Abstracts
The present investigation is aimed to develop self-microemulsifying drug delivery system (SMEDDS) to improve the in vitro dissolution of a BCS (Biopharmaceutical Classification System) class II anti emetic agent, domperidone. Solubility study was performed to identify the ingredients showing highest solubility of domperidone. The ternary phase diagrams were plotted for selected components to identify the area of microemulsion existence. D-optimal mixture experimental design was applied to optimize a liquid SMEDDS using formulation variables; the oil phase X1 (Oleic acid), the surfactant X2 (Labrasol) and the co-surfactant X3 (Transcutol HP). The liquid SMEDDS were evaluated for droplet size, emulsification time, % transmittance and drug release. Stability study was performed at 40 °C/75% RH. Liquid formulation was solidified by adsorption on carrier Aerosil 300. Solid SMEDDS was evaluated and compared with liquid SMEDDS and marketed formulation. Oleic acid was selected as oil, Labrasol as surfactant and Transcutol HP as co-surfactant for formulation of SMEDDS. The optimized batch showed best results in terms of smaller droplet size (<170 nm), emulsification time (<40 s) and drug release (>85% in 15 min) and was stable for 3 months. Solid SMEDDS containing Aerosil 300 showed good flow properties and uniform drug content. XRPD study revealed that the crystalline drug was converted to amorphous form in solid SMEDDS. The rate and extent of drug dissolution from solid SMEDDS was significantly higher than pure drug and commercial tablet formulation. The results demonstrate the potential of SMEDDS as a means of improving solubility, dissolution and hence the bioavailability.
Domperidone/self-microemulsifying delivery; Self-microemulsifying drug delivery system/development; Biopharmaceutical Classification System
O presente estudo teve como objetivo desenvolver sistemas de liberação auto-microemulsificantes (Self-Microemulsifying Drug Delivery System - SMEDDS) de domperidona, agente antiemético, classe II, segundo o sistema de classificação Biofarmacêutica, para melhorar sua dissolução in vitro. Estudo foi realizado para identificar os componentes que revelaram maior solubilidade da domperidona. Determinaram-se os diagramas de fase ternários para esses componentes selecionados tendo em vista a identificação da região de formação da microemulsão. O planejamento experimental foi empregado para otimizar os SMEDDS líquidos, utilizando as seguintes variáveis de formulação: a fase oleosa X1 (ácido oleico), o agente tensoativo X2 (Labrasol) e co-tensoativo X3 (Transcutol HP). Os SMEDDS líquidos foram avaliados quanto às seguintes características: tamanho da gota, tempo de emulsificação,% de transmitância e liberação do fármaco. O estudo de estabilidade foi realizado a 40 °C/75% de umidade relativa. A formulação foi convertida em forma sólida por sua adsorção em Aerosil 300. Os SMEDDS sólidos foram avaliados e comparados com SMEDDS líquidos e a formulação comercializada. O ácido oléico foi selecionado para a fase oleosa, Labrasol como agente tensoativo e Transcutol como co-tensoativo para a formulação de SMEDDS. O lote otimizado mostrou os melhores resultados: menor tamanho de gota (<170 nm), menor tempo de emulsificação (<40 segundos), e de liberação do fármaco (> 85% em 15 min). Além disso, a formulação otimizada manteve-se estável no período de 3 meses. Os SMEDDS sólidos contendo Aerosil 300 apresentaram boas propriedades de fluxo e uniformidade de conteúdo do fármaco. O estudo de difração de raios-X revelou que o fármaco cristalino foi convertido para a forma amorfa, nos SMEDDS sólidos. A velocidade de dissolução do fármaco a partir dos SMEDDS sólidos foi significativamente maior, quando comparado ao fármaco livre e à formulação de comprimidos comercial. Os resultados demonstram o potencial dos SMEDDS como meio para melhorar a solubilidade, a dissolução e, consequentemente, a biodisponibilidade da domperidona.
Domperidona/liberação automicroemulsificante; Sistemas de liberação automicroemulsificante; Sistema de Classificação Biofarmacêutica
INTRODUCTION
Majority of new chemical entities are found to be poorly water soluble in nature. To deliver such drugs in better way, the issue of poor aqueous solubility needs to be addressed by formulation scientist. Use of lipids has been explored in different ways recently to improve the bioavailability of poorly water soluble drugs. The unbeaten examples include simple oily solution, emulsion, microemulsion, nanoemulsion, micellar solution and more recently self-microemulsifying drug delivery systems (SMEDDS) (Hauss, 2007HAUSS, D.J. Oral lipid-based formulations. Adv. Drug Deliv. Rev., v.59, n.7, p.667-676, 2007.). The SMEDDS is advantageous over conventional emulsion in terms of easy manufacturing, scale up and good physical stability.
