Abstracts
The solubility behavior of drugs remains one of the most challenging aspects in formulation development. Solid Dispersion (SD) has tremendous potential for improving drug solubility. Although praziquantel (PZQ) is the first drug of choice in the treatment of schistosomiasis, its poor solubility has restricted its delivery oral route. In spite of its poor solubility, PZQ is well absorbed in the gastrointestinal tract, but large doses are required to achieve adequate concentration at the target sites. The aim of this study was to improve the solubility and dissolution rate of PZQ and to evaluate its intestinal absorption. SDs were formulated with PEG-60 castor oil hydrogenated (CR-60) using a fusion and evaporation method. Pure PZQ and physical mixtures (PM) and PZQ-CR-60 (2:1; 1:1; 1:2 ratios) were compared as regards their solubility, dissolution and intestinal absorption. The experimental results demonstrated the improvement in the solubility, dissolution rate and intestinal absorption. In addition, the solubility behavior showed pH dependency and that the solubility of PZQ was slower in acidic medium than in neutral and basic mediums. The increase in PZQ solubility of the SD with the CR-60 could be attributed to several factors such as improved wettability, local solubilization, drug particle size reduction and crystalline or, interstitial solid solution reduction.
Solid dispersion; Praziquantel; Praziquantel; Praziquantel
A solubilidade de fármacos ainda é um dos principais desafios no desenvolvimento de formulações farmacêuticas. As dispersões sólidas (DS) apresentam grande potencial para melhorar a solubilidade de fármacos. O praziquantel é o fármaco de primeira escolha no tratamento da esquistossomose, contudo a baixa solubilidade em água restringe seu uso à administração pela via oral. Apesar da baixa solubilidade, o PZQ é bem absorvido através do trato gastrintestinal, mas doses orais elevadas são requeridas para garantir concentrações suficientes de fármaco para o tecido alvo. O objetivo deste estudo foi melhorar a solubilidade, a dissolução e avaliar a absorção do PZQ. As DS foram formuladas com óleo de castor hidrogenado - PEG 60 (CR-60), pelo uso dos métodos de fusão e evaporação do solvente. PZQ puro, mistura física (MF) e DS de CR-60-PZQ (1:2; 1:1; 2:1) foram comparados quanto à solubilidade, dissolução e absorção intestinal. Os resultados experimentais mostraram aumento na solubilidade, na taxa de dissolução e na absorção intestinal do PZQ nas DS. A solubilidade do PZQ foi maior em meio ácido, mostrando uma dependência do pH. O aumento na solubilidade do PZQ nas DS com CR-60 foi atribuída a fatores como aumento da molhabilidade, solubilização local, redução granulométrica e redução da cristalinidade ou, ainda, a ocorrência de uma solução sólida intersticial.
Dispersão sólida; Praziquantel; Praziquantel; Praziquantel
ARTICLE
Solid dispersions with hydrogenated castor oil increase solubility, dissolution rate and intestinal absorption of praziquantel
Marco Vinicius ChaudI,II,**Correspondence: M. V. Chaud. Curso de Farmácia, Universidade de Sorocaba. Rodovia Raposo Tavares Km 92,5 - 18023-000 - Sorocaba - SP, Brasil. Tel. 15-21017041; 15-21017124. E-mail: marco.chaud@prof.uniso.br; Pollyanna TamasciaII; Andréa Cristina de LimaIII; Maria Ondina PaganelliII; Maria Palmira Daflon GremiãoIII; Osvaldo de FreitasIV
IDepartment of Pharmaceutical Sciences, University of Sorocaba - UNISO
IIDepartment of Pharmacy , Faculty of Health Sciences, Methodist University of Piracicaba
IIIDepartment of Drugs and Pharmaceutical, Faculty of Pharmaceutical Sciences of Araraquara, São Paulo State University "Júlio de Mesquita Filho"
IVDepartment of Pharmaceutical Sciences of Ribeirão Preto, University of São Paulo
ABSTRACT
The solubility behavior of drugs remains one of the most challenging aspects in formulation development. Solid Dispersion (SD) has tremendous potential for improving drug solubility. Although praziquantel (PZQ) is the first drug of choice in the treatment of schistosomiasis, its poor solubility has restricted its delivery oral route. In spite of its poor solubility, PZQ is well absorbed in the gastrointestinal tract, but large doses are required to achieve adequate concentration at the target sites. The aim of this study was to improve the solubility and dissolution rate of PZQ and to evaluate its intestinal absorption. SDs were formulated with PEG-60 castor oil hydrogenated (CR-60) using a fusion and evaporation method. Pure PZQ and physical mixtures (PM) and PZQ-CR-60 (2:1; 1:1; 1:2 ratios) were compared as regards their solubility, dissolution and intestinal absorption. The experimental results demonstrated the improvement in the solubility, dissolution rate and intestinal absorption. In addition, the solubility behavior showed pH dependency and that the solubility of PZQ was slower in acidic medium than in neutral and basic mediums. The increase in PZQ solubility of the SD with the CR-60 could be attributed to several factors such as improved wettability, local solubilization, drug particle size reduction and crystalline or, interstitial solid solution reduction.
