(12): 1572-1574. http://dx.doi.org/10.1093/bioinformatics/btg180 PMid:12912839.
» http://dx.doi.org/10.1093/bioinformatics/btg180 - Song S, Shao R, Atwell R, Barker S, Vankan D. Phylogenetic and phylogeographic relationships in Ixodes holocyclus and (Acari: Ixodidae) inferred from COX1 and ITS2 sequences. Ixodes cornuatusInt J Parasitol 2011; 41(8): 871-880. http://dx.doi.org/10.1016/j.ijpara.2011.03.008 PMid:21540032.
» http://dx.doi.org/10.1016/j.ijpara.2011.03.008 - Tree Bio. FigTree [online]. London; 2016 [cited 2016 Apr 11]. Available from: http://tree.bio.ed.ac.uk/
» http://tree.bio.ed.ac.uk/ - Wright S. Evolution in mendelian populations. Genetics 1931; 16(2): 97-159. PMid:17246615.
Publication Dates
-
Publication in this collection
29 Aug 2016 -
Date of issue
Jul-Sep 2016
History
-
Received
11 Apr 2016 -
Accepted
20 July 2016