Fundamentally, a SMEDDS is mixture of natural/synthetic oil(s), solid/semisolid surfactant(s) ideally isotropic sometimes containing co-solvent(s) which upon introduction into aqueous phase, readily emulsifies to produce fine oil in water microemulsion. This whole emulsification procedure requires very little agitation, same as the peristaltic motion prevailing in the gut. SMEDDS produce droplets having size less than 100 nm (Colin, 1985COLIN, W.P. Self-emulsifying drug delivery systems: assessment of the efficiency of emulsification. Int. J. Pharm., v.27, n.2-3, p.335-348, 1985.). In comparison to traditional emulsion formulations which are thermodynamically unstable dosage forms and require high energy input, SMEDDS are kinetically stable and spontaneous in emulsion formation. The salient features of SMEDDS include: (a) Enhanced oral bioavailability enabling reduction in dose, (b) More consistent temporal profiles of drug absorption, (c) Selective targeting of drug(s) towards specific absorption window in GIT, (d) Protection of drug(s) from the hostile environment in gut, (e) Control of delivery profiles, (f) Reduced variability including food effects, (g)High drug payloads, (h)possibility of autoclaving(Charman et al., 1992CHARMAN, S.A.; CHARMAN, W.N.; ROGGE, M.C.; WILSON, T.D.; DUTKO, F.J.; POUTON, C.W. Self-emulsifying drug delivery systems: formulation and biopharmaceutic evaluation of an investigational lipophilic compound. Pharm. Res., v.9, n.1, p.87-93, 1992.). Further, the SMEDDS is believed to increase oral absorption via any of the following mechanisms: (a) Retardation of gastric transit time, (b) increase in effective drug solubility in lumen (c), lymphatic transport of the drug, (d) enterocyte based drug transport and, (e) increasing membrane permeability (Poelma, Breãs. Tukker, 1990POELMA, F.G.; BREÄS, R.; TUKKER, J.J. Intestinal absorption of drugs. III. The influence of taurocholate on the disappearance kinetics of hydrophilic and lipophilic drugs from the small intestine of the rat. Pharm. Res., v.7, n.4, p.392-397, 1990.; Poelma et al., 1991POELMA, F.G.; BREÄS, R.; TUKKER, J.J.; CROMMELIN, D.J. Intestinal absorption of drugs: the Influence of mixed micelles on on the disappearance kinetics of drugs from the small intestine of the rat. J. Pharm. Pharmacol., v.43, n.5, p.317-324, 1991.; Shah et al., 1994SHAH, N.H.; CARVAJAL, M.T.; PATEL, C.I.; INFELD, M.H.; MALICK, A.W. Self-emulsifying drug delivery systems (SEDDS) with polyglycolized glycerides for improving in vitro dissolution and oral absorption of lipophilic drugs. Int. J. Pharm., v.106, p.15-23, 1994.; Porter, Charman, 2001PORTER, C.J.; CHARMAN, W.N. Lipid-based formulations for oral administration: opportunities for bioavailability enhancement and lipoprotein targeting of lipophilic drugs. J. Recept. Signal Transduct. Res., v.21, n.2-3, p.215-257, 2001.; Porter, Trevaskis, Charman, 2007PORTER, C.J.; TREVASKIS, N.L.; CHARMAN, W.N. Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat. Rev. Drug Discov., v.6, n.3, p.231-248, 2007.).This facet of SMEDDS makes them stand alone in the category of oral lipid based formulations. Several SMEDDS of BCS class II drugs i.e. acyclovir (Patel, Sawant, 2007PATEL, D.; SAWANT, K.K. Oral bioavailability enhancement of Acyclovir by Self-Microemulsifying Drug Delivery Systems (SMEDDS). Drug Dev. Ind. Pharm., v.33, n.12, p.1318-1326, 2007.), carvedilol (Mahmoud, Bendas, Mahmoud, 2009MAHMOUD, E.A.; BENDAS, E.R.; MOHAMED, M.I. Preparation and evaluation of self-nanoemulsifying tablets of Carvedilol. AAPS PharmSciTech, v.10, n.1, p.183-192, 2009.), coenzyme Q10 (Kommuru et al., 2001KOMMURU, T.R.; GURLEY, B.; KHAN, M.A.; REDDY, I.K. Self-emulsifying drug delivery systems (SEDDS) of coenzyme Q10: formulation development and bioavailability assessment. Int. J. Pharm., v.212, n.2, p.233-246, 2001.), ezetimibe (Dixit, Nagarsenker, 2008DIXIT, R.P.; NAGARSENKER, M.S. Self-nanoemulsifying granules of ezetimibe: design, optimization and evaluation. Eur. J. Pharm. Sci., v.35, n.3, p.183-192, 2008.), nimodipine (Kale, Patravale, 2008KALE, A.A.; PATRAVALE, V.B. Design and evaluation of self-emulsifying drug delivery systems (SEDDS) of nimodipine. AAPS PharmSciTech, v.9, n.1, p.191-196, 2008.), simvastatin (Patil, Patil, Paradkar, 2007PATIL, P.; PATIL, V.; PARADKAR, A. Formulation of a self-emulsifying system for oral delivery of simvastatin: in vitro and in vivo evaluation. Acta Pharm., v.57, n.1, p.111-122, 2007.) etc are well reported in various reputed literature.
However, the solid dosage forms have been the favourite dosage form for manufacturers and patients as well. Anything that comes as solid form is well accepted in terms of performance and stability. The liquid SMEDDS pre-concentrate present a problem of leakage of drug from capsule and it may also lead to dehydration of capsule cell. Another issue with liquid SMEDDS is that solubilization of a complete dose of drugs in single capsule volume suitable for oral administration is sometimes not possible. The liquid pre-concentrate can be mixed along with some solid and/or semisolid excipients to prepare solid dispersions. Solid carriers can be microporous inorganic substances, high surface-area colloidal inorganic adsorbent substances, cross-linked polymers or nanoparticle adsorbents. For example, silica, silicates, colloidal silicon dioxide, magnesium trisilicate, magnesium hydroxide, talcum, crospovidone, cross-linked sodium carboxy methyl cellulose and cross linked polymethyl methacrylate are typical solid carriers.
In the present investigation, domperidone, a well-known antiemetic drug with low oral bioavailability (about 15%), was taken as a candidate drug. This is due to poor solubility and extensive first pass metabolism in the gut wall and liver. Furthermore it is reported that, the bioavailability of domperidone is enhanced in normal subjects when taken after a meal, which indicates that fat may enhance absorption through lymphatic system and thus increase bioavailability (Mueller et al., 1994MUELLER, E.A.; KOVARIK, J.M.; VAN BREE, J.B.; GREVEL, J.; LÜCKER, P.W.; KUTZ, K. Influence of a fat-rich meal on the pharmacokinetics of a new oral formulation of Cyclosporine in a crossover comparison with the market formulation. Pharm. Res., v.11, n.1, p.151-155, 1994.). Hence in present study, oil, surfactant and co-surfactant were selected having high drug solubility followed by formulation region optimization by D optimal design. Solidification was done by using suitable adsorbent so as to get advantage of unit dosage form and improved stability.
MATERIAL AND METHODS
Material
Domperidone was gifted by Torrent Research Centre, Ahmedabad. Labrasol, Transcutol HP and Lauroglycol were provided by Gattefosse, France as gift sample. Cremophor EL and Soluphor P were kindly gifted by BASF, Germany. Isopropyl myristate and Oleic acid were purchased from Central Drug House pvt. Ltd., India. All other chemicals and reagents used were of pharmaceutical grades.