Uniterms: Solid dispersion. Praziquantel/dissolution rates. Praziquantel/solubility. Praziquantel/intestinal absorption.
ABSTRACT
A solubilidade de fármacos ainda é um dos principais desafios no desenvolvimento de formulações farmacêuticas. As dispersões sólidas (DS) apresentam grande potencial para melhorar a solubilidade de fármacos. O praziquantel é o fármaco de primeira escolha no tratamento da esquistossomose, contudo a baixa solubilidade em água restringe seu uso à administração pela via oral. Apesar da baixa solubilidade, o PZQ é bem absorvido através do trato gastrintestinal, mas doses orais elevadas são requeridas para garantir concentrações suficientes de fármaco para o tecido alvo. O objetivo deste estudo foi melhorar a solubilidade, a dissolução e avaliar a absorção do PZQ. As DS foram formuladas com óleo de castor hidrogenado - PEG 60 (CR-60), pelo uso dos métodos de fusão e evaporação do solvente. PZQ puro, mistura física (MF) e DS de CR-60-PZQ (1:2; 1:1; 2:1) foram comparados quanto à solubilidade, dissolução e absorção intestinal. Os resultados experimentais mostraram aumento na solubilidade, na taxa de dissolução e na absorção intestinal do PZQ nas DS. A solubilidade do PZQ foi maior em meio ácido, mostrando uma dependência do pH. O aumento na solubilidade do PZQ nas DS com CR-60 foi atribuída a fatores como aumento da molhabilidade, solubilização local, redução granulométrica e redução da cristalinidade ou, ainda, a ocorrência de uma solução sólida intersticial.
Unitermos: Dispersão sólida. Praziquantel/taxa de dissolução. Praziquantel/solubilidade. Praziquantel/absorção intestinal.
INTRODUCTION
With the advent of high throughput screening (HTS) for agents with potential therapeutic value, the number of candidate drugs which are poorly soluble in water has increased considerably. The formulation of these pharmaceuticals for delivery orally or systemically has become one of the greatest challenges faced by the pharmaceutical industry (Karavas et al., 2007). In recent years, studies on solid dispersion (SD) have shown this to be a promising route to improve the oral availability of those pharmaceuticals which have low solubility in water (Leuner, Dreesman, 2000). The term Solid Dispersion (SD) is used to describe a solid system in which the drug is dispersed in a biologically innocuous hydrophilic carrier. These systems are generally centered on the conversion of a candidate pharmaceutical/carrier mix from liquid to solid state (Chiou, Riegelman, 1971; Corrigan, et al., 1979; Saito et al., 2002; Shim, Kim, 2003; Juppo et al., 2003; Liu et al., 2006; Ahuja et al., 2007). The aim of SD in pharmaceutical manufacture is to alter the solid state properties of the candidate pharmaceutical, thereby increasing dispersion rate, improving the solubility coefficient as well as raising stability. Depending on the type of carrier used, this will change the release characteristics of the pharmaceutical (Sethia, Squillante, 2003; Karavas et al., 2007). The technology of solid dispersion involves preparation of co-precipitates, co-evaporates, nanoparticles, microcapsules, microspheres, and other products. One of the first publications on the concepts and advantages of solid dispersion formulas was published by Sekiguchi and Obi in the late 1970s (Sethia, Squillante, 2003). In their publication these authors presented a new method for reducing the size of poorly soluble particles in water by formation of eutectic mixtures. Law et al. (2003), applied the same principle to obtain eutectic microstructures with polyethylene glycol and fenofibrate. In a series of studies published by Goldberg et al. (1965(a), 1965(c), 1966(b), 1966(d)) the experimental details and theoretical advantages of a solution of solids, were presented. Chiou, Riegelman (1970) used polyethyleneglycol as a carrier and showed rapid and almost complete absorption, in vivo, of griseofulvin. Saito et al. (2002), showed an increase in the solubility of griseofulvin using saccharides as carriers. Other carriers such as lactose, micro crystalline cellulose, chroscarmellose sodium, amide of sodium glycolate, polymethacrylates, have also been used to improve the absorption of pharmaceuticals that are poorly soluble in water (El-Arena et al. 1998; Cornier et al. 2000; Chowder et al., 2000; Babu et al. 2002; Law et al., 2003; Mitchell et