Abstracts
Although thousands of scientific articles have been published on the subject of marker-assisted selection (MAS) and quantitative trait loci (QTL), the application of MAS for QTL in plant breeding has been restricted. Among the main causes for this limited use are the low accuracy of QTL mapping and the high costs of genotyping thousands of plants with tens or hundreds of molecular markers in routine breeding programs. Recently, new large-scale genotyping technologies have resulted in a cost reduction. Nevertheless, the MAS for QTL has so far been limited to selection programs using several generations per year, where phenotypic selection cannot be performed in all generations, mainly in recurrent selection programs. Methods of MAS for QTL in breeding programs using self-pollination have been developed.
MAS; QTL; plant breeding; molecular breeding; genomic selection
Embora milhares de artigos científicos tenham sido publicados com os temas de seleção assistida por marcadores (SAM) e quantitative trait loci (QTLs), a utilização de SAM para QTLs tem tido uso limitado nos programas de melhoramento genético. Entre as principais causas para este baixo uso, estão a baixa precisão com que os QTLs são mapeados, e o alto custo da genotipagem de milhares de indivíduos com dezenas ou centenas de marcadores moleculares, nas rotinas dos programas de melhoramento. Recentemente, novas tecnologias de genotipagem em larga escala têm permitido a redução do custo de genotipagem. Ainda assim, atualmente a SAM para QTLs tem sido limitada à seleção em programas que utilizem diversas gerações por ano, onde a seleção fenotípica não pode ser realizada em todas as gerações, especialmente em programas de seleção recorrente. Métodos para SAM para QTLs em programas que utilizem autofecundação têm sido desenvolvidos.
MAS; QTL; melhoramento genético; melhoramento molecular; seleção genômica
ARTICLE
Marker-assisted selection for quantitative traits
Seleção assistida por marcadores para características quantitativas
Ivan Schuster* * E-mail: ivan@coodetec.com.br
Coodetec Cooperativa Central de Pesquisa Agrícola/Unipar Universidade Paranaense, C. P. 301, 85.813-450, Cascavel, PR, Brazil
ABSTRACT
Although thousands of scientific articles have been published on the subject of marker-assisted selection (MAS) and quantitative trait loci (QTL), the application of MAS for QTL in plant breeding has been restricted. Among the main causes for this limited use are the low accuracy of QTL mapping and the high costs of genotyping thousands of plants with tens or hundreds of molecular markers in routine breeding programs. Recently, new large-scale genotyping technologies have resulted in a cost reduction. Nevertheless, the MAS for QTL has so far been limited to selection programs using several generations per year, where phenotypic selection cannot be performed in all generations, mainly in recurrent selection programs. Methods of MAS for QTL in breeding programs using self-pollination have been developed.
Key words: MAS, QTL, plant breeding, molecular breeding, genomic selection.
RESUMO
Embora milhares de artigos científicos tenham sido publicados com os temas de seleção assistida por marcadores (SAM) e quantitative trait loci (QTLs), a utilização de SAM para QTLs tem tido uso limitado nos programas de melhoramento genético. Entre as principais causas para este baixo uso, estão a baixa precisão com que os QTLs são mapeados, e o alto custo da genotipagem de milhares de indivíduos com dezenas ou centenas de marcadores moleculares, nas rotinas dos programas de melhoramento. Recentemente, novas tecnologias de genotipagem em larga escala têm permitido a redução do custo de genotipagem. Ainda assim, atualmente a SAM para QTLs tem sido limitada à seleção em programas que utilizem diversas gerações por ano, onde a seleção fenotípica não pode ser realizada em todas as gerações, especialmente em programas de seleção recorrente. Métodos para SAM para QTLs em programas que utilizem autofecundação têm sido desenvolvidos.
Palavras-chave: MAS, QTL, melhoramento genético, melhoramento molecular, seleção genômica.
INTRODUCTION
Plant breeding in its conventional form is based on the phenotypic selection of superior plants within segregating populations derived from crosses. In this practice, there are numerous difficulties, especially in relation to genotype x environment (GE) interactions. In addition, phenotypic selection procedures are often costly, time-consuming and in some cases impossible, as is the case of selection for tolerance to some abiotic stresses.