Methods
Solubility studies
The solubility of domperidone in various oils, surfactants, and co-surfactants was determined using the method reported by Basalious et al. (2010)CHARMAN, S.A.; CHARMAN, W.N.; ROGGE, M.C.; WILSON, T.D.; DUTKO, F.J.; POUTON, C.W. Self-emulsifying drug delivery systems: formulation and biopharmaceutic evaluation of an investigational lipophilic compound. Pharm. Res., v.9, n.1, p.87-93, 1992.. Two grams of each selected vehicle was added to each vial containing known excess of domperidone (500 mg). After sealing, the mixture was heated at 40 °C in water-bath for 15 min to facilitate the solubilization and mixed using a vortex mixer. Mixtures were shaken on shaker bath at 30±0.5 °C for 48 h. After reaching equilibrium, the mixtures were centrifuged using refrigerated centrifuge (Remi, C 24 BL) at 3000 rpm for 15 min, then 0.5 mL supernatant was taken with glass micropipette, and the content of domperidone was quantified by UV-Visible double beam spectrophotometer (Shimdzu UV 1800 corporation, Japan) at 286 nm after dilution with methanol.
Construction of pseudo-ternary phase diagram
Based on higher drug solubility ternary phase diagram was developed for selected oil, surfactant and co-surfactant (Table I). Three variables (factor) used were oil, water and mix of surfactant and co-surfactant (Smix) in specific ratio (ie.1:1, 1:2, 2:1). Ternary phase diagram was developed using aqueous titration method (Gupta, Mishra et al., 2011GUPTA, A.K.; MISHRA, D.K.; MAHAJAN, S.C. Preparation and in-vitro evaluation of self emulsifying drug delivery system of antihypertensive drug valsartan. Int. J. Pharm. Life Sci., v.2, n.3, p.633-639, 2011.; Kumar et al., 2011KUMAR, M.S.; SHAILAJA, P.; MURTHY, K.R.; PRADESH, A. Improvement of oral bioavailability of nifedipine through self-microemulsifing drug delivery systems. J. Glob. Trends Pharm. Sci., v.2, n.3, p.364-388, 2011.). Slow titration with aqueous phase was done to each weight ratio of oil and Smix and visual observation was carried out for formation of transparent and easily flowable o/w micro emulsion. The physical state of the micro emulsion was marked on a pseudo-three-component phase diagram with one axis representing aqueous phase, the other representing oil and the third representing a mixture of surfactant and co-surfactant at fixed weight ratios (Smix 1:1). The phase boundary was determined by observing the change in sample appearance from transparent to turbid. The phase diagram was constructed by using sigma plot 12 software. Similarly ternary phase diagrams were prepared for other ratios of surfactant and co-surfactant like 1:2, 2:1, 3:1, 4:1, 5:1, 6:1 etc.
Preparation of SMEDDS
A constant amount of drug was dissolved in oil using vortex mixer (Remi Motors Ltd., India). Required amount of surfactant and co-surfactant were added to the mixtures and further mixed using vortex mixer. These mixtures were warmed to 40 °C using a water bath for 30 mins with intermittent shaking to ensure complete mixing. The formulations were evaluated for emulsification time, percent transmittance, droplet size and in vitro drug release.
Evaluation of SMEDDS
• Emulsification time and transmittance
The SMEDDS formulation (0.1 mL) was introduced into 100 mL of 0.1 N HCl under action of propeller stirrer at constant speed of 100 rpm at 37±5 °C temperature. Emulsification time was measured by visual observation and percent transmittance was measured at 650 nm through UV spectrophotometer(Trull et al., 1994TRULL, A.K.; TAN, K.K.; TAN, L. Enhanced absorption of new oral cyclosporin microemulsion formulation, neoral in liver transplant recipients with external biliary diversion. Transplant. Proc., v.26, p.2977-2978, 1994.)
• Droplet size
Hundred milligram of each formulation was introduced into 100 mL of 0.1 N HCl at 25 °C and the contents were gently stirred using a propeller stirrer. The droplet size of the resultant emulsion was determined by photon correlation spectroscopy using a Mastersizer 2000 (Malvern Instruments, UK) which can measure sizes between 10 and 5000 nm (Patil, Joshi, Paradkar, 2004PATIL, P.; JOSHI, P.; PARADKAR, A. Effect of formulation variables on preparation and evaluation of gelled self-emulsifying drug delivery system (SEDDS) of ketoprofen. AAPS PharmSciTech, v.5, n.3, p.E42, 2004.).
• In vitro dissolution studies
In vitro drug release study was carried out using USP type II (Paddle type) dissolution apparatus. SEMDDS containing 10 mg of domperidone was filled in HPMC capsules (size “00”) and introduced into 900 mL of a dissolution medium 0.1 N HCl and maintained at 37±0.5 °C. The Revolution speed of the paddle was kept constant at 50 rpm. The aliquot of 5 mL was withdrawn at 5, 10, 15, 30, 45, and 60 min and filtered through 0.45 µm membrane filters. The concentration of domperidone was determined spectrophotometrically at 284 nm. The dissolution profile of developed optimized batch was compared with pure drug and marketed preparation Domcolic®.
Formulation optimization of SMEDDS using D optimal design
The mixture experimental study was designed based on a three component system: the oil X1 (Oleic acid), the surfactant X2 (Labrasol) and the co-surfactant X3 (Transcutol HP). Based on the previous result obtained from phase diagram, the range of X1 was selected as 10-30% and that of X2 and X3 was selected as 35-45%. Values of independent variables were introduced into the Design-Expert version 8 software and batch matrix was derived. Sixteen batches were prepared as mentioned above and evaluated (Table II). The emulsification time (Y1), mean droplet size (Y2) and cumulative amount of drug released after 15mins (Y3) were used as the responses.
