Abstract
Ornithodoros mimon is an argasid tick that parasitizes bats, birds and opossums and is also harmful to humans. Knowledge of the transcripts present in the tick gut helps in understanding the role of vital molecules in the digestion process and parasite-host relationship, while also providing information about the evolution of arthropod hematophagy. Thus, the present study aimed to know and ascertain the main molecules expressed in the gut of argasid after their blood meal, through analysis on the gut transcriptome of engorged females of O. mimon using 454-based RNA sequencing. The gut transcriptome analysis reveals several transcripts associated with hemoglobin digestion, such as serine, cysteine, aspartic proteases and metalloenzymes. The phylogenetic analysis on the peptidases confirmed that most of them are clustered with other tick genes. We recorded the presence a cathepsin O peptidase-coding transcript in ticks. The topology of the phylogenetic inferences, based on transcripts of inferred families of homologues, was similar to that of previous reports based on mitochondrial genome and nuclear rRNA sequences. We deposited 2,213 sequence of O. mimon to the public databases. Our findings may help towards better understanding of important argasid metabolic processes, such as digestion, nutrition and immunity.
Keywords:
Argasidae; transcriptome; gut; phylogeny; Ornithodoros mimon
Resumo
Ornithodoros mimon é um carrapato argasídeo parasita de morcegos, aves e marsupiais, além de ser bastante agressivo aos humanos. O conhecimento dos transcritos presentes no intestino dos carrapatos auxilia no entendimento do papel de moléculas vitais no processo de digestão e na relação parasito-hospedeiro, além de fornecer também informações sobre a evolução dos artrópodes hematófagos. Desta maneira, o presente estudo teve como objetivo conhecer e identificar as principais moléculas expressas no intestino de uma espécie de carrapato argasídeo após o repasto sanguíneo, através de uma análise transcritômica descritiva do intestino de fêmeas ingurgitadas de O. mimon, utilizando um sequenciamento de RNA de nova geração da plataforma 454. Além de inferir a relação filogenética de carrapatos através de um conjunto de dados transcritômicos. O transcriptoma do intestino revelou diversos transcritos associados com a digestão da hemoglobina, como proteinases das classes serino, cisteína, aspártica e metalo. Registramos a presença de um transcrito de uma cisteína peptidase do tipo catepsina O em carrapatos. A inferência filogenética baseada em conjunto de dados transcritos homólogos tem uma resolução topológica similar a de outros conjuntos de dados moleculares. Foram depositados no banco de dados gênico público 2213 transcritos de O. mimon. Os achados obtidos no presente estudo podem contribuir para compreensão dos importantes processos, como digestão, nutrição e imunidade dos carrapatos da família Argasidae, além de fornecer informações sobre a filogenia da ordem Ixodida.
Palavras-chave:
Argasidae; transcriptoma; intestino; filogenia; Ornithodoros mimon
Introduction
Family Argasidae comprises ticks popularly known as soft ticks. This family differs from the Ixodidae in relation to some morphological features (lack of a dorsal scutum and the ventral position of the capitulum in the nymphal and adult stages), biological features (several nymphal instars and fast feeding) and ecological characteristics (living in nests or caves, as well as buried in sand) (VIAL, 2009Vial L. Biological and ecological characteristics of soft ticks (Ixodida: Argasidae) and their impact for predicting tick and associated disease distribution. Parasite 2009; 16(3): 191-202. PMid:19839264.). Argasid ticks are hematophagous and their hosts comprise a wide variety of vertebrates (amphibians, reptiles, birds and mammals), including humans (HOOGSTRAAL, 1985Hoogstraal H. Argasid and nuttalliellid ticks as parasites and vectors. Adv Parasitol 1985; 24: 135-238. PMid:3904345.; BARROS-BATTESTI et al., 2013Barros-Battesti DM, Ramirez DG, Landulfo GA, Faccini JL, Dantas-Torres F, Labruna MB, et al. Immature argasid ticks: diagnosis and keys for Neotropical region. Rev Bras Parasitol Vet 2013; 22(4): 443-456. PMid:24473867., 2015Barros-Battesti DM, Landulfo GA, Luz HR, Marcili A, Onofrio VC, Famadas KM. Ornithodoros faccinii n. sp. (Acari: Ixodida: Argasidae) parasitizing the frog Thoropa miliaris (Amphibia: Anura: Cycloramphidae) in Brazil. Parasit Vectors 2015; 8: 268. PMid:25963542.).
Considering the uncertainty of the classification of the family Argasidae by the phylogenetic analysis of ticks (LABRUNA et al., 2008Labruna MB, Terassini FA, Camargo LM, Brandão PE, Ribeiro AF, Estrada-Peña A. New reports of Antricola guglielmonei and Antricola delacruzi in Brazil, and a description of a new argasid species (Acari). J Parasitol 2008; 94(4): 788-792. PMid:18576796., 2011Labruna MB, Nava S, Terassini FA, Onofrio VC, Barros-Battesti DM, Camargo LM, et al. Description of adults and nymph, and redescription of the larva, of Ornithodoros marinkellei (Acari:Argasidae), with data on its phylogenetic position. J Parasitol 2011; 97(2): 207-217. PMid:21506769.; NAVA et al., 2009Nava S, Guglielmone AA, Mangold AJ. An overview of systematics and evolution of ticks. Front Biosci 2009; 14: 2857-2877. PMid:19273240.; ESTRADA-PEÑA et al., 2006Estrada-Peña A, Venzal JM, González-Acuña D, Mangold AJ, Guglielmone AA. Notes on New World Persicargas ticks (Acari: Argasidae) with description of female Argas (P.) keiransi. J Med Entomol 2006; 43(5): 801-809. PMid:17017212., 2010Estrada-Peña A, Mangold AJ, Nava S, Venzal JM, Labruna M, Guglielmone AA. A review of the systematics of the tick family Argasidae (Ixodida). Acarologia 2010; 50(3): 317-333.; VENZAL et al., 2012Venzal JM, Nava S, Mangold AJ, Mastropaolo M, Casás G, Guglielmone AA. Ornithodoros quilinensis sp. nov. (Acari, Argasidae), a new tick species from the Chacoan region in Argentina. Acta Parasitol 2012; 57(3): 329-336. PMid:22875683.; BURGER et al., 2014Burger TD, Shao R, Labruna MB, Barker SC. Molecular phylogeny of soft ticks (Ixodida: Argasidae) inferred from mitochondrial genome and nuclear rRNA sequences. Ticks Tick Borne Dis 2014; 5(2): 195-207. PMid:24378079.), we have adopted the classical systematic proposed by Hoogstraal (1985)Hoogstraal H. Argasid and nuttalliellid ticks as parasites and vectors. Adv Parasitol 1985; 24: 135-238. PMid:3904345.. This classification, adopted by Guglielmone et al. (2010)Guglielmone AA, Robbins RG, Apanaskevich DA, Petney TN, Estrada-Peña A, Horak IG, et al. The Argasidae, Ixodidae and Nuttalliellidae (Acari: Ixodida) of the world: a list of valid species names. Zootaxa 2010; 2528: 1-28., considers the following genera as valid to family Argasidae: Antricola, Argas, Nothoaspis, Ornithodoros and Otobius.
Ticks present different strategies and mechanisms for feeding on host’s blood, such as presence of anticoagulants, antiplatelet and modulators of the immune system in saliva and/or presence of peptidases, carrier molecules and proteins of the innate immunity in the gut (CHMELAŘ et al., 2016Chmelař J, Kotál J, Karim S, Kopacek P, Francischetti IM, Pedra JH, et al. Sialomes and mialomes: a systems-biology view of tick tissues and tick–host interactions. Trends Parasitol 2016; 32(3): 242-254. PMid:26520005.). Thus, knowledge about the vital molecular constituents of ticks, such as genes and/or proteins, have become the target of several studies (CASTRO et al., 2016Castro MH, Klerk D, Pienaar R, Latif AA, Rees DJG, Mans BJ. De novo assembly and annotation of the salivary gland transcriptome of Rhipicephalus appendiculatus male and female ticks during blood feeding. Ticks Tick Borne Dis 2016; 7(4): 536-548. PMid:26830274.; CHMELAŘ et al., 2016Chmelař J, Kotál J, Karim S, Kopacek P, Francischetti IM, Pedra JH, et al. Sialomes and mialomes: a systems-biology view of tick tissues and tick–host interactions. Trends Parasitol 2016; 32(3): 242-254. PMid:26520005.; DÍAZ-MARTÍN et al., 2015Díaz-Martín V, Manzano-Román R, Obolo-Mvoulouga P, Oleaga A, Pérez-Sánchez R. Development of vaccines against Ornithodoros soft ticks: an update. Ticks Tick Borne Dis 2015; 6(3): 211-220. PMid:25802033.; HEEKIN et al., 2013Heekin AM, Guerrero FD, Bendele KG, Saldivar L, Scoles GA, Dowd SE, et al. Gut transcriptome of replete adult female cattle ticks, Rhipicephalus (Boophilus) microplus, feeding upon a Babesia bovis-infected bovine host. Parasitol Res 2013; 112(9): 3075-3090. PMid:23749091.; KOTSYFAKIS et al., 2015Kotsyfakis M, Schwarz A, Erhart J, Ribeiro JM. Tissue- and time-dependent transcription in Ixodes ricinus salivary glands and midguts when blood feeding on the vertebrate host. Sci Rep 2015; 5: 9103. PMid:25765539.; PERNER et al., 2016Perner J, Provazník J, Schrenková J, Urbanová V, Ribeiro JM, Kopáček P. RNA-seq analyses of the midgut from blood-and serum-fed Ixodes ricinus ticks. Sci Rep 2016; 6: 36695. PMid:27824139.; RIBEIRO et al., 2017Ribeiro JMC, Slovák M, Francischetti IMB. An insight into the sialome of Hyalomma excavatum. Ticks Tick Borne Dis 2017; 8(2): 201-207. PMid:28049606.; XU et al., 2016Xu XL, Cheng TY, Yang H, Liao ZH. De novo assembly and analysis of midgut transcriptome of Haemaphysalis flava and identification of genes involved in blood digestion, feeding and defending from pathogens. Infect Genet Evol 2016; 38: 62-72. PMid:26705239.). These studies have sought to ascertain and understand the roles of the molecules involved in the tick-host or tick-pathogen relationship. In addition, there is great interest in discovering new molecules with potential for use within biotechnology and pharmacology, which might help in developing new control methods (CHMELAŘ et al., 2016Chmelař J, Kotál J, Karim S, Kopacek P, Francischetti IM, Pedra JH, et al. Sialomes and mialomes: a systems-biology view of tick tissues and tick–host interactions. Trends Parasitol 2016; 32(3): 242-254. PMid:26520005.; DÍAZ-MARTÍN et al., 2015Díaz-Martín V, Manzano-Román R, Obolo-Mvoulouga P, Oleaga A, Pérez-Sánchez R. Development of vaccines against Ornithodoros soft ticks: an update. Ticks Tick Borne Dis 2015; 6(3): 211-220. PMid:25802033.; RIBEIRO et al., 2017Ribeiro JMC, Slovák M, Francischetti IMB. An insight into the sialome of Hyalomma excavatum. Ticks Tick Borne Dis 2017; 8(2): 201-207. PMid:28049606.). The evolutionary context of bloodsucking organisms is another prominent approach that has been studied, through making inferences about the evolution of genes and proteins, thereby helping to improve knowledge about the hematophagic behavior of the arthropods (MANS et al., 2008Mans BJ, Andersen JF, Francischetti IM, Valenzuela JG, Schwan TG, Pham VM, et al. Comparative sialomics between hard and soft ticks: implications for the evolution of blood-feeding behavior. Insect Biochem Mol Biol 2008; 38(1): 42-58. PMid:18070664.).
DNA sequencing technologies, together with bioinformatics tools, allow a broad view of the probable protein constituents of a tissue, by isolating the messenger RNA repertoire and sequencing of the cDNA (complementary DNA), leading to an increasing knowledge of the putative molecules expressed by a tissue, such as, salivary gland and midgut, from different species (CASTRO et al., 2016Castro MH, Klerk D, Pienaar R, Latif AA, Rees DJG, Mans BJ. De novo assembly and annotation of the salivary gland transcriptome of Rhipicephalus appendiculatus male and female ticks during blood feeding. Ticks Tick Borne Dis 2016; 7(4): 536-548. PMid:26830274.; CHMELAŘ et al., 2016Chmelař J, Kotál J, Karim S, Kopacek P, Francischetti IM, Pedra JH, et al. Sialomes and mialomes: a systems-biology view of tick tissues and tick–host interactions. Trends Parasitol 2016; 32(3): 242-254. PMid:26520005.; KOTSYFAKIS et al., 2015Kotsyfakis M, Schwarz A, Erhart J, Ribeiro JM. Tissue- and time-dependent transcription in Ixodes ricinus salivary glands and midguts when blood feeding on the vertebrate host. Sci Rep 2015; 5: 9103. PMid:25765539.; PERNER et al., 2016Perner J, Provazník J, Schrenková J, Urbanová V, Ribeiro JM, Kopáček P. RNA-seq analyses of the midgut from blood-and serum-fed Ixodes ricinus ticks. Sci Rep 2016; 6: 36695. PMid:27824139.; RIBEIRO et al. 2017Ribeiro JMC, Slovák M, Francischetti IMB. An insight into the sialome of Hyalomma excavatum. Ticks Tick Borne Dis 2017; 8(2): 201-207. PMid:28049606.; SCHWARZ et al., 2014Schwarz A, Tenzer S, Hackenberg M, Erhart J, Gerhold-Ay A, Mazur J, et al. A systems level analysis reveals transcriptomic and proteomic complexity in Ixodes ricinus midgut and salivary glands during early attachment and feeding. Mol Cell Proteomics 2014; 13(10): 2725-2735. PMid:25048707.; VALENZUELA, 2004Valenzuela JG. Exploring tick saliva: from biochemistry to ‘sialomes’ and functional genomics. Parasitology 2004; 129(Suppl): S83-S94. PMid:15938506.; XU et al., 2016Xu XL, Cheng TY, Yang H, Liao ZH. De novo assembly and analysis of midgut transcriptome of Haemaphysalis flava and identification of genes involved in blood digestion, feeding and defending from pathogens. Infect Genet Evol 2016; 38: 62-72. PMid:26705239.).