al., 2003, Ohara et al., 2005; Karavas et al., 2007; Kim et al., 2008). Praziquantel (PZQ, 2-cyclohexylcarbonyl [1,2,3,6,7,11] hexahydro-4H-pyrazin [2,1a] isoquinolin-4-one) is known worldwide as the drug of first choice for the treatment of the most common forms of schistosomiasis and cysticercosis. These two diseases affect millions of people worldwide, especially those living in developing countries, and represent a very high social and economic cost to these countries (González-Esquivel et al., 2005). Although it is poorly soluble in water, PZQ is well absorbed in the intestine (Andrews, 1985; González-Esquivel et al., 2005). However, high oral doses are required to guarantee that sufficient concentrations of the pharmaceutical reach the target larval tissues. At present, PZQ is available only for administration the oral route. However, alternative routes of administration and new drug forms could be considered if the problem of aqueous solubility was overcome. The objective of this study was to prepare solid dispersions by fusion and co-precipitation and evaluate, in vitro, the influence of these preparations of PZQ on solubility, dissolution rate, and intestinal absorption.
MATERIAL AND METHODS
Praziquantel (PZQ) was obtained with a purity of 99.6%, from Induken Quimica Ltda (Brazil). The hydrogenate castor oil - PEG 60 (CR-60) was obtained, with pharmaceutical grade, from Henrifarma Química e Farmacêutica Ltda (Brasil). The supplier data had the following characteristics for the CR-60: melting point 45ºC, iodine value 0.26%, saponification number of 45-50mg KOH/g, hydroxyl number of between 65-70 and pH 6.2 for 10% solution. TC-199 is the designation of the solution used to incubate the inverted intestinal sac and is composed of 145.0mM NaCl; 4.56mM KCl; 1.25mM CaCl2.2H2O; 5.0mM Na2HPO4 and 10.0mM of lactose.
Thermal analysis of Praziquantel
Thermograms were obtained using DSC, DT/TGA analyzing instruments (Perkin-Elmer, Norwalk, CT). Temperature was calibrated using indium (156.6ºC) as the standard. The operating conditions in an open-pan system were as follows: sample weight 5-10mg, 10 ºC min-1 under N2 purging 50mL. min-1. The temperature was increased from 25 to 600ºC then allowed to decrease to 20ºC.
Preparations of solid dispersions by fusion method
PZQ and CR-60 were heated separately in a glycerin bath up to 136-138ºC under constant agitation until the mixture solidified by cooling. The SD was forced through a sieve with mesh openings of 0.35mm.
Preparation of solid dispersions by co-precipitation method
PZQ and CR-60 were dissolved separately in ethanol, and mixed. The mixing time was 30 minutes, under mechanical agitation. The solvent was eliminated using a rotary evaporator (TE-210, Tecnal) under reduced pressure.
Preparation of physical mixtures
The physical mixture (PM) of PZQ with CR-60 was produced using a mortar and pestle. The relative proportions of CR-60: PZQ used in this SD and the PMs were 1:2; 1:1 and 2:1. The products were kept shielded from light in a dessicator with a controlled humidity of (18% UR) until Constant weight, and were subsequently passed through another sieve in order to obtain a uniform surface area of the product.
Evaluation of solubility
The SD of PZQ and the PMs were dispersed in a distilled water solution, HCl 0.1N; Buffered phosphate pH 7.4 or TC-199 and maintained horizontally with agitation for 12 hours at a temperature of 27±2ºC. The concentration of PZQ was 1mg/mL in each dispersion medium. After this stage, the samples were first filtered with qualitative paper filters and subsequently passed through a filter with 0.45mm pores. The tests were performed in triplicate and the quantity of PZQ dissolved was determined by spectrometry (λ = 263nm).
Dissolution studies
This test was carried out using dispersion apparatus 2 as described in USP-30, at 37ºC and 75 rpm in 0.1N HCl with the addition of sodium laurel sulphate. The system was maintained in conditions of non-saturation of the dispersion medium. Samples of 5.0mL were taken at times 2, 5, 10, 20, 50, 90 and 120 minutes, and immediately filtered through membranes with 0.45mm pores. The volume of the dissolution medium was maintained constant by the addition of 5.0mL of 0.1N HCl, kept at 37ºC. The concentration of dissolved PZQ at each time point was determined by spectrometry (l = 263nm).
