Abstract
The aim of this study is to detect the presence of tick-borne agents of genera Rickettsia, Borrelia, Babesia, Ehrlichia and Anaplasma in ticks collected from native wild birds in the state of Rio de Janeiro. Birds were captured and observed carefully to find the ectoparasites. DNA detection of hemoparasites was performed by means of the polymerase chain reaction (PCR). The sequences obtained were analyzed and their homologies were compared to the available isolates in the GenBank platform database. A total of 33 birds were captured from 20 different species, of which 14 were parasitized by Amblyomma longirostre (n = 22). There was absence of DNA from agents of the genera Babesia, Anaplasma and Ehrlichia in the evaluated samples. The phylogenetic analysis indicated that one sample had 100% identity with Rickettsia bellii (KJ534309), the other two samples showed 100% identity with Rickettsia sp. Aranha strain and strain AL (EU274654 and AY360216). The positive sample for R. bellii was also demonstrated to be positive for Borrelia sp., which presented a similarity of 91% with Borrelia turcica (KF422815). This is the first description of Borrelia sp. in ticks of the genus Amblyomma in South America.
Keywords:
Conservation units; ectoparasites; rickettisiae; Borrelia
Resumo
Este trabalho teve como objetivo detectar evidências moleculares da presença de agentes dos gêneros Rickettsia, Borrelia, Babesia, Anaplasma e Ehrlichia transmitidos por carrapatos coletados de aves silvestres no estado do Rio de Janeiro. Aves foram capturadas e observadas cuidadosamente a procura de ectoparasitos. A detecção de DNA de hemoparasitos foi realizada por meio da reação em cadeia da polimerase (PCR). As sequências obtidas foram analisadas e sua homologia comparada aos isolados disponíveis na base de dados da plataforma GenBank. Foram capturadas 33 aves, de 20 espécies diferentes das quais 14 estavam parasitadas por Amblyomma longirostre (n = 22). Houve ausência de DNA de agentes dos gêneros Babesia, Anaplasma e Ehrlichia nas amostras avaliadas. A análise filogenética indicou que uma amostra apresentou 100% de identidade com Rickettsia bellii (KJ534309), as outras duas amostras apresentaram 100% de identidade com Rickettsia sp. cepa Aranha e Cepa AL (EU274654 e AY360216.). A amostra positiva para R. bellii também apresentou positividade para Borrelia sp. que apresentou similaridade de 91% com Borrelia turcica (KF422815). Esta é a primeira descrição de Borrelia sp. em carrapatos do gênero Amblyomma na América do Sul.
Palavras-chaves:
Unidades de conservação; ectoparasitos; rickettisiae; Borrelia
Introduction
Birds are host to a wide variety of ticks, especially when the latter are in the larva and nymph stages. Therefore, this plays an important role in the epidemiology of several important diseases for veterinary and public health (LABRUNA et al., 2007Labruna MB, Sanfilippo LF, Demetrio C, Menezes AC, Pinter A, Guglielmone AA, et al. Ticks collected on birds in the state of São Paulo, Brazil. Exp Appl Acarol 2007; 43(2): 147-160. http://dx.doi.org/10.1007/s10493-007-9106-x. PMid:17882514.
http://dx.doi.org/10.1007/s10493-007-910...
; OGRZEWALSKA et al., 2010Ogrzewalska M, Uezu A, Labruna MB. Ticks (Acari: Ixodidae) infesting wild birds in the eastern Amazon, northern Brazil, with notes on rickettsial infection in ticks. Parasitol Res 2010; 106(4): 809-816. http://dx.doi.org/10.1007/s00436-010-1733-1. PMid:20140452.
http://dx.doi.org/10.1007/s00436-010-173...
; SANCHES et al., 2013Sanches GS, Martins TF, Lopes IT, Costa LF, Nunes PH, Camargo-Mathias MI, et al. Ticks infesting birds in Atlantic Forest fragments in Rio Claro, State of Sao Paulo, Brazil. Rev Bras Parasitol Vet 2013; 22(1): 6-12. http://dx.doi.org/10.1590/S1984-29612013000100003. PMid:24252949.
http://dx.doi.org/10.1590/S1984-29612013...
; CAPLIGINA et al., 2014Capligina V, Salmane I, Keiss O, Vilks K, Japina K, Baumanis V, et al. Prevalence of tick-borne pathogens in ticks collected from migratory birds in Latvia. Ticks Tick Borne Dis 2014; 5(1): 75-81. http://dx.doi.org/10.1016/j.ttbdis.2013.08.007. PMid:24246709.
http://dx.doi.org/10.1016/j.ttbdis.2013....
). Moreover, studies worldwide have reported the importance of wild birds in the expansion of tick-borne diseases, such as anaplasmosis, babesiosis, Lyme disease, spotted fever, and tick-borne encephalitis (PAROLA et al., 2013Parola P, Paddock CD, Socolovschi C, Labruna MB, Mediannikov O, Kernif T, et al. Update on tick- borne rickettsioses around the world: A geographic approach. Clin Microbiol Rev 2013; 26(4): 657-702. http://dx.doi.org/10.1128/CMR.00032-13. PMid:24092850.
http://dx.doi.org/10.1128/CMR.00032-13...
).
By limitations in displacement, ticks also use their hosts to overcome numerous geographical barriers. In recent years, studies of tick–bird associations have been increasing exponentially, due to their importance in the dispersal and maintenance of the different tick species, and (consequently) their passengers’ pathogens (HUBÁLEK, 2004Hubálek Z. An annotated checklist of pathogenic microorganisms associated with migratory birds. J Wildl Dis 2004; 40(4): 639-659. http://dx.doi.org/10.7589/0090-3558-40.4.639. PMid:15650082.
http://dx.doi.org/10.7589/0090-3558-40.4...
; LOSS et al., 2016Loss SR, Noden BH, Hamer GL, Hamer SA. A quantitative synthesis of the role of birds in carrying ticks and tick-borne pathogens in North America. Oecologia 2016; 182(4): 947-959. http://dx.doi.org/10.1007/s00442-016-3731-1. PMid:27670413.
http://dx.doi.org/10.1007/s00442-016-373...