The gut, especially the midgut, is an essential organ for tick survival since it is the main responsible for digesting the blood meal and absorbing the nutrients produced through digestion (SONENSHINE, 1991Sonenshine DE. Biology of ticks. 1st ed. New York: Oxford University Press; 1991.). Furthermore, the midgut is the first tissue that microorganisms colonize, and these need to become established there before they can migrate to other organs and tissues (ANDERSON et al., 2008Anderson JM, Sonenshine DE, Valenzuela JG. Exploring the mialome of ticks: an annotated catalogue of midgut transcripts from the hard tick, Dermacentor variabilis (Acari: Ixodidae). BMC Genomics 2008; 9: 552. PMid:19021911.; KONGSUWAN et al., 2010Kongsuwan K, Josh P, Zhu Y, Pearson R, Gough J, Colgrave ML. Exploring the midgut proteome of partially fed female cattle tick (Rhipicephalus (Boophilus) microplus). J Insect Physiol 2010; 56(2): 212-226. PMid:19840806.). Unlike other blood-sucking arthropods, the digestion of blood feeding in ticks is intracellular, occurring within the digestive lysosomal vesicles (equipped with proteinases) of intestinal epithelial cells, resulting in the formation of oligopeptides, dipeptides and free amino acids, which are released to the cell cytoplasm and used for biological processes (SONENSHINE, 1991Sonenshine DE. Biology of ticks. 1st ed. New York: Oxford University Press; 1991.; SOJKA et al., 2013Sojka D, Franta Z, Horn M, Caffrey CR, Mares M, Kopácek P. New insights into the machinery of blood digestion by ticks. Trends Parasitol 2013; 29(6): 276-285. PMid:23664173.). Peptidases such as cathepsins, legumain, aspartic, metallo and serine proteinase are cited as the main proteolytic enzymes actives in the digestive machinery of hemoglobin (SOJKA et al., 2013Sojka D, Franta Z, Horn M, Caffrey CR, Mares M, Kopácek P. New insights into the machinery of blood digestion by ticks. Trends Parasitol 2013; 29(6): 276-285. PMid:23664173.). All these peptidases have been identified and characterized by biochemical, proteomic and transcriptomic studies in different tick species, such as Dermacentor variabilis (Say, 1821) (ANDERSON et al., 2008Anderson JM, Sonenshine DE, Valenzuela JG. Exploring the mialome of ticks: an annotated catalogue of midgut transcripts from the hard tick, Dermacentor variabilis (Acari: Ixodidae). BMC Genomics 2008; 9: 552. PMid:19021911.), Rhipicephalus microplus (Canestrini, 1888) (MENDIOLA et al., 1996Mendiola J, Alonso M, Marquetti MC, Finlay C. Boophilus microplus: multiple proteolytic activities in the midgut. Exp Parasitol 1996; 82(1): 27-33. PMid:8617328.; KONGSUWAN et al., 2010Kongsuwan K, Josh P, Zhu Y, Pearson R, Gough J, Colgrave ML. Exploring the midgut proteome of partially fed female cattle tick (Rhipicephalus (Boophilus) microplus). J Insect Physiol 2010; 56(2): 212-226. PMid:19840806.; HEEKIN et al., 2013Heekin AM, Guerrero FD, Bendele KG, Saldivar L, Scoles GA, Dowd SE, et al. Gut transcriptome of replete adult female cattle ticks, Rhipicephalus (Boophilus) microplus, feeding upon a Babesia bovis-infected bovine host. Parasitol Res 2013; 112(9): 3075-3090. PMid:23749091.), Ixodes ricinus Linneaus, 1758 (SOJKA et al., 2007Sojka D, Hajdušek O, Dvořák J, Sajid M, Franta Z, Schneider EL, et al. IrAE–An asparaginyl endopeptidase (legumain) in the gut of the hard tick Ixodes ricinus. Int J Parasitol 2007; 37(7): 713-724. PMid:17336985., 2008Sojka D, Franta D, Horn M, Hajdušek O, Caffrey CR, Mares M, et al. Profiling of proteolytic enzymes in the gut of the tick Ixodes ricinus reveals an evolutionarily conserved network of aspartic and cysteine peptidases. Parasit Vectors 2008; 1(1): 7. PMid:18348719., 2013Sojka D, Franta Z, Horn M, Caffrey CR, Mares M, Kopácek P. New insights into the machinery of blood digestion by ticks. Trends Parasitol 2013; 29(6): 276-285. PMid:23664173.; HORN et al., 2009Horn M, Nussbaumerová M, Sanda M, Kovárová Z, Srba J, Franta Z, et al. Hemoglobin digestion in blood-feeding ticks: mapping a multipeptidase pathway by functional proteomics. Chem Biol 2009; 16(10): 1053-1063. PMid:19875079.; FRANTA et al., 2010Franta Z, Frantová H, Konvičková J, Horn M, Sojka D, Mareš M, et al. Dynamics of digestive proteolytic system during blood feeding of the hard tick Ixodes ricinus. Parasit Vectors 2010; 3: 119. PMid:21156061.; KOTSYFAKIS et al., 2015Kotsyfakis M, Schwarz A, Erhart J, Ribeiro JM. Tissue- and time-dependent transcription in Ixodes ricinus salivary glands and midguts when blood feeding on the vertebrate host. Sci Rep 2015; 5: 9103. PMid:25765539.; PERNER et al., 2016Perner J, Provazník J, Schrenková J, Urbanová V, Ribeiro JM, Kopáček P. RNA-seq analyses of the midgut from blood-and serum-fed Ixodes ricinus ticks. Sci Rep 2016; 6: 36695. PMid:27824139.), Haemaphysalis longicornis Neumann, 1901 (KOH et al., 1990Koh K, Shiraishi S, Uchida TA. Digestion of bloodmeal protein after detachment in nymphal Haemaphysalis longicornis. J Fac Ag. Kyushu Univ 1990; 34(3): 241-246.; BOLDBAATAR et al., 2006Boldbaatar D, Sikalizyo Sikasunge C, Battsetseg B, Xuan X, Fujisaki K. Molecular cloning and functional characterization of an aspartic protease from the hard tick Haemaphysalis longicornis. Insect Biochem Mol Biol 2006; 36(1): 25-36. PMid:16360947.), Haemaphysalis flava Neumann 1897 (XU et al., 2016Xu XL, Cheng TY, Yang H, Liao ZH. De novo assembly and analysis of midgut transcriptome of Haemaphysalis flava and identification of genes involved in blood digestion, feeding and defending from pathogens. Infect Genet Evol 2016; 38: 62-72. PMid:26705239.), Ornithodoros tholozani (Laboulbène and Mégnin, 1882) (AKOV et al., 1976Akov S, Samish M, Galun R. Protease activity in female Ornithodoros tholozani ticks. Acta Trop 1976; 33(1): 37-52. PMid:13632.), Ornithodoros moubata (Murray, 1877) (GRANDJEAN, 1984Grandjean O. Blood digestion in Ornithodoros moubata Murray Sensu stricto Walton (Ixodoidea: Argasidae) females. I. Biochemical changes in the midgut lumen and ultrastructure of the midgut cell, related to intracellular digestion. Acarologia 1984; 25(2): 147-165.) and Ornithodoros erraticus (Lucas, 1849) (OLEAGA et al., 2015Oleaga A, Obolo-Mvoulouga P, Manzano-Romá R, Pérez-Sánchez R. Midgut proteome of an argasid tick, Ornithodoros erraticus: a comparison between unfed and engorged females. Parasit Vectors 2015; 8: 525. PMid:26459090.).
Recently, Sojka et al. (2016)Sojka D, Pytelková J, Perner J, Horn M, Konvicková JS, Schrenková J, et al. Multienzyme degradation of host serum albumin in ticks. Ticks Tick Borne Dis 2016; 7(4): 604-613. PMid:26724897. compared the proteolytic degradation of serum albumin and hemoglobin within the intestinal cells of I. ricinus and reported differences and similarities between the processes of digestion. Both protein compounds of the blood are degraded under acidic conditions by a set of cysteine and aspartic proteinases, being albumin predominantly digested by the cathepsins.
Nevertheless even with all the current knowledge about the constituents of the gut, mainly the midgut, little is known about the true role and activity of intestinal proteins from ticks (DÍAZ-MARTÍN et al., 2015Díaz-Martín V, Manzano-Román R, Obolo-Mvoulouga P, Oleaga A, Pérez-Sánchez R. Development of vaccines against Ornithodoros soft ticks: an update. Ticks Tick Borne Dis 2015; 6(3): 211-220. PMid:25802033.; OLEAGA et al., 2015Oleaga A, Obolo-Mvoulouga P, Manzano-Romá R, Pérez-Sánchez R. Midgut proteome of an argasid tick, Ornithodoros erraticus: a comparison between unfed and engorged females. Parasit Vectors 2015; 8: 525. PMid:26459090.). This is evidenced by the fact that there are scarce transcriptomic studies of gut tissue of ticks, especially with soft ticks. The gut transcriptome of the species D. variabilis has been explored, thus resulting in identification of 82 transcripts directly involved in the blood meal digestion process (ANDERSON et al., 2008Anderson JM, Sonenshine DE, Valenzuela JG. Exploring the mialome of ticks: an annotated catalogue of midgut transcripts from the hard tick, Dermacentor variabilis (Acari: Ixodidae). BMC Genomics 2008; 9: 552. PMid:19021911.). A gut transcriptome study on females of R. microplus, in which the expression profiles of some genes of infected and non-infected specimens with those of the protozoon Babesia bovis were compared, showed thirty-three genes with higher levels of expression in gut samples from female ticks feeding on a B. bovis-infected calf (HEEKIN et al., 2013Heekin AM, Guerrero FD, Bendele KG, Saldivar L, Scoles GA, Dowd SE, et al. Gut transcriptome of replete adult female cattle ticks, Rhipicephalus (Boophilus) microplus, feeding upon a Babesia bovis-infected bovine host. Parasitol Res 2013; 112(9): 3075-3090. PMid:23749091.). Schwarz et al. (2014)Schwarz A, Tenzer S, Hackenberg M, Erhart J, Gerhold-Ay A, Mazur J, et al. A systems level analysis reveals transcriptomic and proteomic complexity in Ixodes ricinus midgut and salivary glands during early attachment and feeding. Mol Cell Proteomics 2014; 13(10): 2725-2735. PMid:25048707. studied the transcriptome and proteome of the midgut and salivary gland of I. ricinus and identified 217 and 110 genes expressed exclusively in the midgut and in the salivary gland, respectively. Recently, midgut transcriptome analysis of nymphs and adult of I. ricinus revealed several enzymes associated with protein, carbohydrate and lipid digestion; transporters and channels that might be associated with nutrient uptake; and immunity-related transcripts including antimicrobial peptides (KOTSYFAKIS et al., 2015Kotsyfakis M, Schwarz A, Erhart J, Ribeiro JM. Tissue- and time-dependent transcription in Ixodes ricinus salivary glands and midguts when blood feeding on the vertebrate host. Sci Rep 2015; 5: 9103. PMid:25765539.). Xu et al. (2016)Xu XL, Cheng TY, Yang H, Liao ZH. De novo assembly and analysis of midgut transcriptome of Haemaphysalis flava and identification of genes involved in blood digestion, feeding and defending from pathogens. Infect Genet Evol 2016; 38: 62-72. PMid:26705239. analyzed the midgut transcriptome of H. flava and identified transcripts associated with metabolic processes, suggesting that they were involved in the blood meal digestion, such as peptidases, inhibitors peptidases, phospholipase, hydrolases and immunity-related proteins.
However, for argasid ticks, there are not currently transcriptomic studies about the gut. Therefore, more research is needed in this field, especially in relation to the members of the Argasidae family. Díaz-Martín et al. (2015)Díaz-Martín V, Manzano-Román R, Obolo-Mvoulouga P, Oleaga A, Pérez-Sánchez R. Development of vaccines against Ornithodoros soft ticks: an update. Ticks Tick Borne Dis 2015; 6(3): 211-220. PMid:25802033. and Oleaga et al. (2015)Oleaga A, Obolo-Mvoulouga P, Manzano-Romá R, Pérez-Sánchez R. Midgut proteome of an argasid tick, Ornithodoros erraticus: a comparison between unfed and engorged females. Parasit Vectors 2015; 8: 525. PMid:26459090. warn of the need for more transcriptomic studies of gut tissue from soft ticks, for a better understanding of the proteins and genes of biological and pharmacological interest.
Thus, in the present study, we analyzed the gut transcriptome of engorged females of Ornithodoros mimon Kohls, Clifford & Jones (1969) (Acari: Argasidae) through construction of cDNA libraries and next-generation sequencing (NGS), in order to ascertain and describe the main molecules expressed in the gut after the blood meal. In addition, we inferred the phylogenetic relation of ticks through a set of transcriptomic data.
Materials and Methods
Ticks
The ticks used in this study came from a colony of O. mimon that is maintained in the Parasitology Laboratory of the Butantan Institute. This colony was started in 2008, through collection of live specimens from a home in the city of Araraquara, whose residents had injuries resulting from bites. In order to obtain specimens for gut dissection and extraction, sixty females of O. mimon were fed on rabbits (Oryctolagus cuniculus) of New Zealand breed. The weight of engorged females was 3-4 times greater than before feeding (mean weights of unfed and engorged females were 7.5 ± 2 (4.7-10.6) and 21.6 ± 3.3 (16-25) mg, respectively).
The use of rabbits for artificial feeding of ticks was approved and permitted by the Ethics Committee for Animal Use of the Butantan Institute, under protocol no. 925/12. This study was also approved by the Law of Management of Genetic Heritage (CGEN-CNPq No. 010714/2013-8).
Tissue collection and cDNA library construction
After the engorged females had fed, they were dissected at two times (24 h and 48 h after the meal) in order to collect the gut tissue. However, the samples were considered as single pool of gut tissue from engorged females. During the dissection process, phosphate-buffered saline pH 7.4 (PBS) solution was used to clean, hydrate and view the organs. The gut samples were washed in PBS to remove all the luminal contents (vertebrate host blood) from the gut. After cleaned, samples were incubated in cryotubes containing RNA stabilization solution (RNAlater®; Ambion) at 4 °C overnight. After, the samples were removed from RNAlater before storage at -80 °C, and kept there until use. Gut sample mRNA extraction was performed directly from dissected and frozen tissues, using the Dynabeads mRNA DIRECT kit (Invitrogen), in accordance with the manufacturer's instructions. The mRNA extracted from the intestine was quantified through use of fluorescent ligands of the Quant-iT RiboGreen RNA reagent kit (Invitrogen), in accordance with the manufacturer's guidelines. This ribonucleic acid served as a template for double-stranded (ds) complementary DNA (cDNA) synthesis and unique library construction based in the mixture of the gut tissue. The sequencing was carried out by means of pyrosequencing, using the GS-454 Junior system (Roche), and the whole process was performed using kits, in accordance with the manufacturer's instructions for the equipment.
Bioinformatics
The sequences were assembled using the CLC genomics software (CLCBio). Initially, the sequence data was put through a filtration process to remove low-quality, small-sized (less than 40 bp) and ribosomal sequences (rRNA). For removal of rRNA, first we created a database composed of Acari rRNA sequences from data available in GenBank, and then used the CLC genomics software to compare our sequence data with the Acari rRNA database, removing those that showed similarity.
Small-sized (reads below length 40 bp) and low-quality sequences (quality scores below 0.05) were using CLC filtering analyses. De novo assembly of contiguous consensus sequences (contigs) was performed using the filtered sequences and default assembly parameters (word size of 20 nucleotides and minimum contig length of 200 bp). The contigs represented the consensus obtained through grouping (clustering) sequences that related to overlapping regions of the transcriptome. The assembled transcripts were named using the designation Contig followed by an identification number based on the order of the assembly of the CLC genomics program. These contigs were analyzed and compared with the non-redundant (NR) GenBank database, using informatics tools for automated BLAST (BLASTX) searches (ALTSCHUL et al., 1990Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol 1990; 215(3): 403-410. PMid:2231712.) and BLAST2GO (B2G) searches with default parameters (CONESA et al., 2005Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005; 21(18): 3674-3676. PMid:16081474.). Other databases, such as UniprotKB (UniProt Knowledgebase), MGI (Mouse Genome Informatics), ZFIN (Zebrafish Model Organism Database), RGD (Rat Genome Database) and GO (Gene Ontology Consortium) also were used in the BLAST search. The automated functional annotation of the sequences was performed in three phases: search by homology (BLAST); mapping to collect the GO terms associated with the BLAST hits (e-value 10-6); and annotation to assign the information to sequences (CONESA et al., 2005Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005; 21(18): 3674-3676. PMid:16081474.). The annotated transcripts were grouped using the B2G software in accordance with their biological function assignment (CONESA et al., 2005Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005; 21(18): 3674-3676. PMid:16081474.). Predictive cell subcellular localization in the transcripts was also performed using the B2G software, which used the SignalP algorithm (NIELSEN et al., 1997Nielsen H, Engelbrecht J, Brunak S, von Heijne G. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng 1997; 10(1): 1-6. PMid:9051728.) to identify presence of the signal peptide prediction (SignalP) and the TMHMM algorithm (KROGH et al., 2001Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 2001; 305(3): 567-580. PMid:11152613.) to verify of transmembrane regions (TMHMM).
After the automated search, we manually analyzed all contigs and regrouped them into categories according to the functions and biological process that had been assigned to them. We identified the putative conserved domain protein by means of the tool Conserved domains within a protein or nucleotide coding sequence (NCBI, 2017National Center for Biotechnology Information – NCBI. Search for Conserved Domains within a protein or coding nucleotide sequence [online]. Bethesda: NCBI; 2017 [cited 2017 Mar 1]. Available from: http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi
http://www.ncbi.nlm.nih.gov/Structure/cd...
; MARCHLER-BAUER & BRYANT, 2004Marchler-Bauer A, Bryant SH. CD-Search: protein domain annotations on the fly. Nucleic Acids Res 2004; 32(S2): 327-331. PMid:15215404.). The peptidase genes were classified and blasted in accordance with the MEROPS database, in order to identify the type of catalytic class (RAWLINGS & BARRETT, 1993Rawlings ND, Barrett AJ. Evolutionary families of peptidases. Biochem J 1993; 290(1): 205-218. PMid:8439290.). We used the measurement Reads per kilobase per million (RPKM) mapped reads to compare expression levels among the transcripts.
The raw data was deposited to the Sequence Read Archives (SRA) of the National Center for Biotechnology Information (NCBI) under bioproject number PRJNA310605, biosample SAMN04452891 and run SRR3146437. The contigs of O. mimon were deposited to the Transcriptome Shotgun Assembly Sequence Database (TSA) portal NCBI. This Transcriptome Shotgun Assembly project has been deposited at DDBJ/ENA/GenBank under the accession GEIB00000000. The version described in this paper is the first version, GEIB01000000. The contigs received the version accession number from GEIB1000001.1 to GEIB1002213.1.
Phylogeny
Phylogenetic analyses were performed in order to infer the relationships between the major cysteine peptidases deduced (cathepsins and midgut cysteine proteases) of the transcriptome with the sequences deposited in peptidase databases; and also to verify the usefulness of the whole dataset of transcripts for inferring evolutionary relationships among ticks.
For the first phylogenetic inference a set of metazoan peptidase sequences obtained from public databases and from our transcriptome (cysteine peptidases) was used for the phylogenetic analysis. Multiple alignment was conducted by means of the MAFFT algorithm (KATOH et al., 2002Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 2002; 30(14): 3059-3066. PMid:12136088., 2005Katoh K, Kuma K, Toh H, Miyata T. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res 2005; 33(2): 511-518. PMid:15661851.) with default parameters. IQTree v1.3.10 (NGUYEN et al., 2015Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 2015; 32(1): 268-274. PMid:25371430.) was used for maximum likelihood (ML) inferences, with the option of testing the best protein model on demand, and afterwards letting the program use this best model to infer the ML tree. The ultrafast bootstrap method (which was set at 1,000 cycles) was used as branch support. The tree was viewed and edited in the FigTree v.1.4.1 software. The group of the aspartic peptidase was used as an outgroup in the phylogenetic inference of cysteine peptidase.