Evaluation of intestinal absorption
Intestinal absorption of PZQ was evaluated based on the SD of CR-60:PZQ in a 2:1 proportion and compared with the absorption of pure PZQ and PM. This experiment was conducted using male adult Wistar rats weighing 270 ± 20g. The animals were deprived of solid food for 8 hours before the start of the experiment. The rats were then anesthetized with Thiopental and the intestine dissected and immediately inverted with the aid of a cylindrical flexible rod, for exposure of the mucosal surface. An intestinal segment of about 8.0 cm in length from the pylorus was gently washed with TC-199 without glucose (10ºC). The intestinal segment thus formed was closed at one end with a suture, and filled with a solution of TC-199 without glucose. The other end was closed forming an inverted intestinal sac, which was immediately incubated in a previously oxygenated (95%O2:5%CO2) solution of TC-199 (37ºC), with the addition of PZQ at a concentration of 1mg/mL. The incubation medium was kept under mild agitation while oxygenation was maintained by an oxygen pump. The oxygen concentration of the medium was controlled with an oximeter (Oxygen Meter, Model OX). The quantity of PZQ absorbed was determined after 5, 10, 20, 40, 80 and 120 min. of incubation. After these times, the intestinal segment was removed from the incubation medium and thoroughly washed in a physiologic solution. The contents of the intestinal sac were filtered in a 0.22mm pore membrane. The concentration of PZQ in the medium was determined by spectrometry (λ=263nm). Permeability of the membrane was evaluated according to the transfer of glucose from the external to the internal compartment of the intestinal sac, as described by Barthe et al. (1999).
Statistical analyses
The results are expressed as mean ±standard deviation. Parametric tests were used for the analysis of these results, taking into consideration the distribution of the values and the variability of the measures obtained. The level of significance was set at 5% (p<0.05) for rejection of the null hypothesis, where (*) indicates non significant intragroup values, and (#) indicates non-significant intergroup values.
RESULTS AND DISCUSSION
The intestinal absorption of solid state pharmaceuticals depends upon two processes which should occur in a consecutive fashion: (1) dispersion of the pharmaceutical in the gastrointestinal tract, and (2) transport of the dispersed pharmaceutical through the blood. When the rate of dispersion of the pharmaceutical is significantly lower than the absorption rate, this becomes the limiting step in the absorption process. In order to choose techniques or procedures which can increase the solubility, a preliminary study of absorption needs to be carried out. The inverted intestinal sac model, when used in the phases of development of a product, allows a comparative evaluation of the behavior of the pharmaceutical compound during the permeation phase (Balimane et al., 2000).
Basic criteria and prerequisites for the selection of carriers were based on the possibility of increasing the rate of dispersion of the pharmaceutical using SD, pharmacological inertness and pharmacotechnical functionality. It was also important for this study that the carrier had previously been approved for use by the Agência Nacional de Vigilância Sanitária (ANVISA-National Health Surveillance Agency). CR-60 was also chosen for its tensoactive, softening and solubilizing properties. Finding physiologically compatible solvents capable of dissolving both hydrophilic and hydrophobic pharmaceuticals, and previously approved for this use by the respective governmental agencies, is challenging. Ethanol was chosen as a solvent for both the pharmaceutical and the carrier for this study.
The term co-precipitate has been used by various researchers to denote SDs obtained by dispersion of both the pharmaceutical and the carrier. However, a better term for this process would be co-evaporation. Simonelli et al., 1969 showed that these terms are conceptually different and used the term co-precipitation to describe dispersions obtained when a solution of sulphathiazole and PVP in NaOH was precipitated by the addition of HCl.
Thermal analysis of praziquantel
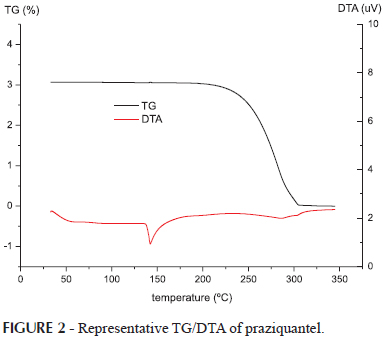
The results of thermal analyses of PZQ are shown in Figures 1 and 2. In the DSC (Figure 1) and DTA (Figure 2), PZQ showed a sharp endothermic peak of 139ºC, corresponding to the melting point of the racemic mixture and melting enthalpy of -96.4J/g. In the TG curve (Figure 2) PZQ has a single stage of thermal decomposition between 200 and 300 º C with a final residual mass of less than 0.3%. These results are similar to those obtained by Liu, et al. (2004) and Torre et al. (1999).