; BUDACHETRI et al., 2017Budachetri K, Williams J, Mukherjee N, Sellers M, Moore F, Karim S. The microbiome of neotropical ticks parasitizing on passerine migratory birds. Ticks Tick Borne Dis 2017; 8(1): 170-173. http://dx.doi.org/10.1016/j.ttbdis.2016.10.014. PMid:27802919.
http://dx.doi.org/10.1016/j.ttbdis.2016....
).
In Brazil, studies on the diversity of ticks on birds have been carried out in several biomes (Atlantic Forest, Amazon, Caatinga, Cerrado, and Pantanal), with the immature stages (larva and nymph) of the genus Amblyomma being found to be the most common (LABRUNA et al., 2007Labruna MB, Sanfilippo LF, Demetrio C, Menezes AC, Pinter A, Guglielmone AA, et al. Ticks collected on birds in the state of São Paulo, Brazil. Exp Appl Acarol 2007; 43(2): 147-160. http://dx.doi.org/10.1007/s10493-007-9106-x. PMid:17882514.
http://dx.doi.org/10.1007/s10493-007-910...
; OGRZEWALSKA et al., 2010Ogrzewalska M, Uezu A, Labruna MB. Ticks (Acari: Ixodidae) infesting wild birds in the eastern Amazon, northern Brazil, with notes on rickettsial infection in ticks. Parasitol Res 2010; 106(4): 809-816. http://dx.doi.org/10.1007/s00436-010-1733-1. PMid:20140452.
http://dx.doi.org/10.1007/s00436-010-173...
; LUZ et al., 2012Luz HR, Faccini JL, Landulfo GA, Berto BP, Ferreira I. Bird ticks in an area of the Cerrado of Minas Gerais State, southeast Brazil. Exp Appl Acarol 2012; 58(1): 89-99. http://dx.doi.org/10.1007/s10493-012-9572-7. PMid:22729500.
http://dx.doi.org/10.1007/s10493-012-957...
; LUGARINI et al., 2015Lugarini C, Martins TF, Ogrzewalska M, de Vasconcelos NC, Ellis VA, de Oliveira JB, et al. Rickettsial agents in avian ixodid ticks in northeast Brazil. Ticks Tick Borne Dis 2015; 6(3): 364-375. http://dx.doi.org/10.1016/j.ttbdis.2015.02.011. PMid:25800099.
http://dx.doi.org/10.1016/j.ttbdis.2015....
; RAMOS et al., 2015Ramos DG, Melo AL, Martins TF, Alves AS, Pacheco TA, Pinto LB, et al. Rickettsial infection in ticks from wild birds from Cerrado and the Pantanal region of Mato Grosso, midwestern Brazil. Ticks Tick Borne Dis 2015; 6(6): 836-842. http://dx.doi.org/10.1016/j.ttbdis.2015.07.013. PMid:26232933.
http://dx.doi.org/10.1016/j.ttbdis.2015....
). Moreover, studies on infectious pathogenic agents in ticks that parasitize birds in Brazil are mainly concentrated in the detection of rickettsial agents (OGRZEWALSKA et al., 2008Ogrzewalska M, Pacheco RC, Uezu A, Ferreira F, Labruna MB. Ticks (Acari: Ixodidae) infesting wild birds in an Atlantic forest area in the state of São Paulo, Brazil, with isolation of Rickettsia from the tick Amblyomma longirostre. J Med Entomol 2008; 45(4): 770-774. http://dx.doi.org/10.1093/jmedent/45.4.770. PMid:18714882.
http://dx.doi.org/10.1093/jmedent/45.4.7...
). Other agents of the genera Borrelia, Anaplasma and Ehrlichia were also detected in ticks that were parasitizing wild birds. In North America and parts of Europe, the DNA of these agents was detected in these arthropods, which were collected from birds (SCOTT et al., 2010Scott JD, Lee MK, Fernando K, Durden LA, Jorgensen DR, Mak S, et al. Detection of Lyme disease spirochete, Borrelia burgdorferi sensu lato, including three novel genotypes in ticks (Acari: Ixodidae) collected from songbirds (Passeriformes) across Canada. J Vector Ecol 2010; 35(1): 124-139. http://dx.doi.org/10.1111/j.1948-7134.2010.00068.x. PMid:20618658.
http://dx.doi.org/10.1111/j.1948-7134.20...
; PALOMAR et al., 2012Palomar AM, Santibáñez P, Mazuelas D, Roncero L, Santibáñez S, Portillo A, et al. Role of birds in dispersal of etiologic agents of tick-borne zoonoses, Spain, 2009. Emerg Infect Dis 2012; 18(7): 1188-1191. http://dx.doi.org/10.3201/eid1807.111777. PMid:22709801.
http://dx.doi.org/10.3201/eid1807.111777...
; ERWIN et al., 2016Erwin JA, Fitak RR, Dwyer JF, Morrison JL, Culver M. Molecular detection of bacteria in the families Rickettsiaceae and Anaplasmataceae in northern crested caracaras (Caracara cheriway). Ticks Tick Borne Dis 2016; 7(3): 470-474. http://dx.doi.org/10.1016/j.ttbdis.2016.01.015. PMid:26837860.
http://dx.doi.org/10.1016/j.ttbdis.2016....
). Moreover, the species of ticks found on birds can also parasitize humans (GUGLIELMONE et al., 2006Guglielmone A, Beati L, Barros-Battesti D, Labruna M, Nava S, Venzal J, et al. Ticks (Ixodidae) on humans in South America. Exp Appl Acarol 2006; 40(2): 83-100. http://dx.doi.org/10.1007/s10493-006-9027-0. PMid:17103085.
http://dx.doi.org/10.1007/s10493-006-902...
). For these reasons, there is a need for studies that characterize organisms that infect ticks found on birds, to enable a better understanding of the diversity and ecology of the zoonoses transmitted by these arthropods. Thus, the objective of the present work is to detect (through molecular techniques) hemoparasites present in ticks from birds of the municipality of Guapimirim and Serra dos Órgãos National Park in the state of Rio de Janeiro, Brazil.
Material and Methods
The study was carried out in the municipality of Guapimirim (latitude: 22º 31' 14”/ longitude: 43º 00' 52”; altitude 256 meters) and in the Serra dos Órgãos National Park (latitude: 22º 31' 43'/ longitude: 43º 00' 12”; altitude 256 meters), both of which are located in the state of Rio de Janeiro, Brazil. Three field visits, each lasting four days, were conducted between March and September 2016 at the place of study.