Formulation of solid SMEDDS
Adsorption on solid carrier is easy and reliable method to convert liquid SMEDDS into solid SMEDDS (Agarwal et al., 2009AGARWAL, V.; SIDDIQUI, A.; ALI, H.; NAZZAL, S. Dissolution and powder flow characterization of solid self-emulsified drug delivery system (SEDDS). Int. J. Pharm., v.366, n.1-2, p.44-52, 2009.). Silicon dioxide shows high adsorption capacity and its permitted safe concentration as per Inactive Ingredients Guide of USFDA is 100 mg. The liquid SMEDDS was added drop wise over the solid adsorbent in a broad porcelain dish. After each addition, the mixture was homogenized using glass rod to ensure uniform distribution of the formulation. The resultant damp mass was passed through sieve no. 120, dried at ambient temperature and evaluated for flow property, compressibility, particle size distribution and in vitro drug release. Optimized formulation was characterized for X-Ray Powder Diffraction study so as to identify the state of drug and also subjected to stability study at 40 °C/70% RH for 3 months (Oh et al., 2011OH, D.H.; KANG, J.H.; KIM, D.W.; LEE, B.-J.; KIM, J.O.; YONG, C.S.; CHOI, H.-G. Comparison of solid self-microemulsifying drug delivery system (solid SMEDDS) prepared with hydrophilic and hydrophobic solid carrier. Int. J. Pharm., v.420, n.2, p.412-418, 2011.).
RESULT AND DISCUSSION
An effective SMEDDS is the one which emulsifies spontaneously to generate oil droplets enclosing the dissolved drug and which also solubilizes the drug in given dissolution medium rapidly and completely. Keeping these criteria in mind, the study was designed in such a way that the results can ensure the behavior of the drug delivery system in vivo. Here, non-ionic surfactants were used in the study since they are known to be less affected by pH and changes in ionic strength.
Solubility study
Result of solubility studies on domperidone in various oils, surfactants and co-surfactants are presented in Table I. Oleic acid showed highest drug solubility (74.9 mg/mL) and no other oil showed comparable solubility and hence only Oleic acid was selected as oil phase for domperidone SMEDDS formulation. Transcutol HP showed highest drug solubility (70.58 mg/mL) and good solubility was also observed in cremophor EL (38.55 mg/mL), Tween-80 (36.6 mg/mL), Labrasol (35.9 mg/mL). Hence Transcutol HP was selected as cosurfactant and the other three were selected as surfactants for development of phase diagram.
Pseudo-ternary phase diagrams
SMEDDS formulation should be simple and safe, prepared using nontoxic surfactants as well as pseudo ternary phase diagrams shows high region of formulation (Kang, Lee et al., 2004KANG, B.K.; LEE, J.S.; CHON, S.K.; JEONG, S.Y.; YUK, S.H.; KHANG, G.; LEE, H.B.; CHO, S.H. Development of self-microemulsifying drug delivery systems (SMEDDS) for oral bioavailability enhancement of simvastatin in beagle dogs. Int. J. Pharm., v.274, n.1-2, p.65-73, 2004.). On the basis of the solubility study of domperidone, oleic acid was used as the oil phase and Transcutol HP was used as the co-surfactant. All three surfactants showed higher monophasic region in 1:1 ratio with Transcutol HP. Figures 1, 2 and 3 show phase diagrams for Cremophor EL, Labrasol and Tween 80 respectively. Labrasol showed higher monophasic region as compared to Cremophor EL and Tween 80 and hence it was further used for formulation of SMEDDS of domperidone.
Pseudo-ternary phase diagrams of Cremophor EL as surfactant in 1:1 ratio with Transcutol HP.
Preparation and evaluation of SMEDDS
From the results of phase diagrams, Oleic acid, Labrasol and Transcutol HP were finalised as oil, surfactant and co-surfactant respectively. As formulation ingredients are selected, SMEDDS was prepared incorporating 10 mg drug. Sixteen batches were prepared and evaluated as showed in Table II. Emulsification time was assessed visually. If formulation is microemulsion, emulsification takes place within a minute on addition of it into water. Emulsion formulation in batches P2, P3, P4, P5, P8, P12, P13 and P14 required time less than a minute which is indicative of micron size of globules. Furthermore the emulsions of these batches were clear with bluish tinge and stable. Generally the formulation can be termed microemulsion only if the globule size in less than 300 nm. Batches P1, P6, P7, P9, P10, P11, P15 and P16 showed higher globule size and low clarity.
Percentage transmittance can be used to reflect clarity and micron size of globule. The average transmittance observed of all the prepared batches was around 80% while batches P2 and P4 showed highest (90%) transmittance.
From the globule size analysis it was concluded that batches P2 and P4 has smaller globule size as compared to other batches, as these batches contained only 10% oil. As the concentration of oil increases the globule size increase whereas increasing the amount of surfactant and co-surfactant leads to decrease in globule size.
In vitro drug release showed that more than 75% drug got released from all batches within 60 min which indicates the solubility enhancing potential of SMEDDS formulations. Drug release at 15min was compared and as expected, batches P2 and P4 showed higher drug release (˃85%) in 15 min due to lower oil content and higher content of surfactant and co-surfactant.