Marker-assisted selection (MAS) is a procedure that has been developed to avoid these problems associated with phenotypic selection, replacing the selection of the phenotype by selection of genes, both directly and indirectly (Francia et al. 2005). Molecular markers are not influenced by the environment and are detected at any stage of plant development (Table 1). With the development of molecular markers and genetic maps, MAS can be used for simple-inherited as well as for quantitative traits.
In the last decade, a number of papers have been published describing the use of MAS in the introgression of quantitative traits through backcrossing programs and strategies to stack favorable alleles by recurrent crossing schemes. For a review of these studies, see Xu and Crouch (2008). This set of theoretical studies has greatly contributed to the understanding of many fundamental genetic parameters related to MAS, such as population type, sample size, genome size and number of markers to be used.
Molecular Marker-Assisted Selection and QTL
MAS is useful in crop breeding programs in four situations that apply to almost all crops: a) when phenotypic selection is unsuitable in view of the cost or time required or due to low penetrance or complex inheritance of the trait; b) when selection depends on specific environmental conditions or developmental stages that influence the expression of the target trait; c) to accelerate the recurrent genome recovering or for the maintenance of recessive alleles in backcrossing programs, d) when pyramiding multiple monogenic traits (resistance to various diseases, quality traits) or several QTL for a trait with complex inheritance (such as drought tolerance or other adaptive traits).
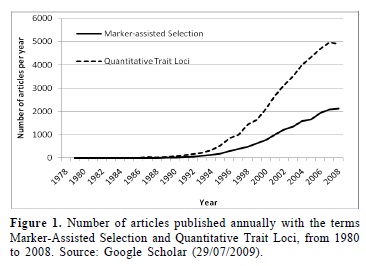
The number of scientific papers published with the term Marker-Assisted Selection currently breaks the barrier of two thousands per year (Figure 1). Similarly, the number of articles published with the term quantitative trait loci is around five thousands per year. However, almost all of these articles demonstrate potential applications of MAS in breeding programs rather than the practical use. Therefore, the effective utility of MAS to develop varieties in breeding programs has been restricted, mostly, to large companies that have developed genomic tools for the species of greatest commercial interest, e.g., maize, soybean, canola, cotton, and sunflower. Breeding programs that use these tools has been developed strategies to generate an ideal genotype, based on the selection of a mosaic of favorable chromosome segments (Xu and Crouch 2008). In breeding programs using these tools the rates of genetic gain have been two times high as the genetic gain by phenotypic selection (Ragot and Lee 2007, Xu and Crouch 2008). It is estimated that in the United States, from 2010 onwards, 12 % of the commercial varieties will be developed through molecular breeding (Fraley 2006, www.monsanto.com/investors/presentations.asp). However, the paper does not mention whether molecular selection was used for characteristics of qualitative or quantitative traits. Most commonly, molecular selection is used for disease resistance genes and for the selection of transgenic traits in breeding programs, both qualitative traits.
MAS for QTL: Theory
The way of applying MAS for QTL depends on the breeding method. Two main breeding methods can be cited: a) recurrent selection to increase the population mean; applied mainly in allogamous populations intolerant to inbreeding. In this method, selection is applied to individual plants that are continually recombined; b) breeding methods that use continuous self-pollination, and sporadic recombination, e.g., the pedigree method and SSD (single-seed descent).
MAS for QTL using recurrent selection
In a recurrent selection program, the breeding value of each plant based on its molecular score can be determined by genotyping plants (or lines) of a breeding population with QTL-associated markers for a quantitative trait of interest, for example yield. Lande and Thompson (1990) called this breeding value "net molecular score". For example, assuming that three QTL have been identified in a mapping population, their effects was estimated and that the additive effect (a) for each locus, defined as half the difference between the two classes of homozygotes (AA and aa), are +10, +5 and -10, respectively. The breeding value of a plant based on the marker locus would be +a for AA plants, zero for Aa plants (since these plants pass favorable and unfavorable alleles to progenies at the same frequency) and -a for aa plants. The net molecular score is the sum of the scores at each locus. Table 2 presents the scores of five plants from a breeding population. Based only on the genotype of the markers plant 2 would be chosen as the best.