The birds were captured between 06:00 and 17:00 hours, using 5–20 ornithological mist nets model (12 m long × 3 m wide, with 16 and 36 mm mesh). The birds were then photographed and identified following the recommendations based on the nomenclature approved by the Brazilian Committee of Ornithological Records (CBRO, 2014Comitê Brasileiro de Registros Ornitológicos – CBRO. Listas das aves do Brasil. 10ª ed. Florianópolis: CBRO; 2014.). Each bird was examined for the presence of ticks. If any were found, they were removed with forceps and placed in 1.5 mL polyethylene tubes containing absolute alcohol for later identification. Samples were initially stored at room temperature for up to four days. After collecting the ticks, the birds were released at the same collection site. All procedures were performed with live birds.
In the laboratory, the ticks were microscopically examined for identification, using a dichotomous key published by Martins et al. (2010)Martins TF, Onofrio VC, Barros-Battesti DM, Labruna MB. Nymphs of the genus Amblyomma (Acari: Ixodidae) of Brazil: descriptions, redescriptions, and identification key. Ticks Tick Borne Dis 2010; 1(2): 75-99. http://dx.doi.org/10.1016/j.ttbdis.2010.03.002. PMid:21771514.
http://dx.doi.org/10.1016/j.ttbdis.2010....
with the aid of a stereoscopic microscope. After identification, the arthropods were stored in RNA stabilization solution and frozen until processed for molecular analysis.
The ticks kept in RNA later were washed in distilled water three times and rehydrated in 200 μL of phosphate buffered saline (PBS). The ticks were then individually placed in 1.5 mL polyethylene tubes. Next, 2 mm zirconium oxide beads and 80 mg of 0.1 mm glass beads were added, both autoclaved, for trituration in Minibeadbeater BIOSPEC® for 1 min.
Cell lysis was performed with the addition of 250 μL of digestion solution (20 mM Tris-HCl, 20 mM EDTA, 400 mM NaCl, 1% sodium dodecyl sulphate, 10mM CaCl2) with 20 μL of proteinase k (20 mg/mL) in incubation overnight at 56 °C. The DNA was extracted by a phenol treatment and another phenol-chloroform treatment followed by precipitation with isopropanol. The DNA pellet (formed after centrifugation of 16000 xg) was washed twice with 70% alcohol and re-suspended in 100 μL of elution buffer (10 mM Tris-HCl, 0,5 mM EDTA pH 9,0) in overnight at 4 °C, as according to Santolin et al. (2013)Santolin IDAC, Famadas KM, McIntosh D. Detecção e identificação de espécies de Rickettsia em carrapatos coletados de aves silvestres no Brasil pela PCR-RFLP. Rev Bras Med Vet 2013; 35(Supl.2): 68-73..
The extracted DNA was tested by a battery of PCR assays that were targeting microorganisms of the genera Rickettsia, Borrelia, Ehrlichia, and Babesia. For this task, specific primers were used for each agent, following the original protocol of each primer (Table 1).
Sequences of the oligonucleotide primers used, along with their respective target genes and the size of the amplified fragment.
Reactions were carried out using reagents from the PROMEGA®, and each reaction contained 3 µL de DNA, 14,2 µL of water, 2 µL de primers (10 µM F+R), 2,5 µL of buffer (10X concentrado), 1,25 µL of MgCl2 (50 mM) and 2 µL of dNTP’s (2,5 mM), and 0,15 µL Taq DNA polimerase (PROMEGA®), in a final volume of 25 µL.
The amplified products were visualized in 1.5% agarose gel, which were stained with ethidium bromide and visualized in a UV-Transilluminator.
Polymerase chain reaction positive samples were submitted to sequencing and phylogenetic analyses. Multiple sequence alignments were performed with the sequences obtained from this study and sequences from GenBank using MUSCLE, using the SeaView v.4 software program (GOUY et al., 2010Gouy M, Guindon S, Gascuel O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol 2010; 27(2): 221-224. http://dx.doi.org/10.1093/molbev/msp259. PMid:19854763.
http://dx.doi.org/10.1093/molbev/msp259...
). The best-fit evolutionary model was determined using MEGA version 7, using the Bayesian information criterion (KUMAR et al., 2016Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol Biol Evol 2016; 33(7): 1870-1874. http://dx.doi.org/10.1093/molbev/msw054. PMid:27004904.
http://dx.doi.org/10.1093/molbev/msw054...
). Phylogenetic relationships were estimated using (a) Maximum likelihood (ML) phylogenetic inference that used PhyML, which was implemented in SeaView (GOUY et al., 2010Gouy M, Guindon S, Gascuel O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol 2010; 27(2): 221-224. http://dx.doi.org/10.1093/molbev/msp259. PMid:19854763.
http://dx.doi.org/10.1093/molbev/msp259...
) and (b) a Bayesian Markov chain Monte Carlo (MCMC) method implemented in MrBayes v.3.2.6 (SOARES et al., 2015Soares HS, Barbieri AR, Martins TF, Minervino AH, de Lima JT, Marcili A, et al. Ticks and rickettsial infection in the wildlife of two regions of the Brazilian Amazon. Exp Appl Acarol 2015; 65(1): 125-140. http://dx.doi.org/10.1007/s10493-014-9851-6. PMid:25273064.
http://dx.doi.org/10.1007/s10493-014-985...
). The MCMC settings consisted of two simultaneous independent runs with four chains each that were run for 10 million generations and sampled every 100th generation, thus yielding 100,000 trees. After eliminating 25% of the samples as burn-in, a consensus tree was built. Statistical support of the clades was measured by a heuristic search, with 1000 bootstrap replicates and the Bayesian posterior probabilities.
The study was evaluated and approved by the Animal Experimentation Ethics Committee of the Federal Rural University of Rio de Janeiro and was conducted with the permission of IBAMA; process num. 43917/3/2505369.