In the second phylogenetic inference we searched in public databases (NCBI) for transcript sequences of tick species from different sources, such as proteins, EST and nucleotides. In total, we found and selected 267,488 sequences from 20 tick species and one mite species (Metaseiulus occidentalis). Coding sequences (CDS) for DNA subjects were extracted using an automated pipeline that searched and obtained CDS from the larger open reading frames of the nucleotide sequences and contigs. These multiple sequences were added to the transcripts of O. mimon and served as input files to the Get Homologues software package (CONTRERAS-MOREIRA & VINUESA, 2013Contreras-Moreira B, Vinuesa P. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl Environ Microbiol 2013; 79(24): 7696-7701. PMid:24096415.), which calculated the number of homologous families using the OMCL option (which uses the OrthoMCL algorithm, LI et al., 2003Li L, Stoeckert CJ Jr, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 2003; 13(9): 2178-2189. PMid:12952885.). For each gene tree, ML was estimated using IQTree (with automatic evolutionary model selection, as mentioned above), each with 1,000 ultrafast bootstraps. Branches with less than 70% support were collapsed using TreeCollapseCL4 (HODCROFT et al., 2014Hodcroft E, Hadfield JD, Fearnhill E, Phillips A, Dunn D, O’Shea S, et al. The contribution of viral genotype to plasma viral set-point in HIV infection. PLoS Pathog 2014; 10(5): e1004112. PMid:24789308.), in order to decrease the likelihood of false positives (i.e. clades that did not really exist).
The tick species tree was inferred using the MulRF software package, which is a tree that minimizes the modified Robinson-Foulds (RF) distance for all input gene trees (CHAUDHARY et al., 2013Chaudhary R, Burleigh JG, Fernández-Baca D. Inferring species trees from incongruent multi-copy gene trees using the Robinson-Foulds distance. Algorithms Mol Biol 2013; 8(1): 28. PMid:24180377., 2015Chaudhary R, Fernández-Baca D, Burleigh JG. MulRF: a software package for phylogenetic analysis using multi-copy gene trees. Bioinformatics 2015; 31(3): 432-433. PMid:25273112.). MulRF infers species trees from multi-copy gene trees using a generalization of RF topological distance, and it works by minimizing the total RF distance from the input multi-labeled trees, being relatively accurate in spite of evolutionary processes affecting single gene trees such as gene duplication and loss, deep coalescence, or lateral gene transfer (given our dataset, the most pervasive are gene duplication and loss). MulRF is able to analyze datasets in which some taxa are absent and/or others are duplicated per gene tree. Its power lies in not assuming any specific evolutionary process during the analysis (i.e. incomplete lineage sorting, gene duplication and/or gene loss).The MulRF method produces more accurate species trees than other gene tree parsimony approaches (CHAUDHARY et al., 2013Chaudhary R, Burleigh JG, Fernández-Baca D. Inferring species trees from incongruent multi-copy gene trees using the Robinson-Foulds distance. Algorithms Mol Biol 2013; 8(1): 28. PMid:24180377.). Nevertheless, because it works like a consensus tree, no nodal supports are available, so this issue must be overcome beforehand; we did this by collapsing branches with less than 70% support in each gene tree using TreeCollapseCL4 (HODCROFT et al., 2014Hodcroft E, Hadfield JD, Fearnhill E, Phillips A, Dunn D, O’Shea S, et al. The contribution of viral genotype to plasma viral set-point in HIV infection. PLoS Pathog 2014; 10(5): e1004112. PMid:24789308.), as mentioned above.
Results
cDNA sequencing
A total of 90,838 reads, with an average size of 420.76 bp, was obtained through pyrosequencing of the gut cDNA of O. mimon from the unique library. Reads of small size and low quality, and those with sequences homologous to ribosomal genes were discarded, thus leaving 87,666 reads. A total of 5,457 contigs were assembled using 44,912 of the sequences (51.2% of the reads obtained); the remaining 42,754 reads were considered to be singlets or singletons. We discarded contigs that had been assembled from fewer than five reads, and thus the number of initial contigs assembled was reduced to 2,235. For the NCBI were deposited 2,213 assembled sequences (contigs), because 22 contigs not passed by the submission processing of TSA.
Automated annotation: function, gene ontology and cell sublocalization
Out of the 2,235 sequences, 1,729 contigs (77.40%) presented BLASTX hits (Figure 1), while 506 (22.60%) did not present similarity with any database sequences. A total of 177 contigs were identified by means of BLASTX, but they did not present GO hits in relation to the functional mapping terms. The contigs that showed relevant similarity hits in BLAST and in GO totaled 1,552 annotated complete sequences. The contigs of O. mimon presented similarity to sequences of mites, mammals, mollusks, insects and other organisms deposited in the databases, but mainly to other tick sequences, such as Ixodes scapularis Say 1821, I. ricinus, R. microplus, D. variabilis, Amblyomma variegatum (Fabricius, 1794), H. longicornis, Argas monolakensis Schwan, Corwin and Brown, 1992, O. moubata, Ornithodoros coriaceus Koch 1844, Ornithodoros capensis Neumann, 1901 and Ornithodoros parkeri Cooley, 1936. Most of the GO terms were retrieved from UniProtKB and MGI.

Summary results from BLAST (BLASTX hits) and annotation in the BLAST2GO (B2G). The sequences that presented BLASTX hit were represented in the With BLAST hit group. Annotated complete comprises the group of sequences that showed relevant similarity hits in BLAST and in Gene Ontology (GO). Without GO comprises the sequences that were identified by means of BLASTX, but they did not present GO hits in relation to the functional mapping terms. Without BLAST hit is the group of sequences that did not present similarity with any database sequences.
Functional assignment
Catalytic, binding and transporter activity were the most representative functions, with 780, 709 and 106 contigs, respectively. The remainder of the annotated contigs was grouped into other functional activities, such as structural activity, antioxidant activity, molecular function regulator and electron carrier (Figure 2).

Principal functional activities assigned to the contigs. The annotated transcripts (contigs) were grouped in different functional activities in accordance with their biological function assignment.
Predictive cell sublocalization and categorization of contigs
As a result of predictive cell sublocalization, 18.2% (406) of the contigs were categorized as belonging to the secretory pathway and 16% (359) as potential integral membrane proteins. Among the annotated contigs, the main GO terms for localization of cell components were cell, organelle and membrane with 718, 552 and 376 sequences, respectively.
All the contigs were grouped into 31 categories, according to the functions and biological processes that had been assigned to them (Table 1). Catalytic activity, transporter channels and unknown categories were the most representative of these, accounting for 17.45% (390), 11% (246) and 37% (825) of the sequences, respectively (Table 1). These categories also reported the highest accumulated levels of RPKM. A total of 825 contigs were grouped in the category of unknown function, because these sequences did not present BLASTX and GO hits with any sequence of databases, or because they showed similarity to hypothetical protein sequences. Other categories, such as energy metabolism (2.8%), nuclear regulation (2.4%), oxidative metabolism (3%), peptidase (3%), cytoskeleton (2.3%), transcription mechanism (2.8%) and translation mechanism (2.7%) were also represented, although with smaller numbers of associated transcripts.
Categorization of the transcripts of female gut of O. mimon, according to functional and biological assignment. In bold are highlighted the categories of higher-level expression.
The distribution of the reads into each category was similar to the proportions of contig distribution and the accumulated RPKM levels in the categories, such that 27%, 14% and 12% of the reads belonged to the “unknown”, catalytic activity and transporter channel groups, respectively (Table 1).
Contigs with the highest RPKM level
Several gut transcripts of O. mimon with high RPKM are enzymes encoded by mitochondrial genes (cytochrome oxidase, ATP synthase and dehydrogenase), histones, Hsp90, β-tubulin, elongation factor, actin, beta 2c, ribosomal proteins and cyclophilin. The functions of these transcripts are required for cell activities that are essential for existence of a cell, such as replication, transcription, nuclear export, translation, mitosis, energy metabolism, nuclear regulation and others. Fifteen contigs with high RPKM levels were grouped in the unknown category and some these transcripts exhibited conserved domains or had hits relating to hypothetical proteins that lacked functional annotation, while others did not have hit any sequence databases.
Transcripts associated with digestion of the blood meal and nutrient uptake
Transcripts of O. mimon potentially involved with gut functions (Table 2), such as digestion, uptake, detoxification and immunity, will be highlighted in the next two sections: Digestive enzymes; and other transcripts associated with the functional activities of the gut.
Digestive enzymes
We identified 103 digestive transcripts, which were divided into three categories: peptidases (67), lipid metabolism (17) and carbohydrate metabolism (19) (Table 1). Possible participation in the metabolic processes of lipids, carbohydrates and proteins was assigned to these enzymes.
Peptidases
We found cDNA peptidases belonging to the serine, cysteine, aspartic and metallo classes, which totaled 67 protease-coding genes. For all these contigs, the proteolytic function was assigned.
Cysteine peptidases were the most abundant category, with 32 transcripts (48% of the peptidases). Genes encoding for caspases, calpains, cathepsins and legumains constituted the cysteine peptidase group. Participation in mediated programmed cell death (apoptosis) was attributed to the O. mimon caspase and apoptotic cysteine genes, because these contigs exhibited conserved caspase and cysteine aspartase catalytic domains (cd00032, pfam00656 and smart00115). Transcripts of calpains exhibited the conserved protein domain cd00044, which led us to characterize them as calcium-dependent cytoplasmic cysteine proteinase. These proteases have different functions, such as cytoskeletal remodeling, cell differentiation, apoptosis and signal transduction.
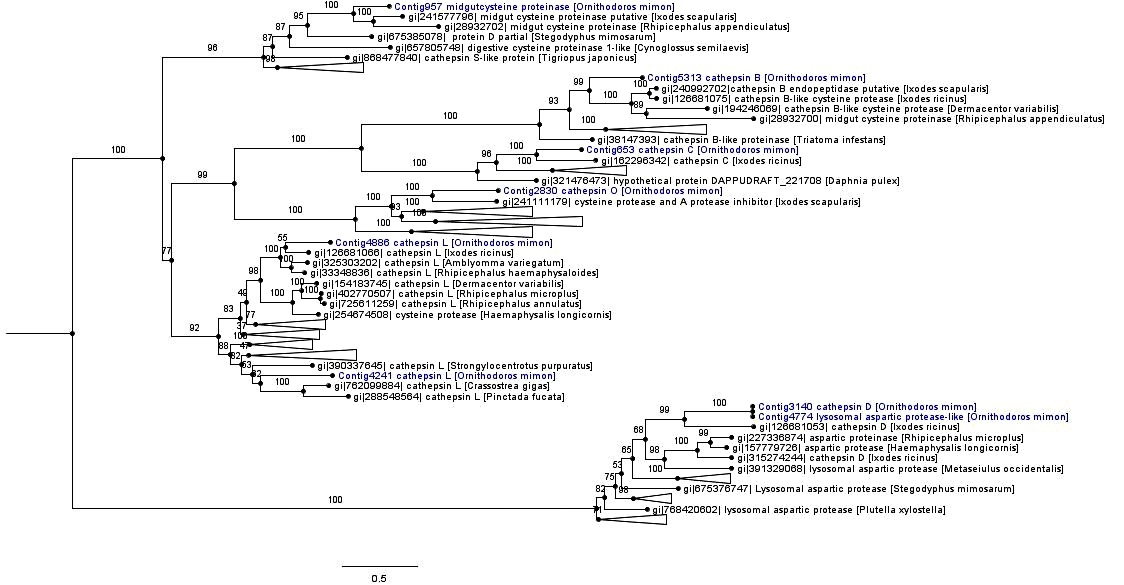
The cathepsins group was represented by cysteine peptidases B, C, L and O. The enzyme code (E.C) 3.4.22.1, which was assigned to Contig5313, and the presence of cathepsin B conserved catalytic domain (cd02620 and pfam00112) indicate that this contig is a protease that belongs to the papain cysteine endopeptidase family (Table 2). The transcriptome of engorged females of O. mimon was enriched for five cathepsin B peptidase-coding transcripts, of which only one (Contig5218) showed a predictive signal for peptide secretion (SignalP). Two genes coding for cathepsin L (Contig4241 and Contig4886) also showed prediction of secretion (SignalP). The Contig4241 transcript presented the best hit in relation to cathepsin L of Strongylocentrotus purpuratus Brandt, 1835 (Echinoida: Strongylocentrotidae: sea urchin), and it did not have any similarity to tick cathepsin L. Differently, the Contig4886 transcript displayed hits mainly in relation to cathepsin L of ticks. We identified a single full-length cDNA of cathepsin C (Contig653) by means of BLASTX (Table 2).
We identified a single cathepsin O enzyme-coding transcript (Contig2830), which had higher similarity to a cysteine protease of I. scapularis and to cathepsin O-like cysteine peptidase of scorpions (Tityus serrulatus) (Table 2). This contig displayed the papain-family peptidase C1A (cd002248, pfam00112 and COG4870) conserved domain, which classified it as a cysteine endopeptidase belonging to the C1 family and C1A subfamily. The protein coded by this contig (JAR87535.1) has a conserved motif of 12 amino acid residues (QLNCGACWAFST) that is characteristic of the active site of eukaryotic thiol (cysteine) proteases. In the alignment of cathepsin O sequences we can observe six identical residues in the conserved motif between the species (Figure 3).

Multiple alignment to compare the cathepsin O-like of O. mimon with selected cathepsin O sequences that showed hits in the BLAST search, highlighting (black rectangle) the conserved motif of eukaryotic thiol (cysteine) protease active site of cathepsin O. These conserved motifs of the protein coded have 12 amino acid residues, and the asterisk (*) represents the conserved peptides residues between the protein coded sequences. Alignments were conducted using MAFFT algorithm with default parameters.
The Contig957 contig showed 66% identification with the putative midgut cysteine proteinase of I. scapularis (Table 2) and exhibited papain-family cysteine protease conserved domain (cd02248 and pfam00112). We found four cDNA molecules for legumain-coding transcripts (Contig5200, Contig4866, Contig1966 and Contig5258). All of these legumains exhibited peptidase C13 family conserved domain (pfam01650), which defined them as asparaginyl peptidases (Table 2).
Thirteen transcripts were correlated with the functional activity of serine endopeptidase, according to sequence similarity, and included in the serine group. Among these, tick serine proteinase, chymotrypsin, lysosomal protein, carboxypeptidase and other serine proteinases can be highlighted (Table 2). The Contig5086 transcript is a tick serine proteinase, which presented signal peptide prediction and a conserved protein domain of trypsin. The Contig3496 and Contig4814 transcripts contained the serine carboxypeptidase conserved domain (pfam00450), and thus, the functional activity of carboxypeptidase was assigned to them (Table 2). Two well-expressed cDNA molecules coding for chymotrypsins (Contig4026 and Contig4983) were identified and had similarity to serine protease-like protein of O. moubata. These chymotrypsin enzyme-coding transcripts exhibited a conserved domain of the catalytic site from serine protease-like trypsin (cd00190, pfam00089 and smart00020) and presented the enzymatic code (EC) 3.4.21.0, which classified it as a hydrolase that acts on peptide bonds.
The metallopeptidase genes were composed of endopeptidases and exopeptidases, such as oligopeptidase, dipeptidase, dipeptidyl peptidase and aminopeptidase, representing 28% (19 contigs) of the intestinal peptidases. Five cDNA molecules with aminopeptidase enzyme-coding (Contig621, Contig5173, Contig5085, Contig4027 and Contig5249) had EC 3.4.11.1 and a GO functional hit relating to exopeptidase, as well as exhibiting an aminopeptidase conserved domain (pfam05195) (Table 2). On the other hand, the Contig3475 and Contig2224 are metalloendopeptidases to which EC 3.4.24 was attributed (Table 2). Contig2690 and Contig3707 presented the type M19 metallopeptidase domain and were classified as dipeptidyl peptidase.
We identified three aspartic endopeptidase protease-coding transcripts (Contig3140, Contig4774 and Contig3706). The Contig3140 transcript was identified as cathepsin D and showed a conserved catalytic aspartic acid domain. This transcript presented signal peptide (SignalP) and transmembrane region (TMHMM) prediction. Contig4774 and Contig3706 were identified as lysosomal aspartic protease-like and aspartic protease, respectively. All these transcripts had similarity mainly to aspartic proteinases of ticks, such as I. ricinus and H. longicornis.
Carbohydrate metabolism
The carbohydrate metabolism category was composed of 19 contigs that represented genes for chitinase, galactosidase, maltase, fucosidase, phosphatase and glycosidase. Two contigs, Contig4231 and Contig4692, exhibited a chitinase conserved domain (cd02872 and COG3325) and EC 3.2.1.14, which classified them as genes coding for chitinase proteins. The Contig4231 presented SignalP, while Contig4692 showed TMHMM. Three cDNA molecules (Contig571, Contig4163 and Contig3170) with possible functions relating to intestinal maltase were identified in the transcriptome of O. mimon (Table 2). These contigs presented the catalytic α-amylase conserved domain (cd11328), which is found in maltase proteins.