The highest efficiency of MAS for QTL is achieved with a combination of molecular and phenotypic data. Lande and Thompson (1990) proposed a selection index (I) using molecular and phenotypic marker-based information:
I = bzz + bmm
where z is the column vector of phenotypic data, which can be expressed as deviations from the mean; m is the column vector of the net molecular scores; bz and bm are the weights assigned to phenotypic and molecular data, respectively.
If one assume bz = 1, the optimal weight for the genotypic data will be:
bm = (1/h2 - 1)/(1 - p)
where h2 is the narrow-sense heritability of the trait (proportion of the additive genetic variance of all QTL in relation to the phenotypic variance), and p is the proportion of genetic variance associated with the marker locus.
The relative efficiency (RE) of index I can be estimated by:

As heritability decreases and the proportion of genetic variance explained by the QTL increases, the weight of molecular data regarding phenotypic data increases (Table 3). Likewise, the RE of index I increases in the same direction.
When QTL for multiple traits will be selected, a multivariate index can be used.
I=bz1z1 + bz2z2 +...+ bznzn+ bm1m1 + bm2m2 +...+ bmnmn
where zi, mi, are the vectors with molecular and phenotypic data, and bzi e bmi are the respective weights for each trait i.
Another way to use a combination of molecular and phenotypic data for multiple traits is to calculate the index separately for each trait, and sum the indexes, weighted by the economic value of the trait, to obtain a single selection index.
As heritability increase, the efficiency of MAS decreases (Table 3), and may not be justified for traits with heritability higher than 50 %. Still, MAS for these traits may be adequate when more than one generation of recombination per year is used, i.e., one or more generations of recombination are grown in off season nurseries. In this case, phenotypic selection is not possible, and only molecular selection is used. In one of the generations of recombination, grown in the main growing season of the species, the combined index of phenotypic and molecular data is used. Thus, one generation of phenotypic selection can be alternated with up to three generations of only molecular selection, resulting in four generations of recombination per year (Eathington et al. 2007). By this way, one can obtain a higher rate of gain per unit time, as more cycles of recombination are performed per unit time.
This model of recurrent selection which uses molecular and phenotypic data in one generation and only molecular data in generations where phenotypic selection cannot be used is suitable for the use of Genomewide selection (Meuwissen et al. 2001, Bernardo and Yu 2007). In this case, knowledge on the markerQTL associations is not required. The plants of a population are genotyped for a large number of markers, e.g., thousands of single nucleotide polymorphism (SNP) markers. In the generation in which phenotypic data are obtained, the breeding value of each marker is estimated. Based on these estimates, the markers are used for selection in generations with molecular selection only.
MAS for QTL in self-pollination breeding programs
In commercial breeding programs of autogamous species, the objective is not to increase the population mean, but to develop the best possible endogamous genotype from self-pollinating plants in a population. Likewise, for the development of commercial hybrids, the objective is to get the best possible hybrid by crossing two inbred genotypes, which in turn were also obtained by selfing. In breeding programs where self-pollinated plants are selected, selection should target plants capable of producing the highest possible number of progenies of the superior genotype for the next generation, by self-pollination.
A simple way to use MAS for QTL in segregating populations is to identify plants that carry favorable alleles for the QTL under selection, and produce as many progenies of these plants as possible. If we consider the MAS for 10 non-related QTL in an F2 generation, the probability of obtaining a plant with all homozygous QTL is 0.2510, or one plant in a million. It is more reasonable to select homozygous as well as heterozygous QTL. In this case, the proportion of plants containing at least one favorable allele in each of the 10 QTL is 0.7510, or one plant in every 18. In this case, it is advisable to focus efforts on assessing the progenies of fewer plants (those with the highest number of favorable alleles for the QTL evaluated), since these plants has the highest probability to produce progenies with better genotype.
The above strategy assumes that all QTL contribute to the same extent to the characteristic, which is not true. Liu et al. (2004) proposed an index to rank the plants based on the weighted sum of all possible genotypic values.