Optimization of SMEDDS
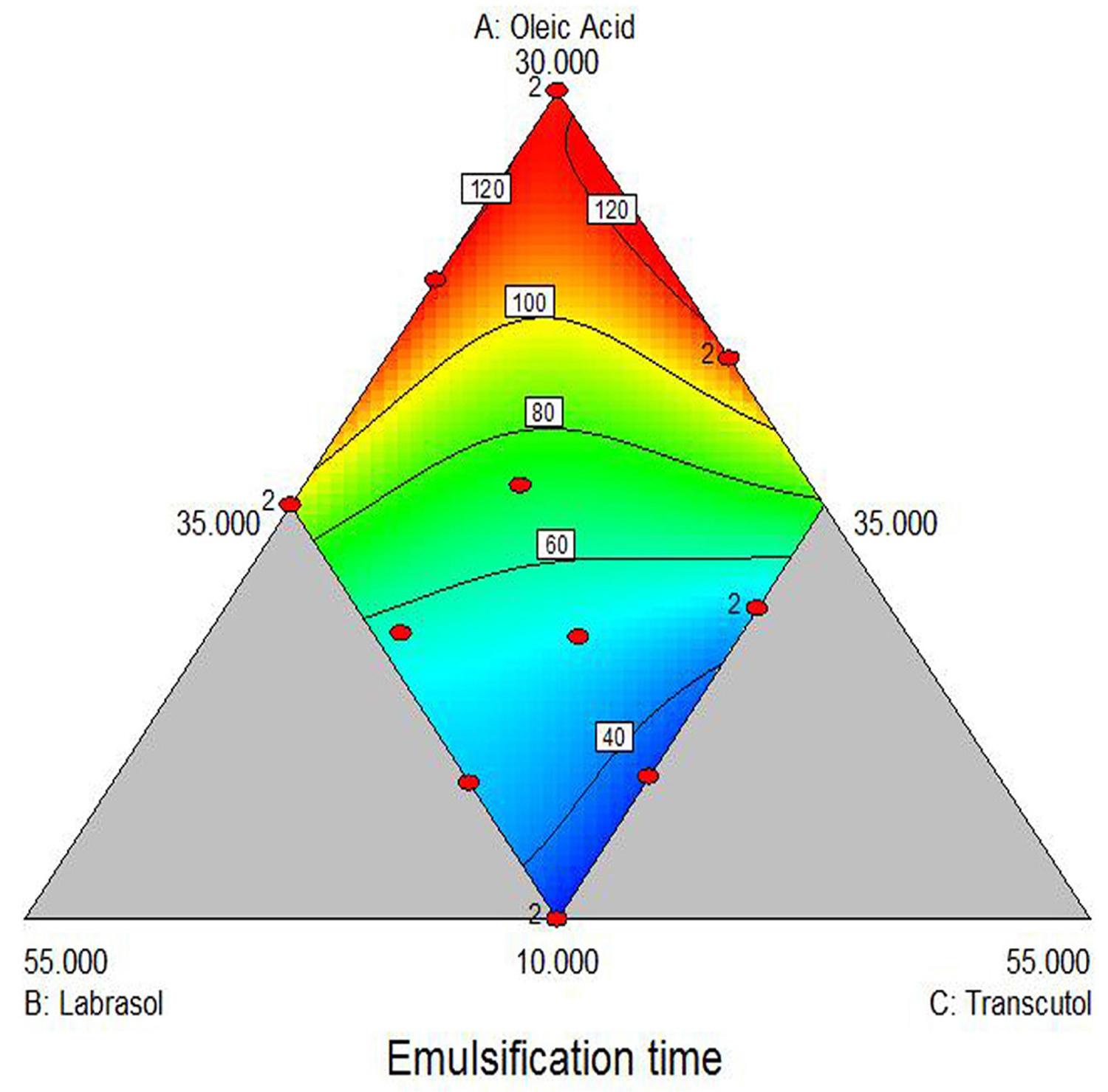
The emulsification time of all sixteen batches are presented in Table II. The emulsification time was ranged from 29s to 120s, which indicate that all the batches quickly converted into microemulsion on exposure to aqueous media. The selected special quadratic model was used to generate the following equation for the emulsification time:
Y1 = 116.70X1 – 29.14X2 – 163.46X3 + 202.16X1X2 + 405.42X1X3 + 508.57X2X3 – 2653.28X12X2X3 – 1015.06X1X22X3 – 141.67X1X2X32 (1)
Figure 4 indicates the effect of Oleic acid, Labrasol and Transcutol HP on emulsification time. It can be observed from the plot that emulsification time may increase with the increase in amount of Oleic acid and may decrease with the increase in amount of Labrasol and Transcutol HP. The P value of 0.05 for any factor in analysis of variance (ANOVA) indicates significant effect of the corresponding factor on the emulsification time (Y1). It can be inferred that the interaction term X1X2, X1X3, X2X3, X1X22X3 and X1X2X32 have non-significant effect on the emulsification time. The only interaction term X12X2X3 has a significant antagonistic effect on emulsification time as indicated by the negative value of the coefficient. Thus we can conclude that emulsification time increase by high concentration of oil (X12).
Response surface plot of emulsification time (Y1) versus three factors (X1 = Oleic acid, X2 = Labrasol, X3 = Transcutol HP).