Results and Discussion
Detailed data of the tick–bird association that was reported in the current study can be found in Table 2. In total, 33 birds were captured, representing three orders, seven families, and 20 species, of which 14 (42%) of 9 species (45%) were parasitized by 22 immature forms of Amblyomma spp. Birds of the order Passeriformes were the most frequent, with 27 (82%) specimens captured, divided into 5 families and 15 species, corroborating with the data published in previous literature (LUZ & FACCINI, 2013Luz HR, Faccini JLH. Parasitismo por carrapatos em anuros no Brasil. Revisão. Vet Zootec 2013; 20: 100-111.; OGRZEWALSKA & PINTER, 2016Ogrzewalska M, Pinter A. Ticks (Acari: Ixodidae) as ectoparasites of Brazilian wild birds and their association with rickettsial diseases. Braz J Vet Res Anim Sci 2016; 53(1): 1-31. http://dx.doi.org/10.11606/issn.1678-4456.v53i1p1-31.
http://dx.doi.org/10.11606/issn.1678-445...
). No ticks were recorded on the following bird species: Passeriformes - Myiozetetes similis, Coereba flaveola, Sicalis flaveola, Tangara cayana, Tangara sayaca, Troglodytes musculus; Columbiformes - Leptotila rufaxilla, Geotrygon montana, Columbina talpacoti; Cuculiformes - Piaya cayana. However, there are numerous records of these species in association with ticks in different biomes in Brazil (LABRUNA et al., 2007Labruna MB, Sanfilippo LF, Demetrio C, Menezes AC, Pinter A, Guglielmone AA, et al. Ticks collected on birds in the state of São Paulo, Brazil. Exp Appl Acarol 2007; 43(2): 147-160. http://dx.doi.org/10.1007/s10493-007-9106-x. PMid:17882514.
http://dx.doi.org/10.1007/s10493-007-910...
; LUGARINI et al., 2015Lugarini C, Martins TF, Ogrzewalska M, de Vasconcelos NC, Ellis VA, de Oliveira JB, et al. Rickettsial agents in avian ixodid ticks in northeast Brazil. Ticks Tick Borne Dis 2015; 6(3): 364-375. http://dx.doi.org/10.1016/j.ttbdis.2015.02.011. PMid:25800099.
http://dx.doi.org/10.1016/j.ttbdis.2015....
; LUZ et al., 2016Luz HR, Faccini JLH, Landulfo GA, Costa Neto SF, Famadas KM. New records for Amblyomma sculptum (Ixodidae) on non-passerine birds in Brazil. Rev Bras Parasitol Vet 2016; 25(1): 124-126. http://dx.doi.org/10.1590/S1984-29612016004. PMid:27007247.
http://dx.doi.org/10.1590/S1984-29612016...
; OGRZEWALSKA & PINTER, 2016Ogrzewalska M, Pinter A. Ticks (Acari: Ixodidae) as ectoparasites of Brazilian wild birds and their association with rickettsial diseases. Braz J Vet Res Anim Sci 2016; 53(1): 1-31. http://dx.doi.org/10.11606/issn.1678-4456.v53i1p1-31.
http://dx.doi.org/10.11606/issn.1678-445...
).
Prevalence and mean intensity of infestation by ticks collected in birds in the Serra dos Órgãos National Park and Guapimirim.
After the laboratory analysis, all ticks were identified as nymphs of Amblyomma longirostre. In general, the infestations found in the birds were low, and were not exceeding the average intensity of 2.5/tick per bird. All the ticks were collected in the head and neck regions, and similar parasitic intensities have also been reported in the Atlantic Forest (LUZ & FACCINI, 2013Luz HR, Faccini JLH. Parasitismo por carrapatos em anuros no Brasil. Revisão. Vet Zootec 2013; 20: 100-111.). The immature parasitism of A. longirostre in wild birds has been reported throughout the neo-tropical region, especially on birds from the Passeriformes sub-group (LABRUNA et al., 2007Labruna MB, Sanfilippo LF, Demetrio C, Menezes AC, Pinter A, Guglielmone AA, et al. Ticks collected on birds in the state of São Paulo, Brazil. Exp Appl Acarol 2007; 43(2): 147-160. http://dx.doi.org/10.1007/s10493-007-9106-x. PMid:17882514.
http://dx.doi.org/10.1007/s10493-007-910...
; NAVA et al., 2010Nava S, Venzal JM, Labruna MB, Mastropaolo M, González EM, Mangold AJ, et al. Hosts, distribution and genetic divergence (16S rDNA) of Amblyomma dubitatum (Acari: Ixodidae). Exp Appl Acarol 2010; 51(4): 335-351. http://dx.doi.org/10.1007/s10493-009-9331-6. PMid:20084537.
http://dx.doi.org/10.1007/s10493-009-933...
; LUZ & FACCINI, 2013Luz HR, Faccini JLH. Parasitismo por carrapatos em anuros no Brasil. Revisão. Vet Zootec 2013; 20: 100-111., 2016). There are also reports of this species of tick on birds in the neartic region, but these arthropods do not have populations established in this region (GUGLIELMONE et al., 2014Guglielmone AA, Robbins RG, Apanaskevich DA, Petney TN, Estrada-Peña A, Horak IG. The hard ticks of the world (Acari: Ixodida: Ixodidae). 1ª ed. London: Springer; 2014. http://dx.doi.org/10.1007/978-94-007-7497-1.
http://dx.doi.org/10.1007/978-94-007-749...
). These findings reinforce the importance of wild birds in the maintenance and dispersion of this ectoparasite, as they are the main group of hosts for immature forms of A. longirostre in the wild environment. On the other hand, the adult stage of A. longirostre has been recorded mainly to be on neo-tropical porcupines of the family Erethizontidae, followed by occasional records on a variety of wild and domestic mammals of the families Cervidae, Canidae, Mustelidae, Phyllostomidae, Equidae, Bradypodidae and Sciuridae (BARROS-BATTESTI et al., 2006Barros-Battesti DM, Arzua M, Bechara GH. Carrapatos de importância médico-veterinária da região neotropical: um guia ilustrado para identificação de espécies. 1a ed. São Paulo: Vox/ICTTD-3/Butantan, 2006.; GUGLIELMONE et al., 2014Guglielmone AA, Robbins RG, Apanaskevich DA, Petney TN, Estrada-Peña A, Horak IG. The hard ticks of the world (Acari: Ixodida: Ixodidae). 1ª ed. London: Springer; 2014. http://dx.doi.org/10.1007/978-94-007-7497-1.
http://dx.doi.org/10.1007/978-94-007-749...
).