where Ik is the index for plant k, pik is the probability that plant k has genotype i for the QTL, fj(l) is the frequency of superior homozygous genotypes j which are produced by self-pollination of a plant with genotype i for the QTL; gj is the genotypic value of the superior homozygous genotype j; iflis the frequency of the l-th gametic stage of genotype i of the QTL.Σl ifl = 1; i = 1,...,3n, n = number of QTL; j = 1 ,..., t, t = number of superior homozygous genotypes to be selected; l = 1,...,2m-1; 2< m <n. The probability pik is estimated based on data from the markers flanking the QTL, and phenotypic data obtained during selection. Liu et al. (2003) proposed a Bayesian approach to estimate this probability.
By this method, all plants of a breeding population are genotyped with markers flanking n QTL and the t plants most likely to produce the highest proportion of progenies with the highest QTL value in homozygosity after self-pollination are selected. Data simulated by Liu et al. (2004) illustrate that the frequency of superior homozygous genotypes for the main t genotypes is five times higher than that obtained by phenotypic selection in early generations (F2 to F4) and three times higher than phenotypic selection in the more advanced generations (F5 to F7). In the SSD method, where phenotypic selection is performed only in advanced generations, MAS used in the early as well as in advanced stages present frequency of superior homozygous genotypes for the main t genotypes three times higher than phenotypic selection in advanced phases.
MAS for QTL: Practice
The success of implementing a MAS program depends on several factors (Holland 2004): a) a genetic map with molecular markers linked to genes controlling qualitative or quantitative traits of agronomic interest; b) a close association between markers and genes or QTL; c) appropriate recombinations between the markers associated with the trait(s) of interest and the rest of the genome; d) the possibility of analyzing a large number of plants with reasonable time and cost investment.
The conversion of the information published in the scientific literature in practical applications for large-scale breeding programs requires some practical, logistic and genetic considerations. First, published molecular markers should be validated, in many cases, in a large number of populations, representing the routinely selected plant breeding material. Then a technical procedure must be developed which is simple, fast and inexpensive for the stages of tissue sampling, DNA extraction, genotyping and data collection and feasible and accurate when applied routinely on a large scale. Moreover, breeders need to develop an integrated system with data traceability and control systems that ensure the integration of genotyping in breeding programs. Finally, a breeding system must be outlined that will optimize the decision-making tools to support breeders with quick, but accurate decisions on selection (Xu and Crouch 2008).
In the near future, one of the main points to make MAS effective in large breeding populations is the availability of large-scale genotyping methods at a reasonable cost. New tools for large-scale genotyping, with chips containing thousands of SNPs (5, 10 and 50 thousand) may improve accuracy in QTL detection with smaller confidence intervals, and even identify markers that are the proper QTL alleles. This may reduce one of the limitations of MAS for QTL, which is the low accuracy of QTL detection. Although at this level of large-scale genotyping the cost per data point is low, the cost per sample is still high. If one considers a chip with 5000 SNPs, and a cost of only U$ 0.02 per data point, the cost per sample is still U$ 100.00.
For routine use in breeding programs, where large populations are evaluated every year, genotyping with a smaller number of markers should be more feasible. After identifying markers nearly associated with QTL, from a map saturated with thousands of markers, a selected set of markers will be used to compose a chip for use in routine MAS. Genotyping with a chip containing 384 SNPs, currently at a cost of U$ 0.09 per data point, totals U$ 34.56 per sample. For a breeding program that uses the pedigree method or SSD, with F2 populations of 500 plants, genotyping will cost U$ 17,280.00 per population. Together with the DNA extraction, plant identification in the field and sampling, the costs exceed U$ 20,000 per population. If the breeding program monitors 500 F2 populations, the cost of molecular analysis will be more than U$ 10 million per year. If we reduce the number of markers to 48 the cost will be reduced by half (8-fold reduction in the number of markers and half the cost). This consideration demonstrates that the cost of genotyping has yet to be greatly reduced to become this strategy feasible.