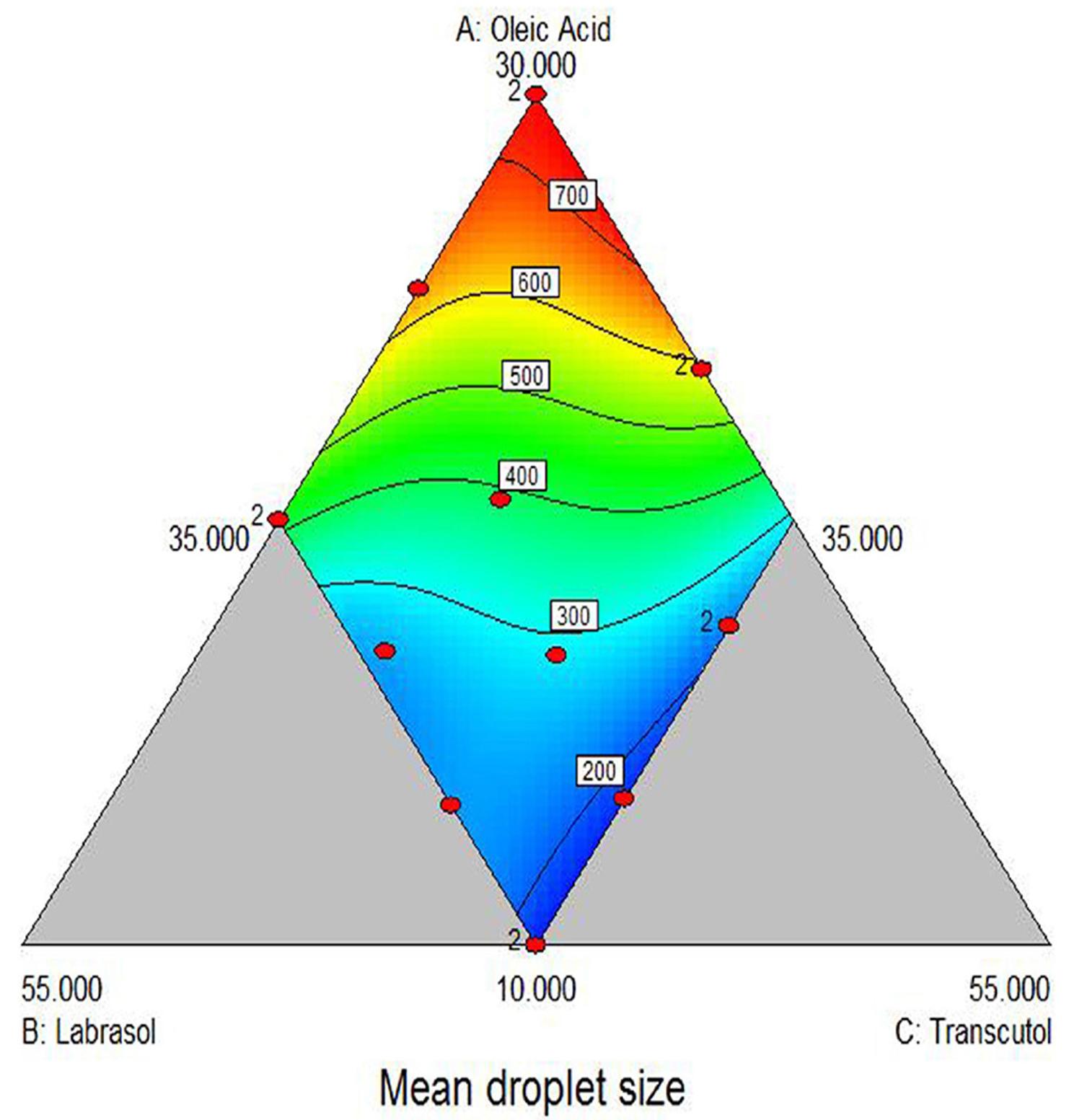
The mean droplet size was selected as another response and is presented in Table II. The mean droplet size was ranged between 150 nm to 721.56 nm, which indicates that the response was sensitive towards the studied factor. The equation for the mean droplet size is as mentioned underneath:
Y2 = 715.75X1 – 314.15X2 – 1454.35X3 + 894.08X1X2 + 1613.06X1X3 + 4197.63X2X3 – 12553.09X12X2X3 – 10897.00X1X22X3 – 5484.62X1X2X32 (2)
As indicated in Figure 5, it was observed that the mean droplet size may increase with the increase in amount of Oleic acid and may decrease with increase in the amount of Labrasol and Transcutol HP. From the P value of 0.05 in ANOVA, it can be inferred that the interaction term X1X2, ,X1X22X3 and X1X2X32 have non-significant effect on the mean droplet size. The interaction term X1X3 and X2X3 have a significant positive synergistic effect on mean droplet size and term X12X2X3 has a significant antagonistic effect on mean droplet size indicated by the negative value of the coefficient. Thus we can conclude that droplet size also increase with increase in oil concentration.
Response surface plot of mean droplet size (Y2) versus three factors (X1 = Oleic acid, X2 = Labrasol, X3 = Transcutol HP).
The drug released at 15min was ranged between 63.69 % and 87.96 %, which indicated that it is also affected by the concentration of all three ingredients. The equation for the drug released at 15min is as below:
Y3 = 65.48X1 + 53.72X2 + 103.83X3 + 10.81X1X2 + 18.85X1X3 + 34.95X2X3 – 680.67X12X2X3 – 712.17X1X22X3 – 516.88X1X2X32 (3)
It can be observed from the Figure 6 that drug released after 15 mins may decrease with the increase in amount of Oleic acid and may increase with the increase in the amount of Labrasol and Transcutol HP. Results of ANOVA show that the (P value >0.05) interaction term X1X3, X2X3, X1 X22X3 and X1X2 X32 have non-significant effect on the drug released after 15 mins. The interaction term X1X2 have a significant positive synergistic effect on drug released after 15 mins and X12X2X3 have a significant antagonistic effect on drug released after 15 mins indicated by the negative value of the coefficient.
Response surface plot of drug released after 15 min (Y3) versus three factors (X1 = Oleic acid, X2 = Labrasol, X3 = Transcutol HP).
It is clear from the developed equations that optimum response in terms of lower emulsification time (<40 s), smaller droplet size (<170 nm) and higher drug release (>85% in 15 mins) was achieved at low concentrations of oil and higher concentrations of surfactants and co surfactant. Hence optimized batches, P2 and P4 containing 10% of oil 45% of surfactant and co-surfactant showed the responses (Y1=32 and 29, Y2=150 and 168, Y3=87.96 and 87.45) which were in close agreement with the predicted ones (30.862, 165.32 and 87.503 respectively). Therefore the developed model was found reliable.