The arboreal habits of its primary hosts may justify the presence of the immature forms on birds that tend to share similar habits, suggesting an arboreal cycle for A. longirostre (LABRUNA et al., 2007Labruna MB, Sanfilippo LF, Demetrio C, Menezes AC, Pinter A, Guglielmone AA, et al. Ticks collected on birds in the state of São Paulo, Brazil. Exp Appl Acarol 2007; 43(2): 147-160. http://dx.doi.org/10.1007/s10493-007-9106-x. PMid:17882514.
http://dx.doi.org/10.1007/s10493-007-910...
).
Molecular analysis by the CS239/CS1069 primers revealed a 838 bp amplification of the gltA gene of Rickettsia spp. in three samples of A. longirostre that were collected on birds of three different species: Saltator similis, Turdus leucomelas, and Tangara seledon. The products sequenced from one sample had a 100% identity rate with the Rickettsia bellii isolated H3 (access in the GenBank: KJ534309). With the other two, the identity was 100% with Rickettsia sp. strain AL and the Rickettsia sp. Aranha strain (access in the GenBank: EU274654 and AY360216, respectively) both currently correlated with Rickettsia amblyommatis (OGRZEWALSKA et al., 2011Ogrzewalska M, Uezu A, Labruna MB. Ticks (Acari: Ixodidae) infesting wild birds in the Atlantic Forest in northeastern Brazil, with notes on rickettsial infection in ticks. Parasitol Res 2011; 108(3): 665-670. http://dx.doi.org/10.1007/s00436-010-2111-8. PMid:20953629.
http://dx.doi.org/10.1007/s00436-010-211...
; KARPATHY et al., 2016Karpathy SE, Slater KS, Goldsmith CS, Nicholson WL, Paddock CD. Rickettsia amblyommatis sp. nov., a spotted fever group Rickettsia associated with multiple species of Amblyomma ticks in North Central and South America. Int J Syst Evol Microbiol 2016; 66(12): 5236-5243. http://dx.doi.org/10.1099/ijsem.0.001502. PMid:27638476.
http://dx.doi.org/10.1099/ijsem.0.001502...
), as shown in Figure 1. These agents have already been described in Brazil by Ogrzewalska et al. (2008)Ogrzewalska M, Pacheco RC, Uezu A, Ferreira F, Labruna MB. Ticks (Acari: Ixodidae) infesting wild birds in an Atlantic forest area in the state of São Paulo, Brazil, with isolation of Rickettsia from the tick Amblyomma longirostre. J Med Entomol 2008; 45(4): 770-774. http://dx.doi.org/10.1093/jmedent/45.4.770. PMid:18714882.
http://dx.doi.org/10.1093/jmedent/45.4.7...
in the same tick species, A. longirostre. However, it was on birds of different species.
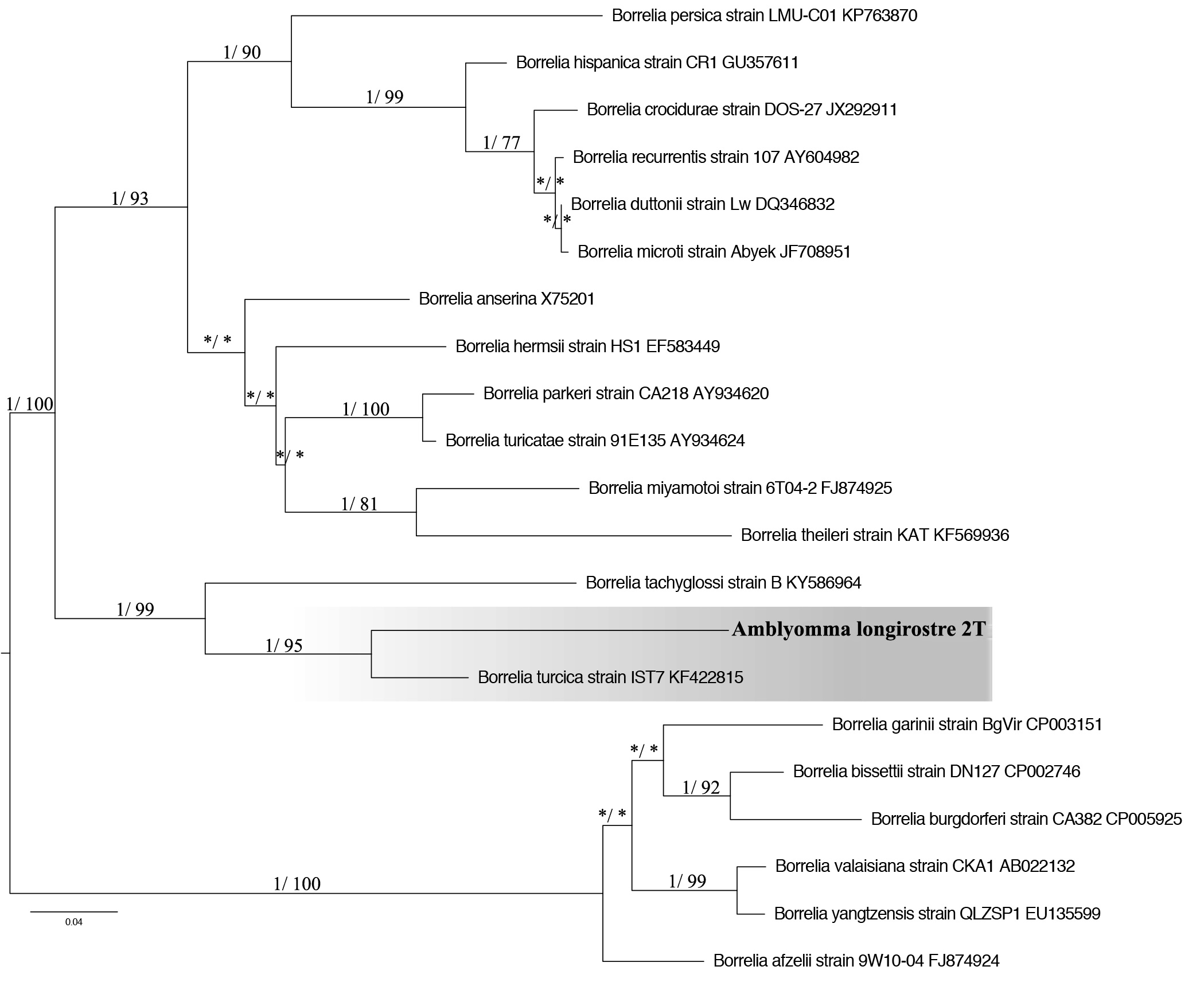
Phylogenetic tree based on the gltA gene (734nt) sequences of Rickettsia, using ML and Bayesian methods. Numbers (>0.7/>70%) above the branches indicate posterior node probabilities or bootstrap values (MrBayes/ML). *Indicate values below 0.7/70. †Exhibited difference between ML and MrBayes tree-building method topology. The scale bars indicate an evolutionary distance of 0.04 substitutions per position in the sequence and the branch labels include GenBank accession numbers. The Tamura 3-parameter model with gamma-distributed heterogeneity (T92 + G) was selected as the best-fit evolutionary model.