The transcripts Contig79 and Contig3343 are predicted to code for enzymes that metabolize fructose, because they exhibited a fructose-bisphosphate-aldolase conserved domain (cd00344 and cd00354). The E.C. 4.1.2.13 assigned to Contig79 and E.C. 3.1.3.11 to Contig3343, classified them as fructose-bisphosphate-aldolase and fructose-bisphosphate, respectively. The functional activity of α-glycosidase and the E.C. 3.2.1.20 were assigned to the Contig4153 and Contig3020, thus identifying them as glycosidase genes. The Contig39 transcript was identified as a glycogen phosphorylase exhibiting the E.C. 2.4.1.1. The conserved domain of α-l-fucosidase was identified in two cDNA molecules (Contig103 and Contig5179), representing the intestinal fucosidase of O. mimon. Glucosyltransferase, α-galactosidase, hexosaminidase and aminotransferase comprise the remaining carbohydrases.
Lipid metabolism
The lipid metabolism category was composed mainly of lipase, phospholipase, reductase, transferase, phosphodiesterase and ligase. All these transcripts were associated with the cell process of lipid metabolism. We identified a single phospholipase b enzyme-coding transcript (Contig1795), which showed best hit with a phospholipase of Limulus polyphemus (Xiphosura: Limulidae) (Lineu, 1758) (Table 2). Another transcript of lipase (Contig2581) had similarity to putative hormone-sensitive lipase hsl, partial of Amblyomma aureolatum Pallas, 1772 (Table 2).
Other transcripts associated with the functional activities of the gut
In this section, we highlight the transcripts of the categories of peptidase inhibitors, immunity, oxidant metabolism and transporter channels, which are associated with the functions of the gut.
Peptidase inhibitor
Seventeen transcripts were grouped in the peptidase inhibitor category (Table 1). Serine protease inhibitor (serpin), cystatins and Kunitz domain inhibitors were the most abundant. The putative conserved domain of serpin (cd00172 and pfam00079) was identified in 12 contigs. Contig4823 had best hit with serine protease inhibitor 5 RmS5 of R. microplus (Table 2). Five transcripts exhibited the Kunitz domain of serpin (cd0019, pfam00014 and smart00131). Of these, only Contig3309 and Contig3457 presented prediction of secretion. The Contig3309 transcript, identified as venom protease inhibitor precursor, displayed hits of 48 to 58% identification with Kunitz inhibitor of snakes. One of these peptidase inhibitors (Contig3184) exhibited macroglobulin conserved domains (cd02897, pfam07677 and pfam07678) and showed its best similarity hit with an α-2-macroglobulin splice variant of O. moubata (Table 2).
The activity of inhibiting cysteine endopeptidases was correlated with the possible products of four contigs, two cystatins (Contig4119 and Contig3897) and two thyropin (Contig5375 and Contig5044). Contig4119 and Contig3897 exhibited a cystatin-conserved domain (cd00042, smart00043 and pfam00031) and had best hits with the O. moubata precursor cystatin (Table 2). Two cDNA molecules coding for thyropin protein and presenting a thyroglobulin conserved protein domain (cd00191) were detected as highly expressed in the gut (Table 2).
Immunity
The immunity category was mainly represented by four contigs, three defensins and one lysozyme. These transcripts were associated with immunity because defensin A and lysozyme presented the GO terms of defense response against gram-negative and positive bacteria and lysosomal activity, respectively. One defensin transcript was highly expressed (Contig5007) and showed 80% identification with O. moubata defensin, while Contig2696 had its best hit with lysozyme of the same species (Table 2).
Oxidant metabolism
The oxidant metabolism category had 70 transcripts associated with it. These are enzymes-coding transcripts possibly related to the oxidation-reduction processes and to the oxidative stress response, such as phospholipid-hydroperoxide glutathione peroxidase (Contig5141, Contig5040, Contig5426 and Contig3924) and glutathione-s-transferase (GST) (Contig5082, Contig5045, Contig5342 and Contig3084). One of the glutathione peroxidase (GSH) transcripts had high levels of expression (Contig5141) and had best hit with the phospholipid-hydroperoxide glutathione peroxidase of R. microplus (Table 2). Other highly expressed transcripts associated with oxidant metabolism were Contig5082 and Contig5045 (Table 2), which exhibited conserved domains of the classes delta and epsilon (cd03045) and mu GST (cd03075), respectively. Contig5045 also exhibited signal peptide and transmembrane region prediction. These transcripts had similarity with GST putative, from I. scapularis and possibly belonged to the cytosolic GST category (Table 2). We identified a transcript of microsomal GST (Contig131), because they presented a Membrane Associated Proteins in Eicosanoid and Glutathione (MAPEG) metabolism conserved domain and signal peptide prediction.
Transporter channels and storage
The transporter channel category grouped 246 contigs. This category is characterized by transport of biological and chemical elements, such as proteins, carbohydrates, lipids, ions and anions. The most abundant transcripts in this category were vitellogenin (10 contigs) and hemelipoglycoprotein (6 contigs). Both transcripts were associated with lipid transporter function. The Contig4329 transcript, identified as vitellogenin-B, presented similarity hits with vitellogenin-B of H. longicornis (Table 2). This transcript exhibited a von Willebrand factor conserved domain (pfam00094 and smart00216) that was also found in other vitellogenins of O. mimon.
Contig4611 (ferritin) and Contig5201 (hemelipoglycoprotein) are probably associated with the nutritional process of ticks. The Contig4611 transcript presented similarity with ferritins of O. moubata, exhibited a ferroxidase-conserved protein domain (cd01056, COG1528, PRK1034 and pfam00210) and was classified in the storage category (Table 2). Contig5201, named as hemelipoglycoprotein, showed its reported best hit in relation to a vitellogenin of H. longicornis (Table 2). This transcript was included in the transporter channel category, because lipid transport function was assigned to it. Three other contigs (Contig4288, Contig4030 and Contig4250) with high RPKM were identified as transporters of lipid and chemical elements.
Phylogeny
In this section, we present the phylogenetic inference results relating to the digestive peptidases and transcript datasets of the ticks.
Phylogeny of digestive peptidases
The monophyletic cathepsin cysteine clade was formed by cathepsins (B, C, L and O), midgut cysteine and dipeptidyl peptidase. Most of the cathepsin cysteines of O. mimon are closely related to tick cathepsins, thereby forming strongly supported monophyletic groups. The exception was for Contig4241, which grouped together with the cathepsin L branch of amphibians, birds and fish (Figure 4 and Supplementary Material Figure S1). Each cathepsin (B, C, L and O) formed a monophyletic group that was well supported in the cathepsin cysteine clade. Cathepsins B and C occurred in the same branch, while that cathepsin O formed a sister group with endopeptidases B and C. Cathepsin L was divided in two distinct clades with strong support. One of those clades was formed only by arthropod sequences, while the other clade was formed by bivalves, echinoderms, arthropods (spider, scorpions and ticks) and vertebrate proteinases. We demonstrated the presence of cathepsin O in ticks, for the first time. This proteinase is phylogenetically close to a protein of I. scapularis that was annotated as cysteine protease and a putative protease inhibitor. The cathepsin O group was also formed by two distinct clades in the inferred phylogeny: one group represented by arthropod peptidases and the other by bird and fish sequences.

Phylogenetic tree of cathepsin peptidases. Phylogenetic comparison of O. mimon peptidases with selected proteinases that showed hits in the BLAST search. The tree was constructed using the maximum likelihood (ML) method and multiple alignment of peptidase amino acid sequences. Some branches were collapsed to reduce the size of the tree. The complete phylogenetic tree of the peptidases can be observed on the Supplementary Material (Figure S1). The O. mimon peptidase are highlighted in blue. Aspartic peptidases group was used as outgroup to rooting the phylogenetic tree.
Aspartic peptidases formed a well-supported monophyletic group represented by cathepsin D and lysosomal aspartic protease genes (A1A family; pepsin). Contig3140 (cathepsin D) and Contig4774 (lysosomal aspartic protease) were seen to be relatively close and they were included in the group of tick aspartic proteinases (Figure 4 and Supplementary Material Figure S1).
Phylogeny of ticks: species tree constructed using transcript datasets
The Get Homologues software identified and selected 425 families of homologous genes from tick transcript datasets. The species tree generated from 425 ML trees of homologous transcripts of tick transcriptomes inferred the phylogenetic relationships of tick species (Figure 5). Two distinct monophyletic clades of the tick families (Argasidae and Ixodidae) were established (Figure 5). The mite M. occidentalis was used as outgroup. In the Argasidae clade, the subfamily Ornithodorinae was composed of species of Ornithodoros and Antricola. The genus Ornithodoros is paraphyletic, forming two groups: the Neotropical Ornithodorinae with the species O. mimon and Antricola delacruzi Estrada-Peña, Barros-Battesti and Venzal, 2004, and Ornithodoros sensu stricto (s.s), which comprises the species Ornithodoros parkeri Cooley, 1936, O. moubata, Ornithodoros rostratus Aragão, 1911 and O. coriaceus. In the Neotropical Ornithodorinae group, O. mimon and A. delacruzi are phylogenetically closely related species, forming a monophyletic group. The subfamily Argasinae is composed of Argas monolakensis.

Phylogenetic relationships of ticks, based on 425 ML trees from transcripts of tick transcriptomes, constructed in MulRF. The blue and red bars represent the Ornithodorinae and Argasinae subfamilies, respectively. Ornithodorinae is divided in two groups, Ornithodoros s.s (brown bar) and Neotropical Ornithodorinae (green bar). Metaseiulus occidentalis mite specie was used as outgroup in the phylogenetic relationships of tick species.
The Ixodidae clade is divided into two groups: Metastriata and Prostriata. The Metastriata group is formed by the subfamilies Rhipicephalinae (R. microplus, Rhipicephalus appendiculatus Neumann 1901, Rhipicephalus sanguineus sensu lato (s.l.) Latreille, 1806, D. variabilis and Dermacentor andersoni Stiles, 1908), Amblyomminae (Amblyomma maculatum Koch, 1844, A. variegatum, Amblyomma americanum (Linnaeus, 1758) and Amblyomma cajennense s.l.) and Haemaphysalinae (H. longicornis). The species A. cajennense was split into 6 taxa (NAVA et al., 2014Nava S, Beati L, Labruna MB, Cáceres AG, Mangold AJ, Guglielmone AA. Reassessment of the taxonomic status of Amblyomma cajennense () with the description of three new species, Amblyomma tonelliae n. sp., Amblyomma interandinum n. sp. and Amblyomma patinoi n. sp., and reinstatement of Amblyomma mixtum, and Amblyomma sculptum (Ixodida: Ixodidae). Ticks Tick Borne Dis 2014; 5(3): 252-276. PMid:24556273.). The species H. longicornis forms an underlying group with other Metastriata ticks. The Prostriata group is represented by only one genus, Ixodes (Ixodinae subfamily).
Discussion
Gut transcriptome analysis on engorged females of O. mimon identified several transcripts that may be associated with the functions of this organ. The transcripts obtained in this study were analyzed and functionally annotated according to the most significant similarity results (hits) in relation to sequences deposited in gene and protein databases such as GenBank and UniProt, using an automated search tool (CONESA et al., 2005Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005; 21(18): 3674-3676. PMid:16081474.). The BLAST search identifies sequences with potential evaluative and functional relationships to the contigs through comparison of similarity with sequences deposited in the databases available. BLAST2GO mapping is the step that retrieves the GO terms associated with each contig. The annotation step performed by this software is a process of assigning functional terms to query sequences from the pool of GO terms gathered in the mapping step (CONESA et al., 2005Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005; 21(18): 3674-3676. PMid:16081474.).
Most of the transcripts were completely annotated using BLAST2GO tool, with the exception of a percentage (22.6%) that did not have any homology with any nucleotide or amino acid sequence in current databases. In the literature, these complete sequences are referred to as “orphan genes”, and their proportion differs considerably from one genome to another (DUJON, 1996Dujon B. The yeast genome project: what did we learn? Trends Genet 1996; 12(7): 263-270. PMid:8763498.; FUKUCHI & NISHIKAWA, 2004Fukuchi S, Nishikawa K. Estimation of the number of authentic orphan genes in bacterial genomes. DNA Res 2004; 11(4): 219-231, 311-313. PMid:15500248.). They are abundant among the transcriptomes of ticks (GIBSON et al., 2013Gibson AK, Smith Z, Fuqua C, Clay K, Colbourne JK. Why so many unknown genes? Partitioning orphans from a representative transcriptome of the lone star tick Amblyomma americanum. BMC Genomics 2013; 14: 135. PMid:23445305.). In the gut transcriptome of D. variabilis, 396 unknown transcripts were identified (47%) (ANDERSON et al., 2008Anderson JM, Sonenshine DE, Valenzuela JG. Exploring the mialome of ticks: an annotated catalogue of midgut transcripts from the hard tick, Dermacentor variabilis (Acari: Ixodidae). BMC Genomics 2008; 9: 552. PMid:19021911.); while in salivary transcriptomes of O. coriaceus and O. parkeri, respectively 59% and 49% of the sequence clusters were classified as unknown (FRANCISCHETTI et al., 2008aFrancischetti IM, Mans BJ, Meng Z, Gudderra N, Veenstra TD, Pham VM, et al. An insight into the sialome of the soft tick, Ornithodoros parkeri. Insect Biochem Mol Biol 2008a; 38(1): 1-21. PMid:18070662., bFrancischetti IM, Meng Z, Mans BJ, Gudderra N, Hall M, Veenstra TD, et al. An insight into the salivary transcriptome and proteome of the soft tick and vector of epizootic bovine abortion, Ornithodoros coriaceus. J Proteomics 2008b; 71(5): 493-512. PMid:18725333.). In the sialome transcriptome of I. scapularis, 34% of its transcripts belonged to unknown categories, while the sialome of A. variegatum showed that most of the genes (466) were of unknown function (RIBEIRO et al., 2006Ribeiro JM, Alarcon-Chaidez F, Francischetti IM, Mans BJ, Mather TN, Valenzuela JG, et al. An annotated catalog of salivary gland transcripts from Ixodes scapularis ticks. Insect Biochem Mol Biol 2006; 36(2): 111-129. PMid:16431279., 2011Ribeiro JMC, Anderson JM, Manoukis NC, Meng Z, Francischetti IMB. A further insight into the sialome of the tropical bont tick, Amblyomma variegatum. BMC Genomics 2011; 12: 136. PMid:21362191.). Approximately 71% of the sequences of the transcriptome of A. americanum lack homology with sequences in databases (GIBSON et al., 2013Gibson AK, Smith Z, Fuqua C, Clay K, Colbourne JK. Why so many unknown genes? Partitioning orphans from a representative transcriptome of the lone star tick Amblyomma americanum. BMC Genomics 2013; 14: 135. PMid:23445305.). The reasons for this lack of homology may be the quality of the sequences, low assembly quality, absence of functional genes, taxonomic isolation of the species and emergence of new genes through duplication and/or transposition mechanisms (TAUTZ & DOMAZET-LOŠO, 2011Tautz D, Domazet-Lošo T. The evolutionary origin of orphan genes. Nat Rev Genet 2011; 12(10): 692-702. PMid:21878963.). We can also suggest that other reasons for the existence of these unknown groups might include the small number of studies conducted on these ticks, or even some particular features of gene composition that are not found in other animals. The transcripts without associated GOs possibly represent genes that have functions that are not fully understood or have simply been poorly annotated.
Predictive analysis on subcellular localization identified that the percentage of secreted and transmembrane proteins was low, thus indicating that most of the gut transcripts of O. mimon are intracellular. Similar results were observed in the gut transcriptome of D. variabilis, for which the percentage of secreted transcripts among the genes expressed was small (ANDERSON et al., 2008Anderson JM, Sonenshine DE, Valenzuela JG. Exploring the mialome of ticks: an annotated catalogue of midgut transcripts from the hard tick, Dermacentor variabilis (Acari: Ixodidae). BMC Genomics 2008; 9: 552. PMid:19021911.). Fifty-three percentage of the transcripts differentially expressed in the gut transcriptome of R. microplus females were classified as intracellular and 18% as membranous (HEEKIN et al., 2013Heekin AM, Guerrero FD, Bendele KG, Saldivar L, Scoles GA, Dowd SE, et al. Gut transcriptome of replete adult female cattle ticks, Rhipicephalus (Boophilus) microplus, feeding upon a Babesia bovis-infected bovine host. Parasitol Res 2013; 112(9): 3075-3090. PMid:23749091.).
In the analysis on functional assignment, the predominant functional activities were binding and catalytic activity, thus indicating that a large proportion of gut genes are enzymes and interactive proteins (such as channels, transporters and receptors). The classification from the automated analysis showed that the category of catalytic activity category was prevalent, thus indicating that many of the digestive tract transcripts were enzymes, such as oxidoreductases, transferases, hydrolases, lyases, isomerases and ligases. In the gut transcriptome of R. microplus, binding and catalytic activity was also identified as the most common functional types of activity (HEEKIN et al., 2013Heekin AM, Guerrero FD, Bendele KG, Saldivar L, Scoles GA, Dowd SE, et al. Gut transcriptome of replete adult female cattle ticks, Rhipicephalus (Boophilus) microplus, feeding upon a Babesia bovis-infected bovine host. Parasitol Res 2013; 112(9): 3075-3090. PMid:23749091.). The same results were observed by Xu et al. (2016)Xu XL, Cheng TY, Yang H, Liao ZH. De novo assembly and analysis of midgut transcriptome of Haemaphysalis flava and identification of genes involved in blood digestion, feeding and defending from pathogens. Infect Genet Evol 2016; 38: 62-72. PMid:26705239., who classified 47 and 42% of the transcripts of H. flava in the functional categories binding and catalytic activity, respectively. In the proteomic study of gut of O. erraticus, Oleaga et al. (2015)Oleaga A, Obolo-Mvoulouga P, Manzano-Romá R, Pérez-Sánchez R. Midgut proteome of an argasid tick, Ornithodoros erraticus: a comparison between unfed and engorged females. Parasit Vectors 2015; 8: 525. PMid:26459090. also identified catalytic activity and binding as the most prevalent activities within their set of proteins, in accordance to our results. Transcripts associated with the transporter channel category were abundant, since several contigs with binding, carrier protein, receptor and channel functions were grouped into this category. The unknown category was the largest in terms of number of transcripts, because many transcripts without well-defined function, and which lacked homology and GO terms, were grouped into this category.