Evaluation of solubility
The ratio of the quantity of CR-60 and PZQ were 1:2; 1:1 and 2:1 (CR-60:PZQ), with the objective in this case being to evaluate the influence of the quantity of the carrier on the solubility. The dissolution profile of PZQ was verified only for the SD which showed optimal solubility. The effect of CR-60 on the solubility of PZQ in water, HCl 0.1N, buffered phosphate pH 7.4 and TC-199 are shown in Tables I-IV. Results are presented as mean±SD for n = 3. The solubility in TC-199 was verified with the aim of establishing a study model for in vitro intestinal absorption studies of PZQ. The solubility of pure PZQ in purified water, HCl 0.1N, buffered phosphate pH 7.4 and TC 199, was 52.63±0.169; 73.51±1.492; 45.43±0.405 and 66.328±0.139 (× 10-1 mg/mL), respectively.
The solubility of PZQ in SDs with CR-60 was statistically significant (p<0.05) in all of the dissolution media tested in this study, irrespective of whether PZQ was used in pure form or in a physical mixture. Solubility of PZQ was greater when the SDs were prepared using the Fusion Method and increased with higher concentration of CR-60. However, the SD obtained by the co-precipitation method was expected to yield superior results because the PZQ and CR-60 would be mixed in the molecular state.
Dissolution studies
The dissolution profile of PZQ in the SDs CR-60: PZQ (2:1) obtained by fusion and dissolution is shown in Figure 3 and compared to the dissolution profile of PZQ in physical mixtures and in the pure state. SDs and PMs presented a significantly greater dissolution rate than did pure PZQ. The result obtained from this dissolution study cannot be directly related with the evaluation of solubility, since it constitutes an exploratory study on the influence of the proportion of carrier (CR-60) on the pharmaceutical agent (PZQ). The dissolution rate of PZQ in an acid medium was higher than the solubility equilibrium rate (TABLES I - IV) where this is influenced by the processing conditions and by the presence of sodium lauril sulphate incorporated into the dissolution medium to maintain the non-saturated conditions of the medium (USP 30).
The improvement in the dissolution and solubility rates using the fusion methods could be ascribed to the saturation of the carrier by the pharmaceutical, associated with the increase in surface area and more rapid release of PZQ. The time to solidification of the liquid mixture can be a critical factor in the saturation of the dispersed system (Chiou, Riegelman, 1971; Allen et al., 1977; Lin, Cham, 1996). Dittgen et al. (1995) evaluated this parameter in the SDs of riboflavin, testosterone and progesterone and stated that the solubility of SD increased with reduced cooling time. In the present study, the dispersions were cooled naturally under constant agitation until solidification. The rate of dissolution of PZQ also proved higher in the SD obtained by the Fusion Method. One of the factors responsible for this result is probably the saturation of the carrier by the pharmaceutical compound, which increased the liberation rate. This did not occur in the dissolution process with evaporation at low pressure, conditions which led to slow release of the solvent. Perhaps rapid removal of the solvent by lyophilization, "spray drying", or under supercritical conditions would increase the solubility of PZQ in the SD obtained by dissolution. The dissolution rate would probably have been higher if the super saturation of the pharmaceutical compound in the matrix were followed by a more rapid evaporation of the solvent. However, in this experimental model, the principal advantage of the dissolution method is the prevention of thermal degradation. Improvement in the dissolution rate of pharmaceutical compounds in SD has been the subject of numerous investigations, and increases of up to 400 fold have been reported (Sethia, Squilante. 2003). In spite of these studies, the mechanisms by which this occurs are poorly understood. In SDs, reductions in granulometry and/or interparticle cohesion forces leading to increase in surface area exposed to the solvent have been considered one of the principle reasons for the increase in the dissolution rate. Reduced particle size has classically been considered the result of the formation of the solid or eutectic solution (Craig, 2002). Notwithstanding, the intrinsic link between the solid state structure and the liberation of the pharmaceutical compound suggests that the particles are held in the dissolution medium as separate physical entities. This reduction in cohesion force, in addition to the wetting properties of CR-60, contributed toward increasing the rate of dissolution. While the mechanism by which the solubility and rate of dissolution of PZQ increases in SD is difficult to elucidate in this experimental model, it appears that the critical factor is the physical-chemical characteristic of CR-60. The tensoactive properties of this carrier appear to predict the mathematical model of Noyes-Whitney for a single-component system in which both the carrier and the pharmaceutical agent dissolve at rates proportional to their soluabilities and coefficients of diffusion. This doubt has raised several questions, such as: what factors determine whether the dissolution is controlled by the carrier or the pharmaceutical? What is occurring to the pharmaceutical during these preparations of SDs? And finally, what are the implications of understanding these mechanisms for the development of a pharmaceutical form and the use of SDs in industrial processes? The answers to these questions are being sought by our group through projects in progress, using infrared spectroscopy, X ray diffraction, differential exploratory calorimetric and process analysis.