CONCLUDING REMARKS
In the last two decades, great advances have been made in understanding the nature of QTL of traits of interest for breeding. Nevertheless, the use of MAS for QTL in genetic improvement programs is limited. MAS has been widely used in marker-assisted backcrossing for selection of the recurrent genome and selection of plants with minimal linkage drag. This has allowed recovering the recurrent genome with a smaller number of backcross generations, as well as an early selection of plants, reducing the number of plants in each generation.
In forward breeding programs, MAS has been applied on a regular basis, especially for traits with high heritability, e.g., MAS for soybean cyst nematode (Concibido et al. 2004) and selection for resistance to other plant diseases. The use of MAS for QTL in breeding programs has been more restricted. The low accuracy with which QTL and their effects have been mapped, and the need to validate these QTL in different genetic backgrounds have been cited as the main cause of this limited use of MAS for QTL. In general, MAS can be used in breeding programs in the following situations: a) Selection for traits of low heritability. In this case, there is a methodological contradiction. The advantage of using MAS rather than phenotypic selection is greatest for low-heritability traits. But the trait heritability is an important parameter for the ability to detect QTL, although QTL for traits with low heritability are generally not detected. b) selection in the off season growing nurseries, where phenotypic selection is not possible. In this case, MAS for QTL may be feasible even for high-heritability traits. Although the selection gain per cycle may be smaller, the gains per unit time are higher, since more than one selection cycle is performed per year.
In addition, the implementation of a MAS program depends on the infrastructure that allows the generation of hundreds of thousands of molecular data at a compatible cost, which has also limited the use of MAS in breeding programs.
Received 1 March 2011
Accepted 20 May 2011
- Bernardo R and Yu Y (2007) Prospects for genomewide selection for quantitative traits in maize. Crop Science 47: 1082-1090.
- Concibido V, Diers BW and Arelli PR (2004) A decade of QTL mapping for cyst nematode resistance in soybean. Crop Science 44: 1121-1131.
- Eathington SR, Crosbie TM, Edwards MD, Reiter RS and Bull JK (2007) Molecular markers in a commercial breeding program. Crop Science 47: S154-S163.
- Francia E, Tacconi G, Crosatti C, Barabaschi D, Bulgarelli D, Dall'Aglio E and Valè G (2005) Marker assisted selection in crop plants. Plant Cell, Tissue and Organ Culture 82: 317-342.
- Holland J (2004) Implementation of molecular markers for quantitative traits in breeding programs change and opportunities. In Fisher T (ed.) New directions for a diverse planet Proc. of the 4th International crop science congress. The Regional Institute Ltd., Gosford. Available at <www.cropscience.org.au/icsc2004/> Accessed in April, 2008.
- Kurzun V (2003) Molecular markers and their applications in cereal breeding. In Marker-assisted selection: A fast track to increase genetic gain in plant and animal breeding? FAO, Rome. Available at <http://www.fao.org/Biotech/docs/Korzun.pdf> Accessed in March, 2011.
- Lande R and Thompson R (1990) Efficiency of marker-assisted selection in the improvement of quantitative traits. Genetics 124: 743-756.
- Liu PY, Zhu J, Lou XY and Lu Y (2003) A method for marker-assisted selection based on QTL with epistatic effects. Genetica 119: 75-86.
- Liu PY, Zhu J and Lu Y (2004) Marker-assisted selection in segregating generations of self-fertilizing crops. Theoretical and Applied Genetics 109: 370-376.
- Meuwissen TH, Hayes BJ and Goddard ME (2001) Prediction of total genetic value using genomewide dense marker maps. Genetics 157: 1819-29.
- Ragot M and Lee M (2007) Marker-assisted selection in maize: current status, potential, limitations and perspectives from the private and public sectors. In Guimarães EP, Ruane J, Scherf BD, Soninno A and Dargie JD (eds.) Marker-assisted selection, current status and future perspectives in crops, livestock, forestry, and fish FAO, Rome, p. 117-150.
- Xu Y and Crouch JH (2008) Marker-assisted selection in plant breeding: from publication to practice. Crop Science 48: 391-407.
Publication Dates
-
Publication in this collection
17 Sept 2012 -
Date of issue
June 2011
History
-
Received
01 Mar 2011 -
Accepted
20 May 2011