Blood meal digestion and nutrient uptake
Based on the mechanisms used by I. ricinus for digestion of hemoglobin that have been described in the literature (HORN et al., 2009Horn M, Nussbaumerová M, Sanda M, Kovárová Z, Srba J, Franta Z, et al. Hemoglobin digestion in blood-feeding ticks: mapping a multipeptidase pathway by functional proteomics. Chem Biol 2009; 16(10): 1053-1063. PMid:19875079.; SOJKA et al., 2013Sojka D, Franta Z, Horn M, Caffrey CR, Mares M, Kopácek P. New insights into the machinery of blood digestion by ticks. Trends Parasitol 2013; 29(6): 276-285. PMid:23664173.; PERNER et al., 2016Perner J, Provazník J, Schrenková J, Urbanová V, Ribeiro JM, Kopáček P. RNA-seq analyses of the midgut from blood-and serum-fed Ixodes ricinus ticks. Sci Rep 2016; 6: 36695. PMid:27824139.), ticks’ intracellular blood meal digestion takes place through the action of lysosomal proteolytic enzymes such as cathepsin, leucine aminopeptidase, legumain and serine peptidase (SOJKA et al., 2013Sojka D, Franta Z, Horn M, Caffrey CR, Mares M, Kopácek P. New insights into the machinery of blood digestion by ticks. Trends Parasitol 2013; 29(6): 276-285. PMid:23664173.). The initial degradation of hemoglobin in ticks is performed by cathepsin D, in association with the auxiliary enzymes cathepsin L and legumain, while cathepsins B and C cleave large hemoglobin fragments to produce dipeptides (HORN et al., 2009Horn M, Nussbaumerová M, Sanda M, Kovárová Z, Srba J, Franta Z, et al. Hemoglobin digestion in blood-feeding ticks: mapping a multipeptidase pathway by functional proteomics. Chem Biol 2009; 16(10): 1053-1063. PMid:19875079.). Other peptidases, such as metallopeptidase and serine peptidase have roles in digestion that remain somewhat unclear, but it has been speculated that serine carboxypeptidase and aminopeptidase might participate in releasing free amino acids in the digestion process (HATTA et al., 2006Hatta T, Kazama K, Miyoshi T, Umemiya R, Liao M, Inoue N, et al. Identification and characterisation of a leucine aminopeptidase from the hard tick Haemaphysalis longicornis. Int J Parasitol 2006; 36(10-11): 1123-1132. PMid:16814790.; ANDERSON et al., 2008Anderson JM, Sonenshine DE, Valenzuela JG. Exploring the mialome of ticks: an annotated catalogue of midgut transcripts from the hard tick, Dermacentor variabilis (Acari: Ixodidae). BMC Genomics 2008; 9: 552. PMid:19021911.; HORN et al., 2009Horn M, Nussbaumerová M, Sanda M, Kovárová Z, Srba J, Franta Z, et al. Hemoglobin digestion in blood-feeding ticks: mapping a multipeptidase pathway by functional proteomics. Chem Biol 2009; 16(10): 1053-1063. PMid:19875079.). Our results indicated that ticks’ digestive mechanisms are present in the transcriptome analysis on engorged females of O. mimon. All the digestive proteinases described above were identified at different levels of expression (RPKM) with the cathepsins (B, D and L), and they are well expressed in the gut of engorged females. These peptidases also were identified in gut transcriptome analyses on I. ricinus, H. flava and D. variabilis, respectively, thus agreeing with our results (KOTSYFAKIS et al., 2015Kotsyfakis M, Schwarz A, Erhart J, Ribeiro JM. Tissue- and time-dependent transcription in Ixodes ricinus salivary glands and midguts when blood feeding on the vertebrate host. Sci Rep 2015; 5: 9103. PMid:25765539.; PERNER et al., 2016Perner J, Provazník J, Schrenková J, Urbanová V, Ribeiro JM, Kopáček P. RNA-seq analyses of the midgut from blood-and serum-fed Ixodes ricinus ticks. Sci Rep 2016; 6: 36695. PMid:27824139.; XU et al., 2016Xu XL, Cheng TY, Yang H, Liao ZH. De novo assembly and analysis of midgut transcriptome of Haemaphysalis flava and identification of genes involved in blood digestion, feeding and defending from pathogens. Infect Genet Evol 2016; 38: 62-72. PMid:26705239.; ANDERSON et al., 2008Anderson JM, Sonenshine DE, Valenzuela JG. Exploring the mialome of ticks: an annotated catalogue of midgut transcripts from the hard tick, Dermacentor variabilis (Acari: Ixodidae). BMC Genomics 2008; 9: 552. PMid:19021911.). Oleaga et al. (2015)Oleaga A, Obolo-Mvoulouga P, Manzano-Romá R, Pérez-Sánchez R. Midgut proteome of an argasid tick, Ornithodoros erraticus: a comparison between unfed and engorged females. Parasit Vectors 2015; 8: 525. PMid:26459090. identified 15 proteins with peptidase activity belonging to four different groups (aspartic-type endopeptidase, metallopeptidase, cysteine-type endopeptidase and serine-type endopeptidase) in the midgut proteome of O. erraticus.
Adding a new member to the known tick digestive enzymes, we have recorded for the first time a sequence coding for a member of the cysteine proteinase family called cathepsin O. Although, this protease may already have been identified in the I. scapularis tick as a cysteine protease putative deposited in the Genbank database, but it was not labeled as cathepsin O. This cysteine peptidase was identified and characterized for the first time from a human breast carcinoma cDNA library, by using a polymerase chain reaction-based cloning strategy (VELASCO et al., 1994Velasco G, Ferrando AA, Puente XS, Sánchez LM, López-Otín C. Human cathepsin O. Molecular cloning from a breast carcinoma, production of the active enzyme in Escherichia coli, and expression analysis in human tissues. J Biol Chem 1994; 269(43): 27136-27142. PMid:7929457.). Recently, Fuzita et al. (2015)Fuzita FJ, Pinkse MW, Patane JS, Juliano MA, Verhaert PD, Lopes AR. Biochemical, transcriptomic and proteomic analyses of digestion in the scorpion Tityus serrulatus: insights into function and evolution of digestion in an ancient arthropod. PLoS One 2015; 10(4): e0123841. PMid:25875018. identified two transcripts of cathepsin O in the transcriptomic and proteomic analyses of digestion in T. serrulatus.
In addition to the digestive mechanisms, ticks have a heme detoxification mechanism based on accumulating heme in specialized organelles of the intestinal epithelium, called hemosomes (LARA et al., 2003Lara FA, Lins U, Paiva-Silva G, Almeida IC, Braga CM, Miguens FC, et al. A new intracellular pathway of haem detoxification in the midgut of the cattle tick Boophilus microplus: aggregation inside a specialized organelle, the hemosome. J Exp Biol 2003; 206(10): 1707-1715. PMid:12682102.). These organelles are formed by transport and delivery of carrier proteins such as hemelipoglycoproteins, which are associated with heme (MAYA-MONTEIRO et al., 2000Maya-Monteiro CM, Daffre S, Logullo C, Lara FA, Alves EW, Capurro ML, et al. HeLp, a heme lipoprotein from the hemolymph of the cattle tick, Boophilus microplus. J Biol Chem 2000; 275(47): 36584-36589. PMid:10964932.; LARA et al., 2005Lara FA, Lins U, Bechara GH, Oliveira PL. Tracing heme in a living cell: hemoglobin degradation and heme traffic in digest cells of the cattle tick Boophilus microplus. J Exp Biol 2005; 208(16): 3093-3101. PMid:16081607.; DUPEJOVA et al., 2011Dupejova J, Sterba J, Vancova M, Grubhoffer L. Hemelipoglycoprotein from the ornate sheep tick, Dermacentor marginatus: structural and functional characterization. Parasit Vectors 2011; 4: 4. PMid:21214898.). Hemelipoglycoprotein in O. mimon constitute a transcript with high levels of RPKM, possibly indicating gene expression in response to digestion of the blood meal, in order to accumulate heme. Another transcript that may be associated with nutrient uptake is the ferritin gene. In response to exposure to iron, insect cells produce ferritin molecules of two types: cytoplasmic and secreted (GEISER et al., 2006Geiser DL, Zhang D, Winzerling JJ. Secreted ferritin: mosquito defense against iron overload? Insect Biochem Mol Biol 2006; 36(3): 177-187. PMid:16503479.). Ferritin was well-expressed in the engorged females of O. mimon, possibly in response to the blood of the host, which is iron-rich. cDNA molecules of ferritin in two tick species (O. moubata and I. ricinus) were characterized and these molecules showed no evidence of being secreted proteins, thus indicating that the protein was cytoplasmic (KOPÁČEK et al., 2003Kopáček P, Zdychová J, Yoshiga T, Weise C, Rudenko N, Law JH. Molecular cloning, expression and isolation of ferritins from two tick species -- Ornithodoros moubata and Ixodes ricinus. Insect Biochem Mol Biol 2003; 33(1): 103-113. PMid:12459205.). These findings are in agreement with our results, given that the O. mimon ferritin did not exhibit any prediction of a signal peptide, according to the SignalP algorithm. Iron is a substance derived from degradation of the hemoglobin present in the blood meal, which results in compounds that serve as nutrients for ticks to feed on, develop and reproduce (KOPÁČEK et al., 2003Kopáček P, Zdychová J, Yoshiga T, Weise C, Rudenko N, Law JH. Molecular cloning, expression and isolation of ferritins from two tick species -- Ornithodoros moubata and Ixodes ricinus. Insect Biochem Mol Biol 2003; 33(1): 103-113. PMid:12459205.). Ferritin is frequently found in the mialome and sialometranscriptome analysis of ticks (ANDERSON et al., 2008Anderson JM, Sonenshine DE, Valenzuela JG. Exploring the mialome of ticks: an annotated catalogue of midgut transcripts from the hard tick, Dermacentor variabilis (Acari: Ixodidae). BMC Genomics 2008; 9: 552. PMid:19021911.; RIBEIRO et al., 2012Ribeiro JMC, Labruna MB, Mans BJ, Maruyama SR, Francischetti IM, Barizon GC, et al. The sialotranscriptome of Antricola delacruzi female ticks is compatible with non-hematophagous behavior and an alternative source of food. Insect Biochem Mol Biol 2012; 42(5): 332-342. PMid:22306723.; KOTSYFAKIS et al., 2015Kotsyfakis M, Schwarz A, Erhart J, Ribeiro JM. Tissue- and time-dependent transcription in Ixodes ricinus salivary glands and midguts when blood feeding on the vertebrate host. Sci Rep 2015; 5: 9103. PMid:25765539.; XU et al., 2016Xu XL, Cheng TY, Yang H, Liao ZH. De novo assembly and analysis of midgut transcriptome of Haemaphysalis flava and identification of genes involved in blood digestion, feeding and defending from pathogens. Infect Genet Evol 2016; 38: 62-72. PMid:26705239.), as well as in the proteome studies (OLEAGA et al., 2015Oleaga A, Obolo-Mvoulouga P, Manzano-Romá R, Pérez-Sánchez R. Midgut proteome of an argasid tick, Ornithodoros erraticus: a comparison between unfed and engorged females. Parasit Vectors 2015; 8: 525. PMid:26459090.).
The gut of ticks also expresses enzymes that digest other blood meal constituents, such as carbohydrates and lipids (MORETI et al., 2013Moreti R, Perrella NN, Lopes AR. Carbohydrate digestion in ticks and a digestive α-L- fucosidase. J Insect Physiol 2013; 59(10): 1069-1075. PMid:23994295.; KOTSYFAKIS et al., 2015Kotsyfakis M, Schwarz A, Erhart J, Ribeiro JM. Tissue- and time-dependent transcription in Ixodes ricinus salivary glands and midguts when blood feeding on the vertebrate host. Sci Rep 2015; 5: 9103. PMid:25765539.; XU et al., 2016Xu XL, Cheng TY, Yang H, Liao ZH. De novo assembly and analysis of midgut transcriptome of Haemaphysalis flava and identification of genes involved in blood digestion, feeding and defending from pathogens. Infect Genet Evol 2016; 38: 62-72. PMid:26705239.). Carbohydrase genes were identified in the digestive tract of O. mimon after the blood meal, among which chitinases and α-glycosidases were the most prevalent. The role of these enzymes within digestion has not been fully understood and there are few studies about it. However, on the other hand, the activity of carbohydrases such as N-acetyl glucosaminidase, chitinase and α-L-fucosidase on the intestinal content of Amblyomma sculptum (cited as cajennense) was recently identified and characterized, and it was suggested that digestion of carbohydrates occurs in the intestinal lumen, unlike the intracellular digestion of proteins (MORETI et al., 2013Moreti R, Perrella NN, Lopes AR. Carbohydrate digestion in ticks and a digestive α-L- fucosidase. J Insect Physiol 2013; 59(10): 1069-1075. PMid:23994295.).
Protease inhibitors are expressed in both the intestine and the salivary glands of ticks (KOPÁČEK et al., 2010Kopáček P, Hajdusek O, Buresová V, Daffre S. Tick innate immunity. Adv Exp Med Biol 2010; 708: 137-162. PMid:21528697.), but their function in ticks is not fully understood. However, other authors have suggested that inhibitors such as serpin can be used by ticks to regulate endopeptidase activity and hemostasis (MULENGA et al., 2001Mulenga A, Sugino M, Nakajim M, Sugimoto C, Onuma M. Tick- Encoded serine proteinase inhibitors (serpins); potential target antigens for tick vaccine development. J Vet Med Sci 2001; 63(10): 1063-1069. PMid:11714020., 2003Mulenga A, Tsuda A, Onuma M, Sugimoto C. Four serine proteinase inhibitors (serpin) from the brown ear tick, Rhipicephalus appendiculatus; cDNA cloning and preliminary characterization. Insect Biochem Mol Biol 2003; 33(2): 267-276. PMid:12535684.; PERNER et al., 2016Perner J, Provazník J, Schrenková J, Urbanová V, Ribeiro JM, Kopáček P. RNA-seq analyses of the midgut from blood-and serum-fed Ixodes ricinus ticks. Sci Rep 2016; 6: 36695. PMid:27824139.; TIRLONI et al., 2014Tirloni L, Seixas A, Mulenga A, Vaz IS Jr, Termignoni C. A family of serine protease inhibitors (serpins) in the cattle tick Rhipicephalus (Boophilus) microplus. Exp Parasitol 2014; 137: 25-34. PMid:24333790.). Peptidase inhibitors have been mentioned as effective antigens for preparing vaccines, because these genes are essential for the survival of ticks, since they are associated with endogenous processes such as regulation of hemolymph coagulation through controlling proteinase activity (IMAMURA et al., 2005Imamura S, Vaz IS Jr, Sugino M, Ohashi K, Onuma M. A serine protease inhibitor (serpin) from Haemaphysalis longicornis as an anti-tick vaccine. Vaccine 2005; 23(10): 1301-1311. PMid:15652673.). Kunitz-like inhibitors of serine peptidase are known to act towards defending the intestine against rickettsial infections and protecting tick cells against acaricide (KOPÁČEK et al., 2010Kopáček P, Hajdusek O, Buresová V, Daffre S. Tick innate immunity. Adv Exp Med Biol 2010; 708: 137-162. PMid:21528697.). In addition to Kunitz inhibitors in ticks, we also found a single transcript that coded for a protein similar to snake venom protease inhibitor, which showed similarity to of Kunitz inhibitors snakes sequences, and may also have a role relating to protease inhibition in ticks.