A sample of A. longirostre from Tangara seledon was positive for Borrelia spp. when using primers BorFlaF1/BorFlaR1 and BorFlaF2/BorFlaR2, which amplifies 740 bp of the flagellin B gene.
This sample presented a 91% identity rate with Borrelia turcica IST7 (access in the genbank: KF422815) (Figure 2), found in Hyalomma aegyptium. The partial sequence of the flaB gene also showed 99% similarity (coverage of 47% e 41%) with Borrelia sp. TX-Amac2 and Borrelia sp. F3 (access number KP861337 and KF395231), and both were found in A. maculatum that infested humans in the United States of America (MITCHELL et al., 2016Mitchell EA, Williamson PC, Billingsley PM, Seals JP, Ferguson EE, Allen MS. Frequency and distribution of Rickettsiae, Borreliae, and Ehrlichiae Detected in Human-Parasitizing Ticks, Texas, USA. Emerg Infect Dis 2016; 22(2): 312-315. http://dx.doi.org/10.3201/eid2202.150469. PMid:26811941.
http://dx.doi.org/10.3201/eid2202.150469...
).

Phylogenetic tree based on the flaB gene (652nt) sequences of Borrelia, using ML and Bayesian methods. Numbers (>0.7/>70%) above the branches indicate posterior node probabilities or bootstrap values (MrBayes/ML). *Indicate values below 0.7/70. The scale bars indicate an evolutionary distance of 0.04 substitutions per position in the sequence. The branch labels include GenBank accession numbers. The Tamura 3-parameter model with gamma-distributed heterogeneity (T92 + G) was selected as the best-fit evolutionary model.
The primers that target the hpt and glpQ genes of Borrelia spp. did not amplify products, probably because it is a conventional PCR and the sample does not have DNA in concentration sufficient for the amplification. Although A. longirostre has been described to infest humans in its immature stages (GUGLIELMONE et al., 2006Guglielmone A, Beati L, Barros-Battesti D, Labruna M, Nava S, Venzal J, et al. Ticks (Ixodidae) on humans in South America. Exp Appl Acarol 2006; 40(2): 83-100. http://dx.doi.org/10.1007/s10493-006-9027-0. PMid:17103085.
http://dx.doi.org/10.1007/s10493-006-902...
), it cannot be said that the transmission may occur, because it is not aware of the pathogenic potential of the bacteria. This finding is of great importance for the literature, because it is the first time a report has pertained to the presence of Borrelia sp. in A. longirostre. There was no DNA from Anaplasma sp., Ehrlichia sp., and protozoa of the order Piroplasmida infecting the ticks in the present study. Therefore, bacteria of the genera Borrelia and Rickettsia can be found in A. longirostre that parasitize birds in the state of Rio de Janeiro.
Acknowledgements
We would like to express our gratitude to the Conselho Nacional de Desenvolvimento Científico e Tecnológico – CNPq (A.H.F. 305480/2013-8) and Fundação de Apoio à pesquisa no estado do Rio de Janeiro – FAPERJ (A.H.F., grant number E 26/201.144/2014), for their financial support.
References
- Almeida AP, Souza TD, Marcili A, Labruna MB. Novel Ehrlichia and Hepatozoon agents infecting the crab-eating fox (Cerdocyon thous) in southeastern Brazil. J Med Entomol 2013; 50(3): 640-646. http://dx.doi.org/10.1603/ME12272 PMid:23802461.
» http://dx.doi.org/10.1603/ME12272 - Barros-Battesti DM, Arzua M, Bechara GH. Carrapatos de importância médico-veterinária da região neotropical: um guia ilustrado para identificação de espécies 1a ed. São Paulo: Vox/ICTTD-3/Butantan, 2006.
- Blanco CM, Teixeira BR, da Silva AG, de Oliveira RC, Strecht L, Ogrzewalska M, et al. Microorganisms in ticks (Acari: Ixodidae) collected on marsupials and rodents from Santa Catarina, Paraná and Mato Grosso do Sul states, Brazil. Ticks Tick Borne Dis 2017; 8(1): 90-98. http://dx.doi.org/10.1016/j.ttbdis.2016.10.003 PMid:27769655.
» http://dx.doi.org/10.1016/j.ttbdis.2016.10.003 - Budachetri K, Williams J, Mukherjee N, Sellers M, Moore F, Karim S. The microbiome of neotropical ticks parasitizing on passerine migratory birds. Ticks Tick Borne Dis 2017; 8(1): 170-173. http://dx.doi.org/10.1016/j.ttbdis.2016.10.014 PMid:27802919.
» http://dx.doi.org/10.1016/j.ttbdis.2016.10.014 - Capligina V, Salmane I, Keiss O, Vilks K, Japina K, Baumanis V, et al. Prevalence of tick-borne pathogens in ticks collected from migratory birds in Latvia. Ticks Tick Borne Dis 2014; 5(1): 75-81. http://dx.doi.org/10.1016/j.ttbdis.2013.08.007 PMid:24246709.
» http://dx.doi.org/10.1016/j.ttbdis.2013.08.007 - Comitê Brasileiro de Registros Ornitológicos – CBRO. Listas das aves do Brasil. 10ª ed. Florianópolis: CBRO; 2014.