Evaluation of intestinal absorption
The inverted intestinal sac model is useful for studying physiological and molecular processes of absorption, providing physical and chemical information on the properties of the pharmaceuticals, and ascertaining the role of the absorption facilitators and inhibitors of intestinal metabolism. In this study, the intestinal inverted sac method was used to evaluate the influence of CR-60 on the intestinal absorption of PZQ. The glucose in the incubation medium (TC-199) was used to evaluate the permeability of the intestinal membrane and the viability of the enterocytes. The segment of small intestine of rats and its inversion was performed carefully using a fine silk covered Rod to avoid morphological damage. Due to the absence of blood and nerves these tissues have low viability. However, when kept at 37ºC in TC-199, these segments were morphologically intact and metabolically active for about 2 hours. In this aspect, our results were similar to those described by Barthe (1999) and Souza et al. (2007) who reported progressive increase in the glucose concentration in the interior of the intestinal sac for a 120-minute period.
Absorption of PZQ was evaluated only for SDs with CR-60 and a ratio of 2:1 (CR-60:PZQ) because this showed optimal solubility, and given the knowledge that the bioavailability of PZQ is limited by its low solubility in water. The absorption of PZQ in SDs was compared with the absorption of pure PZQ and with PM (1:2). Figure 4 shows the profile of intestinal absorption of PZQ over a 120-minute period. The results differed significantly (p<0.05) for SDs after 20 minutes of incubation compared to pure PZQ and PM. The improvement in the intestinal permeation is related to the increase in the dissolution rate of PZQ caused by CR-60 in SDs.
In this study, the use of an inverted intestinal sac demonstrated that CR-60, used in the preparation of the SDs, increases the solubility, dissolution rate and consequently, the absorption of Praziquantel. The solubility profile as well as the velocity and extent of dissolution presented by the SDs, compared to pure PZQ or PM, can be explained by modifications in the physical and chemical characteristics of the particulate system. These modifications have been shown on differential calorimetric studies, X Ray diffraction and infrared spectroscopy (Sarkari et al., 2002; Ahuja et al., 2007; Karavas et al., 2007; Kim et. al., 2008). Among the underlying mechanisms proposed to explain the increase in solubility and dissolution rate is the change from the crystalline state of the pharmaceutical compound to an amorphous state with a larger surface area and increased wettability and solid dissolution (Watanabe et al., 2003; Kim et al., 2008). The factors which led to the final results encountered in this study should be analyzed in conjunction, and may be related to the saturation of the carrier by the pharmaceutical, reduction in granulometry and interfacial tension, the conformational structure between the carrier and pharmaceutical in the SD, or modification of the crystalline state to an amorphous state. This last finding should be confirmed using X ray diffraction spectroscopy and exploratory calorimetry analysis. Studies to evaluate these hypotheses and the stability of solid dispersions are underway.
CONCLUSIONS
Solubility, dissolution profile and in vitro intestinal absorption of PZQ are modified by SD (2:1) with hydrogenated castor oil - PEG 60 (CR-60). The SDs obtained by the Fusion Method showed better results than those obtained by the Co-Precipitation Method. This series of favorable results showed that SD is a useful strategy for increasing the bioavailability of PZQ and could prove equally useful for other pharmaceutical compounds with poor water solubility.
ACKNOWLEDGEMENTS
This study received financial assistance from the National Council of Scientific and Technological Development (Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq)) and the Foundation for aid for Research of the State of São Paulo (Fundação de Amparo a Pesquisa do Estado de São Paulo (FAPESP)).
Received for publication on 23rd October 2008
Accepted for publication on 28th June 2010
- AHUJA, N.; KATARE, O.P.; SINGH, B. Studies on dissolution enhancement and mathematical modeling of drug release of a poorly water-soluble drug using water-soluble carriers. Eur. J. Pharm. Biopharm, v. 65, p. 26-38. 2007.
- ANDREWS, P. Praziquantel: mechanism of anti-schistossomal activity. Pharmacol. Ther. v.29, p-129-156, 1985.