Tick cystatins act as regulators of blood meal digestion and as modulators of the host immune response (SCHWARZ et al., 2012Schwarz A, Valdés JJ, Kotsyfakis M. The role of cystatins in tick physiology and blood feeding. Ticks Tick Borne Dis 2012; 3(3): 117-127. PMid:22647711.). These inhibitors of cysteine peptidases also participate in the innate immunity of ticks by inhibiting specific enzymes of microorganisms (KOPÁČEK et al., 2010Kopáček P, Hajdusek O, Buresová V, Daffre S. Tick innate immunity. Adv Exp Med Biol 2010; 708: 137-162. PMid:21528697.). Two types of cystatins have been found in argasid ticks (GRUNCLOVÁ et al., 2006Grunclová L, Horn M, Vancová M, Sojka D, Franta Z, Mares M, et al. Two secreted cystatins of the soft tick Ornithodoros moubata: differential expression pattern and inhibitory specificity. Biol Chem 2006; 387(12): 1635-1644. PMid:17132111.). Cystatin type I is cytosolic, while type II is secreted, and both substantially diverge from other invertebrate, vertebrate and plant cystatins (GRUNCLOVÁ et al., 2006Grunclová L, Horn M, Vancová M, Sojka D, Franta Z, Mares M, et al. Two secreted cystatins of the soft tick Ornithodoros moubata: differential expression pattern and inhibitory specificity. Biol Chem 2006; 387(12): 1635-1644. PMid:17132111.). The cystatins of O. mimon did not show any prediction of secretion, thus suggesting that they may belong to type I. However, these transcripts showed similarity to sequences of O. moubata cystatins, which according to the literature (GRUNCLOVÁ et al., 2006Grunclová L, Horn M, Vancová M, Sojka D, Franta Z, Mares M, et al. Two secreted cystatins of the soft tick Ornithodoros moubata: differential expression pattern and inhibitory specificity. Biol Chem 2006; 387(12): 1635-1644. PMid:17132111.), are type II (secreted). It is possible that the SignalP algorithm did not identify the signal peptide in the cDNA sequences of cystatins of O. mimon, or that the assembled contig did not include the region where possible signal peptides could occur. The transcripts of thryporin are well expressed, but their function is not fully known.
In the transcriptome of O. mimon, we only found the isoform defensin A, which is common in Ornithodoros species (CHRUDIMSKÁ et al., 2010Chrudimská T, Chrudimský T, Golovchenko M, Rudenko N, Grubhoffer L. New defensins from hard and soft ticks: similarities, differences, and phylogenetic analyses. Vet Parasitol 2010; 167(2-4): 298-303. PMid:19836137.). The main transcripts associated with the immunity category were defensin and lysozyme. Both transcripts have been identified in the intestine of O. moubata, and their antibacterial activity for both Gram-positive and Gram-negative bacteria has been proven (NAKAJIMA et al., 2003Nakajima Y, Ogihara K, Taylor D, Yamakawa M. Antibacterial hemoglobin fragments from the midgut of the soft tick, Ornithodoros moubata (Acari: Argasidae). J Med Entomol 2003; 40(1): 78-81. PMid:12597657.; KOPÁČEK et al., 1999Kopáček P, Vogt R, Jindrák L, Weise C, Safarík I. Purification and characterization of the lysozyme from the gut of the soft tick Ornithodoros moubata. Insect Biochem Mol Biol 1999; 29(11): 989-997. PMid:10560138., 2010Kopáček P, Hajdusek O, Buresová V, Daffre S. Tick innate immunity. Adv Exp Med Biol 2010; 708: 137-162. PMid:21528697.). Four very similar isoforms of defensins (A, B, C and D) that are regulated in response to the blood meal were identified in O. moubata (NAKAJIMA et al., 2001Nakajima Y, van Naters AVG, Taylor D, Yamakawa M. Two isoforms of a member of the arthropod defensin family from the soft tick, Ornithodoros moubata (Acari: Argasidae). Insect Biochem Mol Biol 2001; 31(8): 747-751. PMid:11378409., 2002Nakajima Y, Taylor D, Yamakawa M. Involvement of antibacterial peptide defensin in tick midgut defense. Exp Appl Acarol 2002; 28(1-4): 135-140. PMid:14570123., 2003Nakajima Y, Ogihara K, Taylor D, Yamakawa M. Antibacterial hemoglobin fragments from the midgut of the soft tick, Ornithodoros moubata (Acari: Argasidae). J Med Entomol 2003; 40(1): 78-81. PMid:12597657.). Similarly, the same isoforms were found in other species of Ornithodoros and ixodid ticks, and the number of isoforms varied among the species (CHRUDIMSKÁ et al., 2010Chrudimská T, Chrudimský T, Golovchenko M, Rudenko N, Grubhoffer L. New defensins from hard and soft ticks: similarities, differences, and phylogenetic analyses. Vet Parasitol 2010; 167(2-4): 298-303. PMid:19836137.). Ticks have an efficient protection mechanism inside their gut to combat and inhibit the growth of microorganisms in the blood meal, thereby ensuring their full access to the nutritive resources (GRUNCLOVÁ et al., 2003Grunclová L, Fouquier H, Hypsa V, Kopáček P. Lysozyme from the gut of the soft tick Ornithodoros moubata: the sequence, phylogeny and post-feeding regulation. Dev Comp Immunol 2003; 27(8): 651-660. PMid:12798362.).
A different category that is also involved in tick immunity is oxidative metabolism, as represented by transcripts such as GST and phospholipid hydro peroxide glutathione peroxidase, which were also found in O. mimon. They may act towards cell detoxification caused by acaricide. Thus, these transcripts could participate in resistance mechanisms against chemical control, for example through use of organophosphates (HE et al., 1999He H, Chen AC, Davey RB, Ivie GW, George JE. Characterization and molecular cloning of a glutathione S-transferase gene from the tick, Boophilus microplus (Acari: Ixodidae). Insect Biochem Mol Biol 1999; 29(8): 737-743. PMid:10451925.; COSSÍO-BAYÚGAR et al., 2005Cossío-Bayúgar R, Miranda E, Holman PJ. Molecular cloning of a phospholipid-hydroperoxide glutathione peroxidase gene from the tick, Boophilus microplus (Acari: Ixodidae). Insect Biochem Mol Biol 2005; 35(12): 1378-1387. PMid:16291093.; DUSCHER et al., 2014Duscher GG, Galindo RC, Tichy A, Hummel K, Kocan KM, de la Fuente J. Glutathione S-transferase affects permethrin detoxification in the brown dog tick, Rhipicephalus sanguineus. Ticks Tick Borne Dis 2014; 5(3): 225-233. PMid:24548526.). The GST genes of O. mimon belong to the delta, epsilon and mu classes. The first two classes are specific to the order Insecta, while the mu class is specific to mammals (REDDY et al., 2011Reddy BN, Prasad GBKS, Raghavendra K. In silico analysis of glutathione S-transferase supergene family revealed hitherto unreported insect specific δ-and ɛ-GSTs and mammalian specific μ-GSTs in Ixodes scapularis (Acari: Ixodidae). Comput Biol Chem 2011; 35(2): 114-120. PMid:21514893.). The same classes, as well as other GST genes, have been identified in the genome and gut transcriptome of I. scapularis and I. ricinus, respectively (REDDY et al., 2011Reddy BN, Prasad GBKS, Raghavendra K. In silico analysis of glutathione S-transferase supergene family revealed hitherto unreported insect specific δ-and ɛ-GSTs and mammalian specific μ-GSTs in Ixodes scapularis (Acari: Ixodidae). Comput Biol Chem 2011; 35(2): 114-120. PMid:21514893.; PERNER et al., 2016Perner J, Provazník J, Schrenková J, Urbanová V, Ribeiro JM, Kopáček P. RNA-seq analyses of the midgut from blood-and serum-fed Ixodes ricinus ticks. Sci Rep 2016; 6: 36695. PMid:27824139.). The delta and epsilon classes in ticks may exhibit the same detoxification function as found in mosquitoes against insecticides (REDDY et al., 2011Reddy BN, Prasad GBKS, Raghavendra K. In silico analysis of glutathione S-transferase supergene family revealed hitherto unreported insect specific δ-and ɛ-GSTs and mammalian specific μ-GSTs in Ixodes scapularis (Acari: Ixodidae). Comput Biol Chem 2011; 35(2): 114-120. PMid:21514893.). Perner et al. (2016)Perner J, Provazník J, Schrenková J, Urbanová V, Ribeiro JM, Kopáček P. RNA-seq analyses of the midgut from blood-and serum-fed Ixodes ricinus ticks. Sci Rep 2016; 6: 36695. PMid:27824139. also found a transcript of microsomal GST in the gut transcriptome of I. ricinus, agreeing to our results.
Phylogenetic inferences
The inferred phylogenetic relationships of the digestive peptidases demonstrated that O. mimon peptidases (cysteine and aspartic) are closely related to tick proteinases. This relationship may provide evidence that the digestive transcripts of O. mimon are acting within the mechanism for hemoglobin digestion, especially the cathepsins B, C, D and L. Moreover, these cathepsins were clearly classified in a distinct monophyletic group that was strongly supported. These distinct clades of cathepsins were also identified in an analysis on the digestive activity of the gut of I. ricinus (SOJKA et al., 2008Sojka D, Franta D, Horn M, Hajdušek O, Caffrey CR, Mares M, et al. Profiling of proteolytic enzymes in the gut of the tick Ixodes ricinus reveals an evolutionarily conserved network of aspartic and cysteine peptidases. Parasit Vectors 2008; 1(1): 7. PMid:18348719.). We can suggest that the different cathepsin L genes of O. mimon (Contig4241 and Contig4886) went through distinct evolutionary processes. The Contig4241 transcript may have diverged from the cathepsin L of vertebrates, thus forming a moderately supported clade (bootstrap of 74), while the gene Contig4886 apparently evolved from a common ancestral gene from ticks. The cathepsin O of O. mimon diverged from arthropod cathepsins and formed a monophyletic group with I. scapularis cysteine proteinase. We assume that this protein of I. scapularis can also be a cathepsin O. Therefore, we recorded the presence of this lysosomal papain-like cysteine endopeptidase (cathepsin O) in ticks, for the first time. These cysteine cathepsins belongs to the family C1 (papain) according to Rawlings & Barrett (1993)Rawlings ND, Barrett AJ. Evolutionary families of peptidases. Biochem J 1993; 290(1): 205-218. PMid:8439290. and Barrett & Rawlings (1995Barrett AJ, Rawlings ND. Families and clans of serine peptidases. Arch Biochem Biophys 1995; 318(2): 247-250. PMid:7733651., 1996Barrett AJ, Rawlings ND. Families and clans of cysteine peptidases. Perspect Drug Discov Des 1996; 6(1): 1-11.), that used the term family to describe a group of enzymes in which each member shows an evolutionary relationship (conserved domain of papain). In our phylogenetic analysis, the proteases belonging to the aspartic class form a monophyletic group that belongs to A1 family.
Our phylogenetic analysis on ticks, based on a species tree analysis on the 425 ML trees of homologous transcripts, was quite similar to recent inferences regarding the phylogeny of soft ticks (BURGER et al., 2014Burger TD, Shao R, Labruna MB, Barker SC. Molecular phylogeny of soft ticks (Ixodida: Argasidae) inferred from mitochondrial genome and nuclear rRNA sequences. Ticks Tick Borne Dis 2014; 5(2): 195-207. PMid:24378079.). In that study, molecular phylogeny based on the mitochondrial genome and on nuclear rRNA sequences was constructed. Both of the phylogenetic inferences identified the genus Ornithodoros as paraphyletic, such that Neotropical ticks associated with bats (O. mimon and A. delacruzi) in the Neotropical Ornithodorinae group and of the species of Ornithodoros in the Ornithodoros s. s group. It had already been proposed that Ornithodoros is paraphyletic, from phylogenetic analysis of morphological and developmental characteristics (KLOMPEN & OLIVER, 1993Klompen JSH, Oliver JH Jr. Systematic relationships in the soft ticks (Acari: Ixodida: Argasidae). Syst Entomol 1993; 18(4): 313-331.). The classical systematics and taxonomy of the family Argasidae were modified through elevation of the subgenus Carios to genus status, and this new genus included ticks associated with bats that belonged to the genera Antricola and Nothoaspis, and to the subgenera of Ornithodoros (Alectorobius, Subparmatus and Reticulinasus) and Argas (Chiropterargas and Carios) (KLOMPEN & OLIVER, 1993Klompen JSH, Oliver JH Jr. Systematic relationships in the soft ticks (Acari: Ixodida: Argasidae). Syst Entomol 1993; 18(4): 313-331.). These modifications were adopted by some taxonomists (HORAK et al., 2002Horak IG, Camicas JL, Keirans JE. The Argasidae, Ixodidae and Nuttalliellidae (Acari: Ixodida): a world list of valid tick names. Exp Appl Acarol 2002; 28(1-4): 27-54. PMid:14570115.; BARKER & MURRELL, 2004Barker SC, Murrell A. Systematics and evolution of ticks with a list of valid genus and species names. Parasitology 2004; 129(Suppl): S15-S36. PMid:15938503.; LABRUNA et al., 2008Labruna MB, Terassini FA, Camargo LM, Brandão PE, Ribeiro AF, Estrada-Peña A. New reports of Antricola guglielmonei and Antricola delacruzi in Brazil, and a description of a new argasid species (Acari). J Parasitol 2008; 94(4): 788-792. PMid:18576796.; BARROS-BATTESTI et al., 2011Barros-Battesti DM, Landulfo GA, Onofrio VC, Faccini JL, Marcili A, Nieri-Bastos FA, et al. Carios mimon (Acari: Argasidae): description of adults and redescription of larva. Exp Appl Acarol 2011; 54(1): 93-104. PMid:21161720.), but other researchers questioned the position of Antricola and Nothoaspis, and revalidated them as genera in different phylogenetic analyses (LABRUNA et al., 2008Labruna MB, Terassini FA, Camargo LM, Brandão PE, Ribeiro AF, Estrada-Peña A. New reports of Antricola guglielmonei and Antricola delacruzi in Brazil, and a description of a new argasid species (Acari). J Parasitol 2008; 94(4): 788-792. PMid:18576796., 2011Labruna MB, Nava S, Terassini FA, Onofrio VC, Barros-Battesti DM, Camargo LM, et al. Description of adults and nymph, and redescription of the larva, of Ornithodoros marinkellei (Acari:Argasidae), with data on its phylogenetic position. J Parasitol 2011; 97(2): 207-217. PMid:21506769.; VENZAL et al., 2008Venzal JM, Estrada-Peña A, Mangold AJ, González-Acuña D, Guglielmone AA. The Ornithodoros (Alectorobius) talaje species group (Acari: Ixodida: Argasidae): description of Ornithodoros (Alectorobius) rioplatensis n. sp. from South America. J Med Entomol 2008; 45(5): 832-840. PMid:18826024., 2012Venzal JM, Nava S, Mangold AJ, Mastropaolo M, Casás G, Guglielmone AA. Ornithodoros quilinensis sp. nov. (Acari, Argasidae), a new tick species from the Chacoan region in Argentina. Acta Parasitol 2012; 57(3): 329-336. PMid:22875683.; NAVA et al., 2009Nava S, Guglielmone AA, Mangold AJ. An overview of systematics and evolution of ticks. Front Biosci 2009; 14: 2857-2877. PMid:19273240., 2010Nava S, Venzal JM, Terassini FA, Mangold AJ, Camargo LMA, Labruna MB. Description of a New Argasid tick (Acari: Ixodida) from bat caves in Brazilian. Amazon J Parasitol 2010; 96(6): 1089-1101. PMid:21158616.; ESTRADA-PEÑA et al., 2006Estrada-Peña A, Venzal JM, González-Acuña D, Mangold AJ, Guglielmone AA. Notes on New World Persicargas ticks (Acari: Argasidae) with description of female Argas (P.) keiransi. J Med Entomol 2006; 43(5): 801-809. PMid:17017212., 2010Estrada-Peña A, Mangold AJ, Nava S, Venzal JM, Labruna M, Guglielmone AA. A review of the systematics of the tick family Argasidae (Ixodida). Acarologia 2010; 50(3): 317-333.). Other studies in the Neotropical region have also suggested that there is a close phylogenetic relationship between the genera Antricola and Ornithodoros (Alectorobius) (LABRUNA et al., 2008Labruna MB, Terassini FA, Camargo LM, Brandão PE, Ribeiro AF, Estrada-Peña A. New reports of Antricola guglielmonei and Antricola delacruzi in Brazil, and a description of a new argasid species (Acari). J Parasitol 2008; 94(4): 788-792. PMid:18576796., 2011Labruna MB, Nava S, Terassini FA, Onofrio VC, Barros-Battesti DM, Camargo LM, et al. Description of adults and nymph, and redescription of the larva, of Ornithodoros marinkellei (Acari:Argasidae), with data on its phylogenetic position. J Parasitol 2011; 97(2): 207-217. PMid:21506769.; NAVA et al., 2009Nava S, Guglielmone AA, Mangold AJ. An overview of systematics and evolution of ticks. Front Biosci 2009; 14: 2857-2877. PMid:19273240.; ESTRADA-PEÑA et al., 2006Estrada-Peña A, Venzal JM, González-Acuña D, Mangold AJ, Guglielmone AA. Notes on New World Persicargas ticks (Acari: Argasidae) with description of female Argas (P.) keiransi. J Med Entomol 2006; 43(5): 801-809. PMid:17017212., 2010Estrada-Peña A, Mangold AJ, Nava S, Venzal JM, Labruna M, Guglielmone AA. A review of the systematics of the tick family Argasidae (Ixodida). Acarologia 2010; 50(3): 317-333.; VENZAL et al., 2012Venzal JM, Nava S, Mangold AJ, Mastropaolo M, Casás G, Guglielmone AA. Ornithodoros quilinensis sp. nov. (Acari, Argasidae), a new tick species from the Chacoan region in Argentina. Acta Parasitol 2012; 57(3): 329-336. PMid:22875683.). In all of these inferences, the members of these genera form two distinct monophyletic groups that occur on a common branch. The Ornithodoros s.s group comprises the true Ornithodoros species in the subfamily Ornithodorinae, based in the classification of the Argasidae family (KLOMPEN & OLIVER, 1993Klompen JSH, Oliver JH Jr. Systematic relationships in the soft ticks (Acari: Ixodida: Argasidae). Syst Entomol 1993; 18(4): 313-331.), which, in our phylogenetic analysis, are represented by O. parkeri, O. moubata, O. coriaceus and O. rostratus. Recent inferences regarding the phylogeny of soft ticks (BURGER et al., 2014Burger TD, Shao R, Labruna MB, Barker SC. Molecular phylogeny of soft ticks (Ixodida: Argasidae) inferred from mitochondrial genome and nuclear rRNA sequences. Ticks Tick Borne Dis 2014; 5(2): 195-207. PMid:24378079.) also identified the same species and other Ornithodoros sp. within Ornithodoros s.s., thereby agreeing with our results. Thus, our phylogenetic inference agrees with the proposal of Neotropical Ornithodorinae and Ornithodoros s.s. groups in the family Argasidae.