- Erwin JA, Fitak RR, Dwyer JF, Morrison JL, Culver M. Molecular detection of bacteria in the families Rickettsiaceae and Anaplasmataceae in northern crested caracaras (Caracara cheriway). Ticks Tick Borne Dis 2016; 7(3): 470-474. http://dx.doi.org/10.1016/j.ttbdis.2016.01.015 PMid:26837860.
» http://dx.doi.org/10.1016/j.ttbdis.2016.01.015 - Gouy M, Guindon S, Gascuel O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol 2010; 27(2): 221-224. http://dx.doi.org/10.1093/molbev/msp259 PMid:19854763.
» http://dx.doi.org/10.1093/molbev/msp259 - Guglielmone A, Beati L, Barros-Battesti D, Labruna M, Nava S, Venzal J, et al. Ticks (Ixodidae) on humans in South America. Exp Appl Acarol 2006; 40(2): 83-100. http://dx.doi.org/10.1007/s10493-006-9027-0 PMid:17103085.
» http://dx.doi.org/10.1007/s10493-006-9027-0 - Guglielmone AA, Robbins RG, Apanaskevich DA, Petney TN, Estrada-Peña A, Horak IG. The hard ticks of the world (Acari: Ixodida: Ixodidae). 1ª ed. London: Springer; 2014. http://dx.doi.org/10.1007/978-94-007-7497-1
» http://dx.doi.org/10.1007/978-94-007-7497-1 - Hubálek Z. An annotated checklist of pathogenic microorganisms associated with migratory birds. J Wildl Dis 2004; 40(4): 639-659. http://dx.doi.org/10.7589/0090-3558-40.4.639 PMid:15650082.
» http://dx.doi.org/10.7589/0090-3558-40.4.639 - Karpathy SE, Slater KS, Goldsmith CS, Nicholson WL, Paddock CD. Rickettsia amblyommatis sp. nov., a spotted fever group Rickettsia associated with multiple species of Amblyomma ticks in North Central and South America. Int J Syst Evol Microbiol 2016; 66(12): 5236-5243. http://dx.doi.org/10.1099/ijsem.0.001502 PMid:27638476.
» http://dx.doi.org/10.1099/ijsem.0.001502 - Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol Biol Evol 2016; 33(7): 1870-1874. http://dx.doi.org/10.1093/molbev/msw054 PMid:27004904.
» http://dx.doi.org/10.1093/molbev/msw054 - Labruna MB, Sanfilippo LF, Demetrio C, Menezes AC, Pinter A, Guglielmone AA, et al. Ticks collected on birds in the state of São Paulo, Brazil. Exp Appl Acarol 2007; 43(2): 147-160. http://dx.doi.org/10.1007/s10493-007-9106-x PMid:17882514.
» http://dx.doi.org/10.1007/s10493-007-9106-x - Labruna MB, Whitworth T, Horta MC, Bouyer DH, McBride JW, Pinter A, et al. Rickettsia species infecting Amblyomma cooperi ticks from an area in the state of Sao Paulo, Brazil, where Brazilian spotted fever is endemic. J Clin Microbiol 2004; 42(1): 90-98. http://dx.doi.org/10.1128/JCM.42.1.90-98.2004 PMid:14715737.
» http://dx.doi.org/10.1128/JCM.42.1.90-98.2004 - Loss SR, Noden BH, Hamer GL, Hamer SA. A quantitative synthesis of the role of birds in carrying ticks and tick-borne pathogens in North America. Oecologia 2016; 182(4): 947-959. http://dx.doi.org/10.1007/s00442-016-3731-1 PMid:27670413.
» http://dx.doi.org/10.1007/s00442-016-3731-1 - Lugarini C, Martins TF, Ogrzewalska M, de Vasconcelos NC, Ellis VA, de Oliveira JB, et al. Rickettsial agents in avian ixodid ticks in northeast Brazil. Ticks Tick Borne Dis 2015; 6(3): 364-375. http://dx.doi.org/10.1016/j.ttbdis.2015.02.011 PMid:25800099.
» http://dx.doi.org/10.1016/j.ttbdis.2015.02.011 - Luz HR, Faccini JLH. Parasitismo por carrapatos em anuros no Brasil. Revisão. Vet Zootec 2013; 20: 100-111.
- Luz HR, Faccini JL, Landulfo GA, Berto BP, Ferreira I. Bird ticks in an area of the Cerrado of Minas Gerais State, southeast Brazil. Exp Appl Acarol 2012; 58(1): 89-99. http://dx.doi.org/10.1007/s10493-012-9572-7 PMid:22729500.
» http://dx.doi.org/10.1007/s10493-012-9572-7 - Luz HR, Faccini JLH, Landulfo GA, Costa Neto SF, Famadas KM. New records for Amblyomma sculptum (Ixodidae) on non-passerine birds in Brazil. Rev Bras Parasitol Vet 2016; 25(1): 124-126. http://dx.doi.org/10.1590/S1984-29612016004 PMid:27007247.
» http://dx.doi.org/10.1590/S1984-29612016004 - Martins TF, Onofrio VC, Barros-Battesti DM, Labruna MB. Nymphs of the genus Amblyomma (Acari: Ixodidae) of Brazil: descriptions, redescriptions, and identification key. Ticks Tick Borne Dis 2010; 1(2): 75-99. http://dx.doi.org/10.1016/j.ttbdis.2010.03.002 PMid:21771514.
» http://dx.doi.org/10.1016/j.ttbdis.2010.03.002 - Massung RF, Slater K, Owens JH, Nicholson WL, Mather TN, Solberg VB, et al. Nested PCR assay for detection of granulocytic ehrlichiae. J Clin Microbiol 1998; 36(4): 1090-1095. PMid:9542943.
- McCoy BN, Maïga O, Schwan TG. Detection of Borrelia theileri in Rhipicephalus geigyi from Mali. Ticks Tick Borne Dis 2014; 5(4): 401-403. http://dx.doi.org/10.1016/j.ttbdis.2014.01.007 PMid:24709337.
» http://dx.doi.org/10.1016/j.ttbdis.2014.01.007 - Mitchell EA, Williamson PC, Billingsley PM, Seals JP, Ferguson EE, Allen MS. Frequency and distribution of Rickettsiae, Borreliae, and Ehrlichiae Detected in Human-Parasitizing Ticks, Texas, USA. Emerg Infect Dis 2016; 22(2): 312-315. http://dx.doi.org/10.3201/eid2202.150469 PMid:26811941.