- ALLEN, L.V.; YANCHICK, V. A.; MANESS, D. D. Dissolution rates of corticosteroids utilizing sugar glass dispersions. J. Pharm. Sci., v.249. p.494-497. 1977.
- BABU, G. V. M. M.; PRASAD, C. D. S.; MURTHY, K. V. R. Evaluation of modified gum karaya as carrier for the dissolution enhancement of poorly water-soluble drug nimodipine. Int. J. Pharm., v.234, p.1-17. 2002.
- BALIMANE, P. V.; CHONG, S.; MORRISON, R. A. Current methodologies used for evaluation of intestinal permeability and absorption. J. Pharmacol. Toxicol.Methods, v.44, p.301-312, 2000.
- BARTHE, L.; BESSOUET, M.; WOODLEY, J. F.; HOUIN, G. The improved everted gut sac: a simple method to study intestinal P-glycoprotein. Int. J. Pharm, v.173, p. 255-258. 1998.
- BARTHE, L.; WOODLEY, J.; HOUIN, G. Gastrointestinal absorption of drugs: methods and studies. Fundam. Clin. Pharmacol, v.13, p.154-168. 1999.
- CHIOU, W.L.; RIEGELMAN, S. Absorption characteristics of solid dispersed and micronized griseofulvin in man. J. Pharm. Sci, v.60, p.1376-1980. 1971.
- CHIOU, W.L.; RIEGELMAN, S. Oral absorption of griseofulvin in dogs: increased absorption via solid dispersion in polyethylene glycol 6000. J. Pham. Sci, v.59, p.937-941. 1970.
- CHOWDARY, K. P. R.; RAO, S. S. Investigation of dissolution enhancement of itraconazole by solid dispersion in superdisintegrants. Drug Dev. Ind. Pharm, v.26, p.1207-1211. 2000.
- CORNAIRE, G.; WOODLEY, J. F.; SAIVIN, S.; LEGENDRE, J. Y.; DECOURT, S.; CLOAREC, A.; HOUIN, G. Effect of polyoxyl 35 castor oil and polysorbate 80 on the intestinal absorption of digoxin in vitro. Arzn. Forsch. Drug Res, v.50, p. 576-579. 2000.
- CORRIGAN, O. J.; MURPHY, C. A.; TIMONEY, R. F. Dissolution properties of polyethyleneglycols and polyethyleneglycol-drug systems. Int. J. Pharm., v.4, p.67-74. 1979.
- DITTGEN, M.; FRICKE, S.; GERECKE, H.; OSTERWALD, H. Hot spin mixing- a new technology to manufacture solid dispersions, part 1: Testosterone. Pharmazie, v.50, p.225-226. 1995.
- EL-ARINI, S. K.; LEUENBERGER, H. Dissolution properties of praziquantel-pvp systems. Pharm. Acta Helv., v.73, p.89-94. 1998.
- GOLDBERG, A. H.; GIBALDI, M; KANIG, J. L. Increasing dissolution rates and gastrointestinal absorption of drugs via solid solution and eutectic mixture I. Theoretical consideration and discussion of the literature. J. Pharm. Sci., v.54, p.1145-1148. 1965.
- GOLDBERG, A. H.; GIBALDI, M. Increasing dissolution rates and gastrointestinal absorption of drugs via solid solution and eutectic mixtures IV. Chloranphenicol-urea system. J. Pharm. Sci, v.55, p.581-583. 1966.
- GOLDBERG, A. H.; GIBALDI, M.; KANIG, J. L. Increasing dissolution rates and gastrointestinal absorption of drugs via solid solution and eutectic mixtures. II Experimental evaluation of a eutectic mixture: urea-acetaminophen system. J. Pharm. Sci, v.55, p.482-487. 1965.
- GOLDBERG, A. H.; GIBALDI, M.; KANIG, J. L. Increasing dissolution rates and gastrointestinal absorption of drugs via solid solution and eutectic mixtures. III. Experimental evaluation of griseofulvin-succinic acid solid solution. J. Pharm. Sci, v.55, p.487-492. 1966.
- GONZÁLEZ-ESQUIVEL, D.; RIVIERA, J.; CASTRO, N.; YEPEZ-MULIA, L.; HELGI, J.C. In vitro characterization of some biopharmaceutical properties of praziquantel. Int. J. Pharm, v.295, p.93-95. 2005.
- JUPPO, A. M.; BOISSIER C.; KHOO C. Evaluation of solid dispersion particles prepared with SEDS. Int. J. Pharm, v.250, p.385-401. 2003.