The paraphyletic group of genus Ornithodoros can be an evidence that the subgenus Alectorobius may not belong to the genus, such as is proposed by Klompen & Oliver (1993)Klompen JSH, Oliver JH Jr. Systematic relationships in the soft ticks (Acari: Ixodida: Argasidae). Syst Entomol 1993; 18(4): 313-331.. In other words, if O. mimon is considered as Carios mimon, in accordance with the classification of Klompen & Oliver (1993)Klompen JSH, Oliver JH Jr. Systematic relationships in the soft ticks (Acari: Ixodida: Argasidae). Syst Entomol 1993; 18(4): 313-331., who grouped Ornithodoros/Alectorobius and Antricola to genus Carios sensu lato, then this last genus would be monophyletic, represented by the Neotropical Ornithodorinae group in our phylogenetic inference. On the other hand, if the species O. mimon and A. delacruzi are considered as belonging to two distinct genera, Carios and Antricola, in accordance to Labruna et al. (2008)Labruna MB, Terassini FA, Camargo LM, Brandão PE, Ribeiro AF, Estrada-Peña A. New reports of Antricola guglielmonei and Antricola delacruzi in Brazil, and a description of a new argasid species (Acari). J Parasitol 2008; 94(4): 788-792. PMid:18576796., the members of Neotropical Ornithodorinae group (O. mimon and A. delacruzi) would form two monophyletic clades, which comprise the genera Carios and Antricola (LABRUNA et al., 2008Labruna MB, Terassini FA, Camargo LM, Brandão PE, Ribeiro AF, Estrada-Peña A. New reports of Antricola guglielmonei and Antricola delacruzi in Brazil, and a description of a new argasid species (Acari). J Parasitol 2008; 94(4): 788-792. PMid:18576796.). In both situations, the genus Ornithodoros would also be a monophyletic group, formed only by Ornithodoros s. s. group. Burger et al. (2014)Burger TD, Shao R, Labruna MB, Barker SC. Molecular phylogeny of soft ticks (Ixodida: Argasidae) inferred from mitochondrial genome and nuclear rRNA sequences. Ticks Tick Borne Dis 2014; 5(2): 195-207. PMid:24378079. also suggest that the Neotropical Ornithodorinae clade may be compatible with the genus Carios s. l. of Klompen & Oliver (1993)Klompen JSH, Oliver JH Jr. Systematic relationships in the soft ticks (Acari: Ixodida: Argasidae). Syst Entomol 1993; 18(4): 313-331., although they highlighted that, to confirm this hypothesis, it is required more data, particularly from the type species of Carios, Argas (Carios) vespertilionis (BURGER et al., 2014Burger TD, Shao R, Labruna MB, Barker SC. Molecular phylogeny of soft ticks (Ixodida: Argasidae) inferred from mitochondrial genome and nuclear rRNA sequences. Ticks Tick Borne Dis 2014; 5(2): 195-207. PMid:24378079.).
In the Ixodidae clade, the species of Rhipicephalus (R. microplus, R. appendiculatus and R. sanguineus s.l.) form a sister group with the members of Dermacentor (D. variabilis and D. andersoni), thus supporting the Rhipicephalinae subfamily (BLACK et al., 1997Black WC 4th, Klompen JS, Keirans JE. Phylogenetic relationships among tick subfamilies (Ixodida: Ixodidae: Argasidae) based on the 18S nuclear rDNA gene. Mol Phylogenet Evol 1997; 7(1): 129-144. PMid:9007027.; BARKER & MURRELL, 2002Barker SC, Murrell A. Phylogeny, evolution and historical zoogeography of ticks: a review of recent progress. Exp Appl Acarol 2002; 28(1-4): 55-68. PMid:14570116.). In our phylogenetic analysis, Rhipicephalinae was a sister group of Amblyomminae, while Haemaphysalinae formed an underlying group in Metastriata. There is some controversy regarding the phylogenetic positions of these subfamilies and genera (NAVA et al., 2009Nava S, Guglielmone AA, Mangold AJ. An overview of systematics and evolution of ticks. Front Biosci 2009; 14: 2857-2877. PMid:19273240.). Some phylogenetic studies on ixodids have inferred that there is a close relationship between Rhipicephalinae and Haemaphysalinae, such that Amblyomminae had an underlying position in relation to them (CRAMPTON et al., 1996Crampton A, McKay I, Barker SC. Phylogeny of ticks (Ixodida) inferred from nuclear ribosomal DNA. Int J Parasitol 1996; 26(5): 511-517. PMid:8818731.; BLACK et al., 1997Black WC 4th, Klompen JS, Keirans JE. Phylogenetic relationships among tick subfamilies (Ixodida: Ixodidae: Argasidae) based on the 18S nuclear rDNA gene. Mol Phylogenet Evol 1997; 7(1): 129-144. PMid:9007027.; MANGOLD et al., 1998Mangold AJ, Bargues MD, Mas-Coma S. 18S rRNA gene sequences and phylogenetic relationships of European hard-tick species (Acari: Ixodidae). Parasitol Res 1998; 84(1): 31-37. PMid:9491423.). However, in another analysis based in 16s rDNA, Amblyomminae and Haemaphysalinae were placed on a common branch, but Amblyomminae was paraphyletic, with the species A. americanum and A. sculptum (cited as A. cajennense) close to Rhipicephalinae, and A. variegatum and Amblyomma hebraeum Koch, 1844, correlated with Haemaphysalinae (BLACK & PIESMAN, 1994Black WC 4th, Piesman J. Phylogeny of hard-and soft-tick taxa (Acari: Ixodida) based on mitochondrial 16S rDNA sequences. Proc Natl Acad Sci USA 1994; 91(21): 10034-10038. PMid:7937832.). Amblyomminae was also reported to be paraphyletic in another phylogenetic inference (DOBSON & BARKER, 1999Dobson SJ, Barker SC. Phylogeny of the hard ticks (Ixodidae) inferred from 18S rRNA indicates that the genus Aponomma is paraphyletic. Mol Phylogenet Evol 1999; 11(2): 288-295. PMid:10191073.), in which this subfamily was a sister group of Rhipicephalinae. Our results showed topology similar to that proposed by these last phylogenetic inferences (BLACK & PIESMAN, 1994Black WC 4th, Piesman J. Phylogeny of hard-and soft-tick taxa (Acari: Ixodida) based on mitochondrial 16S rDNA sequences. Proc Natl Acad Sci USA 1994; 91(21): 10034-10038. PMid:7937832.; DOBSON & BARKER, 1999Dobson SJ, Barker SC. Phylogeny of the hard ticks (Ixodidae) inferred from 18S rRNA indicates that the genus Aponomma is paraphyletic. Mol Phylogenet Evol 1999; 11(2): 288-295. PMid:10191073.), thus disagreeing with the remainder of the previous inferences regarding the Ixodidae family.
Thus, we can conclude that transcripts from tick transcriptomes are a viable source for demonstrating, inferring, reevaluating and validating the phylogenetic relationships of ticks, with resolution similar to that of other datasets, such as mitochondrial genome and nuclear rRNA sequences. However, we must highlight that there is a need for more transcriptome studies of ticks, mainly of argasids, to improve the resolution of the transcriptomic datasets and to better knowledge of the roles of the genes of the survival and development of ticks.
Conclusions
In the our transcriptomic analysis, which is the first mialome of soft ticks, we demonstrated that important genes associated with the gut functions, such as digestion of blood meal and nutrition, are expressed on engorged female of O. mimon, contributing to a better understanding of the gut proteins, the possible mechanisms of digestion and the parasite-host relationship. Besides, we demonstrated also that transcriptomic datasets can be a viable source to inference the phylogeny of ticks. Proteomics studies will likely increase the understanding of the biochemical and physiological functions of gut proteins, providing information that could be a starting point for discovering new targets to development alternative tick-control strategies.
Supplementary Material
Supplementary Material accompanies this paper.
Figure S1. Phylogenetic tree of cathepsins.
This material is available as part of the online article from http://www.scielo.br/rbpv
Acknowledgements
This study was supported by the National Council for Scientific and Technological Development (Conselho Nacional de Desenvolvimento Científico e Tecnológico, CNPq; grant 480117/2011-0 to DMBB) and in part by the Research Support Foundation of the State of São Paulo (Fundação de Amparo à Pesquisa do Estado de São Paulo, FAPESP; grant number 2010/52183-3 to DMBB). We are also very grateful to the Genomic and Transcriptomic Facility from Applied Special Toxinology Laboratory (LETA) of the Butantan Institute, especially to Úrsula Castro Oliveira and Mariana Salgado Morone for their technical assistance.
References
- Akov S, Samish M, Galun R. Protease activity in female Ornithodoros tholozani ticks. Acta Trop 1976; 33(1): 37-52. PMid:13632.
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol 1990; 215(3): 403-410. PMid:2231712.
- Anderson JM, Sonenshine DE, Valenzuela JG. Exploring the mialome of ticks: an annotated catalogue of midgut transcripts from the hard tick, Dermacentor variabilis (Acari: Ixodidae). BMC Genomics 2008; 9: 552. PMid:19021911.
- Barker SC, Murrell A. Phylogeny, evolution and historical zoogeography of ticks: a review of recent progress. Exp Appl Acarol 2002; 28(1-4): 55-68. PMid:14570116.
- Barker SC, Murrell A. Systematics and evolution of ticks with a list of valid genus and species names. Parasitology 2004; 129(Suppl): S15-S36. PMid:15938503.
- Barrett AJ, Rawlings ND. Families and clans of serine peptidases. Arch Biochem Biophys 1995; 318(2): 247-250. PMid:7733651.
- Barrett AJ, Rawlings ND. Families and clans of cysteine peptidases. Perspect Drug Discov Des 1996; 6(1): 1-11.
- Barros-Battesti DM, Landulfo GA, Luz HR, Marcili A, Onofrio VC, Famadas KM. Ornithodoros faccinii n. sp. (Acari: Ixodida: Argasidae) parasitizing the frog Thoropa miliaris (Amphibia: Anura: Cycloramphidae) in Brazil. Parasit Vectors 2015; 8: 268. PMid:25963542.
- Barros-Battesti DM, Landulfo GA, Onofrio VC, Faccini JL, Marcili A, Nieri-Bastos FA, et al. Carios mimon (Acari: Argasidae): description of adults and redescription of larva. Exp Appl Acarol 2011; 54(1): 93-104. PMid:21161720.
- Barros-Battesti DM, Ramirez DG, Landulfo GA, Faccini JL, Dantas-Torres F, Labruna MB, et al. Immature argasid ticks: diagnosis and keys for Neotropical region. Rev Bras Parasitol Vet 2013; 22(4): 443-456. PMid:24473867.
- Black WC 4th, Klompen JS, Keirans JE. Phylogenetic relationships among tick subfamilies (Ixodida: Ixodidae: Argasidae) based on the 18S nuclear rDNA gene. Mol Phylogenet Evol 1997; 7(1): 129-144. PMid:9007027.
- Black WC 4th, Piesman J. Phylogeny of hard-and soft-tick taxa (Acari: Ixodida) based on mitochondrial 16S rDNA sequences. Proc Natl Acad Sci USA 1994; 91(21): 10034-10038. PMid:7937832.
- Boldbaatar D, Sikalizyo Sikasunge C, Battsetseg B, Xuan X, Fujisaki K. Molecular cloning and functional characterization of an aspartic protease from the hard tick Haemaphysalis longicornis. Insect Biochem Mol Biol 2006; 36(1): 25-36. PMid:16360947.
- Burger TD, Shao R, Labruna MB, Barker SC. Molecular phylogeny of soft ticks (Ixodida: Argasidae) inferred from mitochondrial genome and nuclear rRNA sequences. Ticks Tick Borne Dis 2014; 5(2): 195-207. PMid:24378079.
- Castro MH, Klerk D, Pienaar R, Latif AA, Rees DJG, Mans BJ. De novo assembly and annotation of the salivary gland transcriptome of Rhipicephalus appendiculatus male and female ticks during blood feeding. Ticks Tick Borne Dis 2016; 7(4): 536-548. PMid:26830274.
- Chaudhary R, Burleigh JG, Fernández-Baca D. Inferring species trees from incongruent multi-copy gene trees using the Robinson-Foulds distance. Algorithms Mol Biol 2013; 8(1): 28. PMid:24180377.
- Chaudhary R, Fernández-Baca D, Burleigh JG. MulRF: a software package for phylogenetic analysis using multi-copy gene trees. Bioinformatics 2015; 31(3): 432-433. PMid:25273112.
- Chmelař J, Kotál J, Karim S, Kopacek P, Francischetti IM, Pedra JH, et al. Sialomes and mialomes: a systems-biology view of tick tissues and tick–host interactions. Trends Parasitol 2016; 32(3): 242-254. PMid:26520005.
- Chrudimská T, Chrudimský T, Golovchenko M, Rudenko N, Grubhoffer L. New defensins from hard and soft ticks: similarities, differences, and phylogenetic analyses. Vet Parasitol 2010; 167(2-4): 298-303. PMid:19836137.
- Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005; 21(18): 3674-3676. PMid:16081474.
- Contreras-Moreira B, Vinuesa P. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl Environ Microbiol 2013; 79(24): 7696-7701. PMid:24096415.
- Cossío-Bayúgar R, Miranda E, Holman PJ. Molecular cloning of a phospholipid-hydroperoxide glutathione peroxidase gene from the tick, Boophilus microplus (Acari: Ixodidae). Insect Biochem Mol Biol 2005; 35(12): 1378-1387. PMid:16291093.
- Crampton A, McKay I, Barker SC. Phylogeny of ticks (Ixodida) inferred from nuclear ribosomal DNA. Int J Parasitol 1996; 26(5): 511-517. PMid:8818731.
- Díaz-Martín V, Manzano-Román R, Obolo-Mvoulouga P, Oleaga A, Pérez-Sánchez R. Development of vaccines against Ornithodoros soft ticks: an update. Ticks Tick Borne Dis 2015; 6(3): 211-220. PMid:25802033.
- Dobson SJ, Barker SC. Phylogeny of the hard ticks (Ixodidae) inferred from 18S rRNA indicates that the genus Aponomma is paraphyletic. Mol Phylogenet Evol 1999; 11(2): 288-295. PMid:10191073.
- Dujon B. The yeast genome project: what did we learn? Trends Genet 1996; 12(7): 263-270. PMid:8763498.
- Dupejova J, Sterba J, Vancova M, Grubhoffer L. Hemelipoglycoprotein from the ornate sheep tick, Dermacentor marginatus: structural and functional characterization. Parasit Vectors 2011; 4: 4. PMid:21214898.
- Duscher GG, Galindo RC, Tichy A, Hummel K, Kocan KM, de la Fuente J. Glutathione S-transferase affects permethrin detoxification in the brown dog tick, Rhipicephalus sanguineus. Ticks Tick Borne Dis 2014; 5(3): 225-233. PMid:24548526.
- Estrada-Peña A, Mangold AJ, Nava S, Venzal JM, Labruna M, Guglielmone AA. A review of the systematics of the tick family Argasidae (Ixodida). Acarologia 2010; 50(3): 317-333.
- Estrada-Peña A, Venzal JM, González-Acuña D, Mangold AJ, Guglielmone AA. Notes on New World Persicargas ticks (Acari: Argasidae) with description of female Argas (P.) keiransi. J Med Entomol 2006; 43(5): 801-809. PMid:17017212.
- Francischetti IM, Mans BJ, Meng Z, Gudderra N, Veenstra TD, Pham VM, et al. An insight into the sialome of the soft tick, Ornithodoros parkeri. Insect Biochem Mol Biol 2008a; 38(1): 1-21. PMid:18070662.
- Francischetti IM, Meng Z, Mans BJ, Gudderra N, Hall M, Veenstra TD, et al. An insight into the salivary transcriptome and proteome of the soft tick and vector of epizootic bovine abortion, Ornithodoros coriaceus. J Proteomics 2008b; 71(5): 493-512. PMid:18725333.
- Franta Z, Frantová H, Konvičková J, Horn M, Sojka D, Mareš M, et al. Dynamics of digestive proteolytic system during blood feeding of the hard tick Ixodes ricinus. Parasit Vectors 2010; 3: 119. PMid:21156061.
- Fukuchi S, Nishikawa K. Estimation of the number of authentic orphan genes in bacterial genomes. DNA Res 2004; 11(4): 219-231, 311-313. PMid:15500248.
