Abstracts
Tissue damage due to oxidative stress is directly linked to development of many, if not all, human morbidity factors and chronic diseases. In this context, the search for dietary natural occurring molecules with antioxidant activity, such as flavonoids, has become essential. In this study, we investigated a set of 41 flavonoids (23 flavones and 18 flavonols) analyzing their structures and biological antioxidant activity. The experimental data were submitted to a QSAR (quantitative structure-activity relationships) study. NMR 13C data were used to perform a Kohonen self-organizing map study, analyzing the weight that each carbon has in the activity. Additionally, we performed MLR (multilinear regression) using GA (genetic algorithms) and molecular descriptors to analyze the role that specific carbons and substitutions play in the activity.
Flavonoids; 13C NMR; Kohonen self-organizing map
Danos aos tecidos devido ao estresse oxidativo estão diretamente ligados ao desenvolvimento de muitos, senão todos, os fatores de sedentarismo e de doenças crônicas. Neste contexto, a busca de moléculas naturais, que participam da nossa dieta e que possuam atividade antioxidante, flavonóides, torna-se de grande interesse. Neste estudo, nós investigamos um conjunto de 41 flavonóides (23 flavonas e 18 flavonóis), relacionando suas estruturas e atividade antioxidante. Os dados experimentais foram submetidos à análise de QSAR (relações quantitativas estrutura-atividade). Dados de RMN 13C foram utilizados para realizar um estudo do mapa auto-organizável de Kohonen, analisando o peso que cada carbono tem na atividade. Além disso, realizamos uma MLR (regressão múltipla) usando GA (algoritmos genéticos) e descritores moleculares para avaliar a influência de carbonos e substituições na atividade.
RMN 13C; Flavonóides; Mapa auto-organizável de Kohonen
ARTICLE
13C NMR spectral data and molecular descriptors to predict the antioxidant activity of flavonoids
Luciana ScottiIV,* * Correspondence: L. Scotti, Laboratório de Tecnologia Farmacêutica, Universidade Federal da Paraíba, Campus I - Cidade Universitária, 58051-900 - João Pessoa - Paraíba, Brasil. E-mail: luciana.scotti@gmail.com ; Mariane Balerine FernandesI; Eric MuramatsuI; Vicente de Paula EmerecianoII; Josean Fechine TavaresIV; Marcelo Sobral da SilvaIV; Marcus Tullius ScottiIII
IFaculty of Pharmaceutical Sciences, University of São Paulo
IIInstitute of Chemistry, University of São Paulo
IIICenter of Applied Sciences and Education, Federal University of Paraiba
IVLaboratory of Pharmaceutical Technology, LTF, University Federal of Paraíba
ABSTRACT
Tissue damage due to oxidative stress is directly linked to development of many, if not all, human morbidity factors and chronic diseases. In this context, the search for dietary natural occurring molecules with antioxidant activity, such as flavonoids, has become essential. In this study, we investigated a set of 41 flavonoids (23 flavones and 18 flavonols) analyzing their structures and biological antioxidant activity. The experimental data were submitted to a QSAR (quantitative structure-activity relationships) study. NMR 13C data were used to perform a Kohonen self-organizing map study, analyzing the weight that each carbon has in the activity. Additionally, we performed MLR (multilinear regression) using GA (genetic algorithms) and molecular descriptors to analyze the role that specific carbons and substitutions play in the activity.
Uniterms: Flavonoids/antioxidant activity. 13C NMR. Kohonen self-organizing map.
RESUMO
Danos aos tecidos devido ao estresse oxidativo estão diretamente ligados ao desenvolvimento de muitos, senão todos, os fatores de sedentarismo e de doenças crônicas. Neste contexto, a busca de moléculas naturais, que participam da nossa dieta e que possuam atividade antioxidante, flavonóides, torna-se de grande interesse. Neste estudo, nós investigamos um conjunto de 41 flavonóides (23 flavonas e 18 flavonóis), relacionando suas estruturas e atividade antioxidante. Os dados experimentais foram submetidos à análise de QSAR (relações quantitativas estrutura-atividade). Dados de RMN 13C foram utilizados para realizar um estudo do mapa auto-organizável de Kohonen, analisando o peso que cada carbono tem na atividade. Além disso, realizamos uma MLR (regressão múltipla) usando GA (algoritmos genéticos) e descritores moleculares para avaliar a influência de carbonos e substituições na atividade.
Unitermos: RMN 13C. Flavonóides/atividade antioxidante. Mapa auto-organizável de Kohonen.
INTRODUCTION
Flavonoids are phenolic compounds isolated from a wide range of vascular plants, with over 8000 individual compounds known (Halliwell et al., 2000). They possess a wide variety of biological activities including antiinflammatory, antimicrobial, antiallergenic, antiviral, vasodilatory action and antioxidant activity. These compounds are able either to suppress free radical formation and chain initiation or to scavenge free radical and chain propagation (Fernández et al., 2005).
These compounds contain a basic structure constituted of 15 carbon atoms arranged in three rings (C6-C3-C6) and have shown the molecular structural requirements for marked radical scavenging activity against the diphenyl picrylhydrazyl (DPPH) radical. The presence of the galloyl group, as well as the number and position of hydroxyl groups, are important and further enhance the radical scavenging activity of flavonoids. On the other hand, the methoxylation or glycosylation of a free hydroxyl group may reduce greatly or even abolish the radical-scavenging capacity of flavonoids (Yokozawa et al., 1998; Scotti et al., 2007, 2009; Rastija et al., 2009; Pasha et al., 2007). The DPPH system is a stable radical-generating procedure that simulates the biological free radical producing mechanism (equation 1) (Molyneux, 2004).