» http://dx.doi.org/10.3201/eid2202.150469 - Nava S, Venzal JM, Labruna MB, Mastropaolo M, González EM, Mangold AJ, et al. Hosts, distribution and genetic divergence (16S rDNA) of Amblyomma dubitatum (Acari: Ixodidae). Exp Appl Acarol 2010; 51(4): 335-351. http://dx.doi.org/10.1007/s10493-009-9331-6 PMid:20084537.
» http://dx.doi.org/10.1007/s10493-009-9331-6 - Ogrzewalska M, Pacheco RC, Uezu A, Ferreira F, Labruna MB. Ticks (Acari: Ixodidae) infesting wild birds in an Atlantic forest area in the state of São Paulo, Brazil, with isolation of Rickettsia from the tick Amblyomma longirostre. J Med Entomol 2008; 45(4): 770-774. http://dx.doi.org/10.1093/jmedent/45.4.770 PMid:18714882.
» http://dx.doi.org/10.1093/jmedent/45.4.770 - Ogrzewalska M, Pinter A. Ticks (Acari: Ixodidae) as ectoparasites of Brazilian wild birds and their association with rickettsial diseases. Braz J Vet Res Anim Sci 2016; 53(1): 1-31. http://dx.doi.org/10.11606/issn.1678-4456.v53i1p1-31
» http://dx.doi.org/10.11606/issn.1678-4456.v53i1p1-31 - Ogrzewalska M, Uezu A, Labruna MB. Ticks (Acari: Ixodidae) infesting wild birds in the eastern Amazon, northern Brazil, with notes on rickettsial infection in ticks. Parasitol Res 2010; 106(4): 809-816. http://dx.doi.org/10.1007/s00436-010-1733-1 PMid:20140452.
» http://dx.doi.org/10.1007/s00436-010-1733-1 - Ogrzewalska M, Uezu A, Labruna MB. Ticks (Acari: Ixodidae) infesting wild birds in the Atlantic Forest in northeastern Brazil, with notes on rickettsial infection in ticks. Parasitol Res 2011; 108(3): 665-670. http://dx.doi.org/10.1007/s00436-010-2111-8 PMid:20953629.
» http://dx.doi.org/10.1007/s00436-010-2111-8 - Palomar AM, Santibáñez P, Mazuelas D, Roncero L, Santibáñez S, Portillo A, et al. Role of birds in dispersal of etiologic agents of tick-borne zoonoses, Spain, 2009. Emerg Infect Dis 2012; 18(7): 1188-1191. http://dx.doi.org/10.3201/eid1807.111777 PMid:22709801.
» http://dx.doi.org/10.3201/eid1807.111777 - Parola P, Paddock CD, Socolovschi C, Labruna MB, Mediannikov O, Kernif T, et al. Update on tick- borne rickettsioses around the world: A geographic approach. Clin Microbiol Rev 2013; 26(4): 657-702. http://dx.doi.org/10.1128/CMR.00032-13 PMid:24092850.
» http://dx.doi.org/10.1128/CMR.00032-13 - Ramos DG, Melo AL, Martins TF, Alves AS, Pacheco TA, Pinto LB, et al. Rickettsial infection in ticks from wild birds from Cerrado and the Pantanal region of Mato Grosso, midwestern Brazil. Ticks Tick Borne Dis 2015; 6(6): 836-842. http://dx.doi.org/10.1016/j.ttbdis.2015.07.013 PMid:26232933.
» http://dx.doi.org/10.1016/j.ttbdis.2015.07.013 - Sanches GS, Martins TF, Lopes IT, Costa LF, Nunes PH, Camargo-Mathias MI, et al. Ticks infesting birds in Atlantic Forest fragments in Rio Claro, State of Sao Paulo, Brazil. Rev Bras Parasitol Vet 2013; 22(1): 6-12. http://dx.doi.org/10.1590/S1984-29612013000100003 PMid:24252949.
» http://dx.doi.org/10.1590/S1984-29612013000100003 - Santolin IDAC, Famadas KM, McIntosh D. Detecção e identificação de espécies de Rickettsia em carrapatos coletados de aves silvestres no Brasil pela PCR-RFLP. Rev Bras Med Vet 2013; 35(Supl.2): 68-73.
- Schwan TG, Raffel SJ, Schrumpf ME, Policastro PF, Rawlings JA, Lane RS, et al. Phylogenetic analysis of the spirochetes Borrelia parkeri and Borrelia turicatae and the potential for tick-borne relapsing fever in Florida. J Clin Microbiol 2005; 43(8): 3851-3859. http://dx.doi.org/10.1128/JCM.43.8.3851-3859.2005 PMid:16081922.
» http://dx.doi.org/10.1128/JCM.43.8.3851-3859.2005 - Scott JD, Lee MK, Fernando K, Durden LA, Jorgensen DR, Mak S, et al. Detection of Lyme disease spirochete, Borrelia burgdorferi sensu lato, including three novel genotypes in ticks (Acari: Ixodidae) collected from songbirds (Passeriformes) across Canada. J Vector Ecol 2010; 35(1): 124-139. http://dx.doi.org/10.1111/j.1948-7134.2010.00068.x PMid:20618658.
» http://dx.doi.org/10.1111/j.1948-7134.2010.00068.x - Seo MG, Yun SH, Choi SK, Cho GJ, Park YS, Cho KH, et al. Molecular and phylogenetic analysis of equine piroplasms in the Republic of Korea. Res Vet Sci 2013; 94(3): 579-583. http://dx.doi.org/10.1016/j.rvsc.2013.01.014 PMid:23415067.
» http://dx.doi.org/10.1016/j.rvsc.2013.01.014 - Soares HS, Barbieri AR, Martins TF, Minervino AH, de Lima JT, Marcili A, et al. Ticks and rickettsial infection in the wildlife of two regions of the Brazilian Amazon. Exp Appl Acarol 2015; 65(1): 125-140. http://dx.doi.org/10.1007/s10493-014-9851-6 PMid:25273064.
» http://dx.doi.org/10.1007/s10493-014-9851-6
Publication Dates
-
Publication in this collection
06 June 2019 -
Date of issue
Apr-Jun 2019
History
-
Received
26 Nov 2018 -
Accepted
01 Mar 2019