- Fuzita FJ, Pinkse MW, Patane JS, Juliano MA, Verhaert PD, Lopes AR. Biochemical, transcriptomic and proteomic analyses of digestion in the scorpion Tityus serrulatus: insights into function and evolution of digestion in an ancient arthropod. PLoS One 2015; 10(4): e0123841. PMid:25875018.
- Geiser DL, Zhang D, Winzerling JJ. Secreted ferritin: mosquito defense against iron overload? Insect Biochem Mol Biol 2006; 36(3): 177-187. PMid:16503479.
- Gibson AK, Smith Z, Fuqua C, Clay K, Colbourne JK. Why so many unknown genes? Partitioning orphans from a representative transcriptome of the lone star tick Amblyomma americanum. BMC Genomics 2013; 14: 135. PMid:23445305.
- Grandjean O. Blood digestion in Ornithodoros moubata Murray Sensu stricto Walton (Ixodoidea: Argasidae) females. I. Biochemical changes in the midgut lumen and ultrastructure of the midgut cell, related to intracellular digestion. Acarologia 1984; 25(2): 147-165.
- Grunclová L, Fouquier H, Hypsa V, Kopáček P. Lysozyme from the gut of the soft tick Ornithodoros moubata: the sequence, phylogeny and post-feeding regulation. Dev Comp Immunol 2003; 27(8): 651-660. PMid:12798362.
- Grunclová L, Horn M, Vancová M, Sojka D, Franta Z, Mares M, et al. Two secreted cystatins of the soft tick Ornithodoros moubata: differential expression pattern and inhibitory specificity. Biol Chem 2006; 387(12): 1635-1644. PMid:17132111.
- Guglielmone AA, Robbins RG, Apanaskevich DA, Petney TN, Estrada-Peña A, Horak IG, et al. The Argasidae, Ixodidae and Nuttalliellidae (Acari: Ixodida) of the world: a list of valid species names. Zootaxa 2010; 2528: 1-28.
- Hatta T, Kazama K, Miyoshi T, Umemiya R, Liao M, Inoue N, et al. Identification and characterisation of a leucine aminopeptidase from the hard tick Haemaphysalis longicornis. Int J Parasitol 2006; 36(10-11): 1123-1132. PMid:16814790.
- He H, Chen AC, Davey RB, Ivie GW, George JE. Characterization and molecular cloning of a glutathione S-transferase gene from the tick, Boophilus microplus (Acari: Ixodidae). Insect Biochem Mol Biol 1999; 29(8): 737-743. PMid:10451925.
- Heekin AM, Guerrero FD, Bendele KG, Saldivar L, Scoles GA, Dowd SE, et al. Gut transcriptome of replete adult female cattle ticks, Rhipicephalus (Boophilus) microplus, feeding upon a Babesia bovis-infected bovine host. Parasitol Res 2013; 112(9): 3075-3090. PMid:23749091.
- Hodcroft E, Hadfield JD, Fearnhill E, Phillips A, Dunn D, O’Shea S, et al. The contribution of viral genotype to plasma viral set-point in HIV infection. PLoS Pathog 2014; 10(5): e1004112. PMid:24789308.
- Hoogstraal H. Argasid and nuttalliellid ticks as parasites and vectors. Adv Parasitol 1985; 24: 135-238. PMid:3904345.
- Horak IG, Camicas JL, Keirans JE. The Argasidae, Ixodidae and Nuttalliellidae (Acari: Ixodida): a world list of valid tick names. Exp Appl Acarol 2002; 28(1-4): 27-54. PMid:14570115.
- Horn M, Nussbaumerová M, Sanda M, Kovárová Z, Srba J, Franta Z, et al. Hemoglobin digestion in blood-feeding ticks: mapping a multipeptidase pathway by functional proteomics. Chem Biol 2009; 16(10): 1053-1063. PMid:19875079.
- Imamura S, Vaz IS Jr, Sugino M, Ohashi K, Onuma M. A serine protease inhibitor (serpin) from Haemaphysalis longicornis as an anti-tick vaccine. Vaccine 2005; 23(10): 1301-1311. PMid:15652673.
- Katoh K, Kuma K, Toh H, Miyata T. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res 2005; 33(2): 511-518. PMid:15661851.
- Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 2002; 30(14): 3059-3066. PMid:12136088.
- Klompen JSH, Oliver JH Jr. Systematic relationships in the soft ticks (Acari: Ixodida: Argasidae). Syst Entomol 1993; 18(4): 313-331.
- Koh K, Shiraishi S, Uchida TA. Digestion of bloodmeal protein after detachment in nymphal Haemaphysalis longicornis. J Fac Ag. Kyushu Univ 1990; 34(3): 241-246.
- Kongsuwan K, Josh P, Zhu Y, Pearson R, Gough J, Colgrave ML. Exploring the midgut proteome of partially fed female cattle tick (Rhipicephalus (Boophilus) microplus). J Insect Physiol 2010; 56(2): 212-226. PMid:19840806.
- Kopáček P, Hajdusek O, Buresová V, Daffre S. Tick innate immunity. Adv Exp Med Biol 2010; 708: 137-162. PMid:21528697.
- Kopáček P, Vogt R, Jindrák L, Weise C, Safarík I. Purification and characterization of the lysozyme from the gut of the soft tick Ornithodoros moubata. Insect Biochem Mol Biol 1999; 29(11): 989-997. PMid:10560138.
- Kopáček P, Zdychová J, Yoshiga T, Weise C, Rudenko N, Law JH. Molecular cloning, expression and isolation of ferritins from two tick species -- Ornithodoros moubata and Ixodes ricinus. Insect Biochem Mol Biol 2003; 33(1): 103-113. PMid:12459205.
- Kotsyfakis M, Schwarz A, Erhart J, Ribeiro JM. Tissue- and time-dependent transcription in Ixodes ricinus salivary glands and midguts when blood feeding on the vertebrate host. Sci Rep 2015; 5: 9103. PMid:25765539.
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 2001; 305(3): 567-580. PMid:11152613.
- Labruna MB, Nava S, Terassini FA, Onofrio VC, Barros-Battesti DM, Camargo LM, et al. Description of adults and nymph, and redescription of the larva, of Ornithodoros marinkellei (Acari:Argasidae), with data on its phylogenetic position. J Parasitol 2011; 97(2): 207-217. PMid:21506769.
- Labruna MB, Terassini FA, Camargo LM, Brandão PE, Ribeiro AF, Estrada-Peña A. New reports of Antricola guglielmonei and Antricola delacruzi in Brazil, and a description of a new argasid species (Acari). J Parasitol 2008; 94(4): 788-792. PMid:18576796.
- Lara FA, Lins U, Bechara GH, Oliveira PL. Tracing heme in a living cell: hemoglobin degradation and heme traffic in digest cells of the cattle tick Boophilus microplus. J Exp Biol 2005; 208(16): 3093-3101. PMid:16081607.
- Lara FA, Lins U, Paiva-Silva G, Almeida IC, Braga CM, Miguens FC, et al. A new intracellular pathway of haem detoxification in the midgut of the cattle tick Boophilus microplus: aggregation inside a specialized organelle, the hemosome. J Exp Biol 2003; 206(10): 1707-1715. PMid:12682102.
- Li L, Stoeckert CJ Jr, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 2003; 13(9): 2178-2189. PMid:12952885.
- Mangold AJ, Bargues MD, Mas-Coma S. 18S rRNA gene sequences and phylogenetic relationships of European hard-tick species (Acari: Ixodidae). Parasitol Res 1998; 84(1): 31-37. PMid:9491423.
- Mans BJ, Andersen JF, Francischetti IM, Valenzuela JG, Schwan TG, Pham VM, et al. Comparative sialomics between hard and soft ticks: implications for the evolution of blood-feeding behavior. Insect Biochem Mol Biol 2008; 38(1): 42-58. PMid:18070664.
- Marchler-Bauer A, Bryant SH. CD-Search: protein domain annotations on the fly. Nucleic Acids Res 2004; 32(S2): 327-331. PMid:15215404.
- Maya-Monteiro CM, Daffre S, Logullo C, Lara FA, Alves EW, Capurro ML, et al. HeLp, a heme lipoprotein from the hemolymph of the cattle tick, Boophilus microplus. J Biol Chem 2000; 275(47): 36584-36589. PMid:10964932.
- Mendiola J, Alonso M, Marquetti MC, Finlay C. Boophilus microplus: multiple proteolytic activities in the midgut. Exp Parasitol 1996; 82(1): 27-33. PMid:8617328.
- Moreti R, Perrella NN, Lopes AR. Carbohydrate digestion in ticks and a digestive α-L- fucosidase. J Insect Physiol 2013; 59(10): 1069-1075. PMid:23994295.
- Mulenga A, Sugino M, Nakajim M, Sugimoto C, Onuma M. Tick- Encoded serine proteinase inhibitors (serpins); potential target antigens for tick vaccine development. J Vet Med Sci 2001; 63(10): 1063-1069. PMid:11714020.
- Mulenga A, Tsuda A, Onuma M, Sugimoto C. Four serine proteinase inhibitors (serpin) from the brown ear tick, Rhipicephalus appendiculatus; cDNA cloning and preliminary characterization. Insect Biochem Mol Biol 2003; 33(2): 267-276. PMid:12535684.
- Nakajima Y, Ogihara K, Taylor D, Yamakawa M. Antibacterial hemoglobin fragments from the midgut of the soft tick, Ornithodoros moubata (Acari: Argasidae). J Med Entomol 2003; 40(1): 78-81. PMid:12597657.
- Nakajima Y, Taylor D, Yamakawa M. Involvement of antibacterial peptide defensin in tick midgut defense. Exp Appl Acarol 2002; 28(1-4): 135-140. PMid:14570123.
- Nakajima Y, van Naters AVG, Taylor D, Yamakawa M. Two isoforms of a member of the arthropod defensin family from the soft tick, Ornithodoros moubata (Acari: Argasidae). Insect Biochem Mol Biol 2001; 31(8): 747-751. PMid:11378409.
- National Center for Biotechnology Information – NCBI. Search for Conserved Domains within a protein or coding nucleotide sequence [online]. Bethesda: NCBI; 2017 [cited 2017 Mar 1]. Available from: http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi
» http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi - Nava S, Beati L, Labruna MB, Cáceres AG, Mangold AJ, Guglielmone AA. Reassessment of the taxonomic status of Amblyomma cajennense () with the description of three new species, Amblyomma tonelliae n. sp., Amblyomma interandinum n. sp. and Amblyomma patinoi n. sp., and reinstatement of Amblyomma mixtum, and Amblyomma sculptum (Ixodida: Ixodidae). Ticks Tick Borne Dis 2014; 5(3): 252-276. PMid:24556273.
- Nava S, Guglielmone AA, Mangold AJ. An overview of systematics and evolution of ticks. Front Biosci 2009; 14: 2857-2877. PMid:19273240.
- Nava S, Venzal JM, Terassini FA, Mangold AJ, Camargo LMA, Labruna MB. Description of a New Argasid tick (Acari: Ixodida) from bat caves in Brazilian. Amazon J Parasitol 2010; 96(6): 1089-1101. PMid:21158616.
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 2015; 32(1): 268-274. PMid:25371430.
- Nielsen H, Engelbrecht J, Brunak S, von Heijne G. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng 1997; 10(1): 1-6. PMid:9051728.
- Oleaga A, Obolo-Mvoulouga P, Manzano-Romá R, Pérez-Sánchez R. Midgut proteome of an argasid tick, Ornithodoros erraticus: a comparison between unfed and engorged females. Parasit Vectors 2015; 8: 525. PMid:26459090.
- Perner J, Provazník J, Schrenková J, Urbanová V, Ribeiro JM, Kopáček P. RNA-seq analyses of the midgut from blood-and serum-fed Ixodes ricinus ticks. Sci Rep 2016; 6: 36695. PMid:27824139.
- Rawlings ND, Barrett AJ. Evolutionary families of peptidases. Biochem J 1993; 290(1): 205-218. PMid:8439290.
- Reddy BN, Prasad GBKS, Raghavendra K. In silico analysis of glutathione S-transferase supergene family revealed hitherto unreported insect specific δ-and ɛ-GSTs and mammalian specific μ-GSTs in Ixodes scapularis (Acari: Ixodidae). Comput Biol Chem 2011; 35(2): 114-120. PMid:21514893.
- Ribeiro JM, Alarcon-Chaidez F, Francischetti IM, Mans BJ, Mather TN, Valenzuela JG, et al. An annotated catalog of salivary gland transcripts from Ixodes scapularis ticks. Insect Biochem Mol Biol 2006; 36(2): 111-129. PMid:16431279.
- Ribeiro JMC, Anderson JM, Manoukis NC, Meng Z, Francischetti IMB. A further insight into the sialome of the tropical bont tick, Amblyomma variegatum. BMC Genomics 2011; 12: 136. PMid:21362191.
- Ribeiro JMC, Labruna MB, Mans BJ, Maruyama SR, Francischetti IM, Barizon GC, et al. The sialotranscriptome of Antricola delacruzi female ticks is compatible with non-hematophagous behavior and an alternative source of food. Insect Biochem Mol Biol 2012; 42(5): 332-342. PMid:22306723.
- Ribeiro JMC, Slovák M, Francischetti IMB. An insight into the sialome of Hyalomma excavatum. Ticks Tick Borne Dis 2017; 8(2): 201-207. PMid:28049606.
- Schwarz A, Tenzer S, Hackenberg M, Erhart J, Gerhold-Ay A, Mazur J, et al. A systems level analysis reveals transcriptomic and proteomic complexity in Ixodes ricinus midgut and salivary glands during early attachment and feeding. Mol Cell Proteomics 2014; 13(10): 2725-2735. PMid:25048707.
- Schwarz A, Valdés JJ, Kotsyfakis M. The role of cystatins in tick physiology and blood feeding. Ticks Tick Borne Dis 2012; 3(3): 117-127. PMid:22647711.
- Sojka D, Franta D, Horn M, Hajdušek O, Caffrey CR, Mares M, et al. Profiling of proteolytic enzymes in the gut of the tick Ixodes ricinus reveals an evolutionarily conserved network of aspartic and cysteine peptidases. Parasit Vectors 2008; 1(1): 7. PMid:18348719.
- Sojka D, Franta Z, Horn M, Caffrey CR, Mares M, Kopácek P. New insights into the machinery of blood digestion by ticks. Trends Parasitol 2013; 29(6): 276-285. PMid:23664173.
- Sojka D, Hajdušek O, Dvořák J, Sajid M, Franta Z, Schneider EL, et al. IrAE–An asparaginyl endopeptidase (legumain) in the gut of the hard tick Ixodes ricinus. Int J Parasitol 2007; 37(7): 713-724. PMid:17336985.
- Sojka D, Pytelková J, Perner J, Horn M, Konvicková JS, Schrenková J, et al. Multienzyme degradation of host serum albumin in ticks. Ticks Tick Borne Dis 2016; 7(4): 604-613. PMid:26724897.
- Sonenshine DE. Biology of ticks 1st ed. New York: Oxford University Press; 1991.
- Tautz D, Domazet-Lošo T. The evolutionary origin of orphan genes. Nat Rev Genet 2011; 12(10): 692-702. PMid:21878963.
- Tirloni L, Seixas A, Mulenga A, Vaz IS Jr, Termignoni C. A family of serine protease inhibitors (serpins) in the cattle tick Rhipicephalus (Boophilus) microplus. Exp Parasitol 2014; 137: 25-34. PMid:24333790.
- Valenzuela JG. Exploring tick saliva: from biochemistry to ‘sialomes’ and functional genomics. Parasitology 2004; 129(Suppl): S83-S94. PMid:15938506.
- Velasco G, Ferrando AA, Puente XS, Sánchez LM, López-Otín C. Human cathepsin O. Molecular cloning from a breast carcinoma, production of the active enzyme in Escherichia coli, and expression analysis in human tissues. J Biol Chem 1994; 269(43): 27136-27142. PMid:7929457.
- Venzal JM, Estrada-Peña A, Mangold AJ, González-Acuña D, Guglielmone AA. The Ornithodoros (Alectorobius) talaje species group (Acari: Ixodida: Argasidae): description of Ornithodoros (Alectorobius) rioplatensis n. sp. from South America. J Med Entomol 2008; 45(5): 832-840. PMid:18826024.
- Venzal JM, Nava S, Mangold AJ, Mastropaolo M, Casás G, Guglielmone AA. Ornithodoros quilinensis sp. nov. (Acari, Argasidae), a new tick species from the Chacoan region in Argentina. Acta Parasitol 2012; 57(3): 329-336. PMid:22875683.
- Vial L. Biological and ecological characteristics of soft ticks (Ixodida: Argasidae) and their impact for predicting tick and associated disease distribution. Parasite 2009; 16(3): 191-202. PMid:19839264.
- Xu XL, Cheng TY, Yang H, Liao ZH. De novo assembly and analysis of midgut transcriptome of Haemaphysalis flava and identification of genes involved in blood digestion, feeding and defending from pathogens. Infect Genet Evol 2016; 38: 62-72. PMid:26705239.
Publication Dates
-
Publication in this collection
Apr-Jun 2017
History
-
Received
01 Mar 2017 -
Accepted
17 Apr 2017