Solidification of SMEDDS
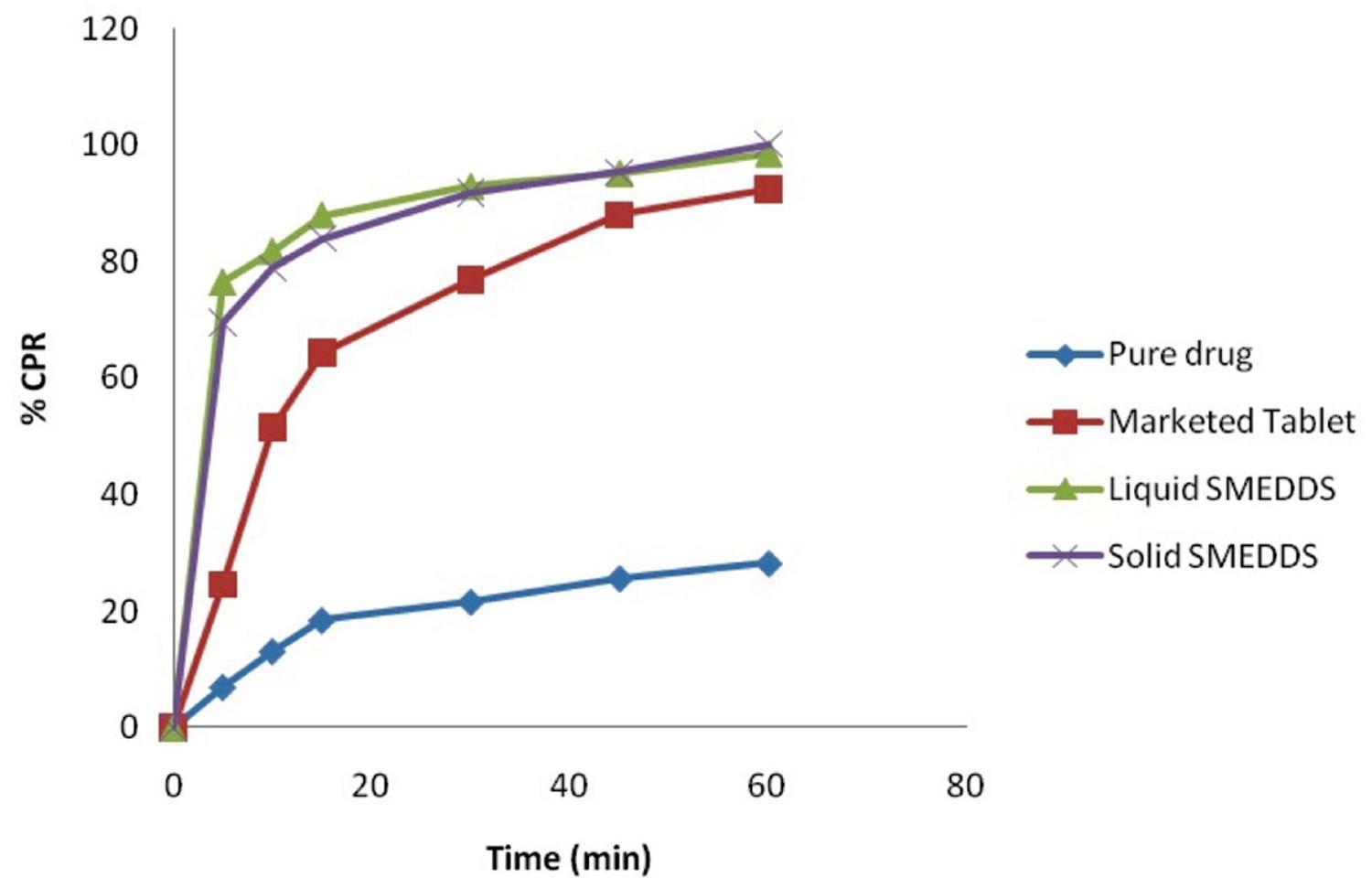
In present study two grades of colloidal silicon dioxide, Aerosil 200 (20% to 30%) and Aerosil 300 (20% to 40%) were used. Five batches were prepared and evaluated (Table III). Batches SP1 and SP3 containing 20% of Aerosil 200 and Aerosil 300 respectively were adhesive and showed poor flow property. Thus 20% of carrier is not enough for formulation of free flowing solid mass. Batches SP2 and SP4 containing 30% carrier showed good compressibility, flow property and dry mass produced can easily be filled in “size 00” HPMC capsule. Hence it was concluded that 30% carrier was enough for formation of solid SMEDDS of domperidone. In comparison to batch SP2, batch SP4 containing Aerosil 300 showed better flow properties which indicate that Aerosil 300 is having better adsorption capacity. Batch SP5 containing 40% adsorbent showed that the mass produced cannot be filled in “size 00” HPMC capsule. Further it increases the cost of formulation and hence we can conclude that concentration of carrier should be optimized. Batch SP4 showed 200.6 nm mean droplet size after emulsion formation and 84.79% transmittance which are well accepted and comparable to liquid SMEDDS. Further batch SP4 was also evaluated for in vitro drug release and compared with liquid SMEDDS, pure drug and marketed product (Figure 7). Comparative drug release profile shows the dissolution enhancement potential of SMEDDS formulations. Further we can conclude that solidification did not affect the selected responses specially the in vitro drug release profile.
Comparative drug release profile of pure drug, marketed product, liquid SMEDDS (batch P2) and solid SMEDDS (batch SP4).
Batch SP4 and pure drug were analysed to check the crystallinity by X-Ray Powder Diffraction (Figure 8). It can be concluded from the result that pure drug is in crystalline form and it is converted into amorphous state when formulated in SMEDDS which may be responsible for higher drug solubility. Accelerated stability study after 3 months showed comparable drug release and assay. Further the study should continue for 6 months to conclude about the stability of formulated domperidone solid SMEDDS.
CONCLUSION
Solid SMEDDS is one of the recent approaches for formulation of unit dosage form for drugs with low aqueous solubility. Selection of oil and surfactant, co surfactant blend is crucial and vary from drug to drug based on solubility study. The liquid SMEDDS pre concentrate was converted into solid by adsorption on to a carrier (Aerosil 300). The optimized solid SMEDDS formulation of domperidone showed significant increase in dissolution rate compared to marketed tablet (Domcolic®) and pure drug indicates the potential of SMEDDS. We can conclude from this study that solid SMEDDS formulation is capable to enhance solubility and dissolution of poorly water soluble drugs like domperidone which may result in improved therapeutic performance.
ACKNOWLEDGMENT
The authors are thankful to Gattefosse, France and BASF, Germany for providing gift samples. The authors are also thankful to Dr. Ruchi Sawhney for her kind support in preparation of the manuscript.
REFERENCES
- AGARWAL, V.; SIDDIQUI, A.; ALI, H.; NAZZAL, S. Dissolution and powder flow characterization of solid self-emulsified drug delivery system (SEDDS). Int. J. Pharm., v.366, n.1-2, p.44-52, 2009.