- KARAVAS, E.; EMMANUEL, G.; SIGALAS, M. P.; AVGOUSTAKIS, K., BIKIARIS, D. Investigation of release mechanism of sparingly water-soluble drug from solid dispersion in hydrophilic carriers based on physical state of drug, particle size distribution and drug-polymer interactions. Eur. J. Pharm. Biopharm, v. 66, p. 334-347. 2007.
- KIM, M. S.; JIN, S. J.; KIM, J. S.; PARK, H. J.; SONG, H. S.; NEUBERT, R. H. H.; HWANG., S. J. Preparation, characterization and in vivo evaluation of amorphous atorvastatin calcium nanoparticles using supercrtitical antisolvent (SAS process). Eur. J. Pharm. Biopharm, v.69, p.454-465. 2008.
- LAW, D.; WANG, W.; SCHMITT, E. A.; QIU, Y.; KRILL, S. L.; FORT, J. J. Properties of rapidly dissolving eutectic mixtures of poly(ethylene glycol) and fenofibrate: The eutectic microstructure. J. Pharm. Sci, v.92, p.505-515. 2003.
- LEUNER, C.; DRESSMAN, J. Improving rug solubility for oral delivery using solid dispersions. Eur. J. Pharm. Biopharm, v.50, p.47-60. 2000.
- LIN, C. W.; CHAM, T. M. Effect of particle size on the available surface area of nifedipine from nifedipine-polyethyleneglycol 6000 solid dispersion. Int. J. Pharm. Sci, v.127, p.261-272. 1996.
- LIU, Y.; WANG, X.; WANG, J., CHING, C.B. Structural characterization and enantioseparation of the chiral compound Praziquantel. J. Pharm. Sci., v.93, p.3039-3046. 2004.
- LIU, C.; ZHU, S. J.; ZHOU, Y.; WEI, Y. P.; PEI, Y. Y. Enhanced of dissolution of cyclosporineA using solid dispersion with polyoxyethylene (40) stearate. Pharmazie, v. 61, p.681-684., 2006.
- MITCHELL, S. A.; REYNOLDS, T. D.; DASBACH, T. P. A compaction process to enhance dissolution of poorly water-soluble drugs using hydroxypropyl methylcellulose. Int. J. Pharm, v.250, p 3-11, 2003.
- OHARA, T.; KITAMURA, S.; KITAGAWA, T.; TERADA, K. Dissolution mechanism of poorly water-soluble drug from extended release solid dispersion system with ethylcellulose and hydroxypropylmethylcellulose. Int. J. Pharm., v.302, p.95-102. 2005.
- SARKARI, M.; BROWN, J.; CHEN, X.; SWINNEA, S.; WILLIAMMS, R. O.; JOHNSTON, K. P. Enhanced drug dissolution using evaporative precipitation into aqueous solution. Int. J. Pharm., v. 243, p.17-31. 2002.
- SAITO, M.; UGAJIN, T.; NOZAWA, Y.; SADZUKA, Y.; MIYAGISHIMA, A.; SONOBE, T. Preparation and dissolution characteristics of griseofulvin solid dispersions with saccharides. Int. J. Pharm, v.249, p.71-79. 2002.
- SETHIA, S.; SQUILLANTE, E. Solid dispersion: revival with greater possibilities and applications in oral drug delivery. Crit. Rev. Ther. Drug Carrier Syst, v.20, p.215-247. 2003.
- SHIM, S. C.; KIM, J. Physicochemical characterization of solid dispersion of furosemide with TPGS. Int. J. Pharm, v.251, p.79-84. 2003.
- SIMONELLI, A. P.; MEHTA, S. C; HIGUCHI, W. I. Dissolution rates of high energy polyvinylpyrrolidone-sulfathiazole coprecipitates. J. Pharm. Sci, v.58, p. 538-549. 1969.
- TORRE, P.; TORRADO, S.; TORRADO, S. Preparation, dissolution and characterization of praziquantel solid dispersion. Chem. Pharm. Bull, v.47, p.1629-1633. 1999.
- UNITED STATES PHARMACOPEIA. National Formulary. USP 30, NF 25. Rockeville: United States Pharmacopeial Convention Inc., 2007. p.3590
- WATABE, T.; HASEGAWA, S.; WAKIYAMA, N.; KUSAI, A.; SENNA, M. Comparison between polyvinylpyrrolidone and silica nanoparticles as carriers for indomethacin in a solid state dispersion. Int. J. Pharm, v.250, p. 283-286. 2003.
Publication Dates
-
Publication in this collection
12 Nov 2010 -
Date of issue
Sept 2010
History
-
Accepted
28 June 2010 -
Received
23 Oct 2008