- BASALIOUS, E.B.; SHAWKY, N.; BADR-ELDIN, S.M. SNEDDS containing bioenhancers for improvement of dissolution and oral absorption of lacidipine. I: Development and optimization. Int. J. Pharm., v.391, n.1-2, p.203-211, 2010.
- CHARMAN, S.A.; CHARMAN, W.N.; ROGGE, M.C.; WILSON, T.D.; DUTKO, F.J.; POUTON, C.W. Self-emulsifying drug delivery systems: formulation and biopharmaceutic evaluation of an investigational lipophilic compound. Pharm. Res., v.9, n.1, p.87-93, 1992.
- COLIN, W.P. Self-emulsifying drug delivery systems: assessment of the efficiency of emulsification. Int. J. Pharm., v.27, n.2-3, p.335-348, 1985.
- DIXIT, R.P.; NAGARSENKER, M.S. Self-nanoemulsifying granules of ezetimibe: design, optimization and evaluation. Eur. J. Pharm. Sci., v.35, n.3, p.183-192, 2008.
- GUPTA, A.K.; MISHRA, D.K.; MAHAJAN, S.C. Preparation and in-vitro evaluation of self emulsifying drug delivery system of antihypertensive drug valsartan. Int. J. Pharm. Life Sci., v.2, n.3, p.633-639, 2011.
- HAUSS, D.J. Oral lipid-based formulations. Adv. Drug Deliv. Rev., v.59, n.7, p.667-676, 2007.
- KALE, A.A.; PATRAVALE, V.B. Design and evaluation of self-emulsifying drug delivery systems (SEDDS) of nimodipine. AAPS PharmSciTech, v.9, n.1, p.191-196, 2008.
- KANG, B.K.; LEE, J.S.; CHON, S.K.; JEONG, S.Y.; YUK, S.H.; KHANG, G.; LEE, H.B.; CHO, S.H. Development of self-microemulsifying drug delivery systems (SMEDDS) for oral bioavailability enhancement of simvastatin in beagle dogs. Int. J. Pharm., v.274, n.1-2, p.65-73, 2004.
- KOMMURU, T.R.; GURLEY, B.; KHAN, M.A.; REDDY, I.K. Self-emulsifying drug delivery systems (SEDDS) of coenzyme Q10: formulation development and bioavailability assessment. Int. J. Pharm., v.212, n.2, p.233-246, 2001.
- KUMAR, M.S.; SHAILAJA, P.; MURTHY, K.R.; PRADESH, A. Improvement of oral bioavailability of nifedipine through self-microemulsifing drug delivery systems. J. Glob. Trends Pharm. Sci., v.2, n.3, p.364-388, 2011.
- MAHMOUD, E.A.; BENDAS, E.R.; MOHAMED, M.I. Preparation and evaluation of self-nanoemulsifying tablets of Carvedilol. AAPS PharmSciTech, v.10, n.1, p.183-192, 2009.
- MUELLER, E.A.; KOVARIK, J.M.; VAN BREE, J.B.; GREVEL, J.; LÜCKER, P.W.; KUTZ, K. Influence of a fat-rich meal on the pharmacokinetics of a new oral formulation of Cyclosporine in a crossover comparison with the market formulation. Pharm. Res., v.11, n.1, p.151-155, 1994.
- OH, D.H.; KANG, J.H.; KIM, D.W.; LEE, B.-J.; KIM, J.O.; YONG, C.S.; CHOI, H.-G. Comparison of solid self-microemulsifying drug delivery system (solid SMEDDS) prepared with hydrophilic and hydrophobic solid carrier. Int. J. Pharm., v.420, n.2, p.412-418, 2011.
- PATEL, D.; SAWANT, K.K. Oral bioavailability enhancement of Acyclovir by Self-Microemulsifying Drug Delivery Systems (SMEDDS). Drug Dev. Ind. Pharm., v.33, n.12, p.1318-1326, 2007.
- PATIL, P.; JOSHI, P.; PARADKAR, A. Effect of formulation variables on preparation and evaluation of gelled self-emulsifying drug delivery system (SEDDS) of ketoprofen. AAPS PharmSciTech, v.5, n.3, p.E42, 2004.
- PATIL, P.; PATIL, V.; PARADKAR, A. Formulation of a self-emulsifying system for oral delivery of simvastatin: in vitro and in vivo evaluation. Acta Pharm., v.57, n.1, p.111-122, 2007.
- POELMA, F.G.; BREÄS, R.; TUKKER, J.J. Intestinal absorption of drugs. III. The influence of taurocholate on the disappearance kinetics of hydrophilic and lipophilic drugs from the small intestine of the rat. Pharm. Res., v.7, n.4, p.392-397, 1990.
- POELMA, F.G.; BREÄS, R.; TUKKER, J.J.; CROMMELIN, D.J. Intestinal absorption of drugs: the Influence of mixed micelles on on the disappearance kinetics of drugs from the small intestine of the rat. J. Pharm. Pharmacol., v.43, n.5, p.317-324, 1991.
- PORTER, C.J.; CHARMAN, W.N. Lipid-based formulations for oral administration: opportunities for bioavailability enhancement and lipoprotein targeting of lipophilic drugs. J. Recept. Signal Transduct. Res., v.21, n.2-3, p.215-257, 2001.
- PORTER, C.J.; TREVASKIS, N.L.; CHARMAN, W.N. Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat. Rev. Drug Discov., v.6, n.3, p.231-248, 2007.
- SHAH, N.H.; CARVAJAL, M.T.; PATEL, C.I.; INFELD, M.H.; MALICK, A.W. Self-emulsifying drug delivery systems (SEDDS) with polyglycolized glycerides for improving in vitro dissolution and oral absorption of lipophilic drugs. Int. J. Pharm., v.106, p.15-23, 1994.
- TRULL, A.K.; TAN, K.K.; TAN, L. Enhanced absorption of new oral cyclosporin microemulsion formulation, neoral in liver transplant recipients with external biliary diversion. Transplant. Proc., v.26, p.2977-2978, 1994.
Publication Dates
-
Publication in this collection
Jan-Mar 2014
History
-
Received
27 Feb 2013 -
Accepted
22 Aug 2013