Quantitative structure-activity relationships (QSAR) of flavonoids are important due to their ease of analysis, since they are natural active substances that show small plane molecules with few functional groups, thus it is sometimes possible to directly find a relationship between the structure and the activity by analyzing the different functional groups attached to the flavonoid molecule. In this sense, the application of graph theory to chemical structures results in several molecular descriptors that are used to better comprehend important molecular aspects.
In addition to the descriptors that characterize the molecular structure, the 13C NMR can also be used to describe the molecular structure. These data yield rich information about the molecular structure and are sufficiently sensitive to detect small differences in the molecule. These differences are measured by the variation of chemical shifts, and the values can be used to associate the chemical structure with the respective biological activity since this association can help understand the influence of the chemical environment on the biological activity selected. Thus, the 13C NMR data may be useful in QSAR studies (Berger, 2006; Masui et al., 2006; Vanderhoeven et al., 2004).
Artificial Neural Networks (ANNs) are one of the most used approaches to achieve computer classification and pattern recognition. ANNs are an option for data set analysis that have unknown correlations, such as the prediction of the biological activity, taking into account the 13C NMR. The most used ANN architecture for pattern recognition is the Kohonen network also known as the Self-Organizing Map (SOM) (Lawrence, 1994; Kohonen, 1989,1997,1998; Kohonen, Oja, 1998). Each neurone in the grid is associated to a weight, and similar patterns stimulate neurones with similar weight, so that similar patterns are mapped near each other, aiding QSAR analysis by simplifying visualisation and understanding (Gasteiger et al., 1996).
The aim of this paper was to predict the antioxidant activity of flavonoids measured by the DPPH system using two methodologies. The first approach is based on the molecular information given by the 13C NMR data, where the ANNs were used to predict the biological activity of the flavonoids, sorting them into active or inactive compounds. In the second approach, several molecular descriptors were computed and utilized to predict the antioxidant activity of flavonoids through genetic algorithms.
METHODOLOGY
Data set
In this study, a data set of 41 flavonoids (23 flavones and 18 flavonols), whose activities were reported in the literature by Yokozawa et al., 1998, was employed. In the paper, antioxidant activity was measured spectrophotometrically by the DPPH method (Table I). The activity was reported as IC50 that indicates the concentration required to inhibit the DPPH radical by 50% (decrease of 50% in absorbance). Since the radical scavenging activity varied by orders of magnitude, as well as its respective errors, it was transformed into natural log values (pIC50 = -logIC50).
In order to design training and prediction sets, the complete data set was processed using cluster analysis. The compounds were first divided into two subsets: one training set composed of 20 molecules, and one external test set composed of 6 compounds (compounds 06, 10, 25, 29, 34, 36) (see Table I), suitable to analyze predictive performance. The size of the test set was around 20% of the whole set, taking care that the test set contains representative samples of the trained group and included the range of activity values of the training group.
The active compounds, with pIC50 values, were selected to generate regression models utilizing genetic algorithms. Models for antiradical activity were constructed based on the training set and then validated internally (cross validation - leave one out) and externally.
Molecular optimisation
All structures were extracted from the SISTEMAT database (Gastmans et al., 1990a,b; Ferreira et al., 2001). An in-house program for data extraction was written in Java and subsequently used to select the flavonoid compounds.
Molecular modeling computations were performed on SPARTAN for Windows v.4.0 software (Wavefunction, Inc., Irvine, Calif.). The molecules were subjected to geometry optimization and conformational analysis (systematic analysis with dihedral angle rotated every 30º). The semi-empirical quantum chemical method used was AM1 (Austin Model1) (Dewar et al., 1985, 1990), which is suitable for this analysis (Bringmann et al., 2000), despite the existence of more recent methods, and the root mean square (RMS) gradient value of 0.001 kcal/mol was used as the termination condition. Energy minimized molecules were saved as MDL MolFiles for computing various molecular descriptors using DRAGON Professional version 5.4 (Dragon, 2010).
Molecular descriptors
DRAGON computer software (Dragon, 2010; Todeschini et al., 2000) was employed to calculate 1664 molecular descriptors that were used for performing Multilinear regression (MLR) analysis. For all descriptors, the constant variables were excluded, just retaining those that presented a different value. For the remaining descriptors, pairwise correlation (r < 0.99) analysis was performed to exclude those which were highly correlated. Thus, the number of DRAGON descriptors used in our calculations was reduced to 548.
Genetic algorithm analysis
The MobyDigs program was used for the calculation of regression models utilizing genetic algorithms (GA). GA is a class of methods based on biological evolution roles. The first step is to create a population of linear regression models. These regression models mate with each other, mutate, cross-over, reproduce, and then evolve through successive generations towards an optimum solution (Leardi, 2001; Kubinyi, 1994a,b).
The search for the best subsets of models is calculated by using Ordinary Least Squares regression (OLS) under the Genetic Algorithm (GA) approach, i.e. by the Variable Subset Selection - Genetic Algorithm (VSS-GA) method. Thus, by examining the regression coefficients, the standard deviations, significances and number of variables in the equation, the significance of the models could be determined.
13C NMR data
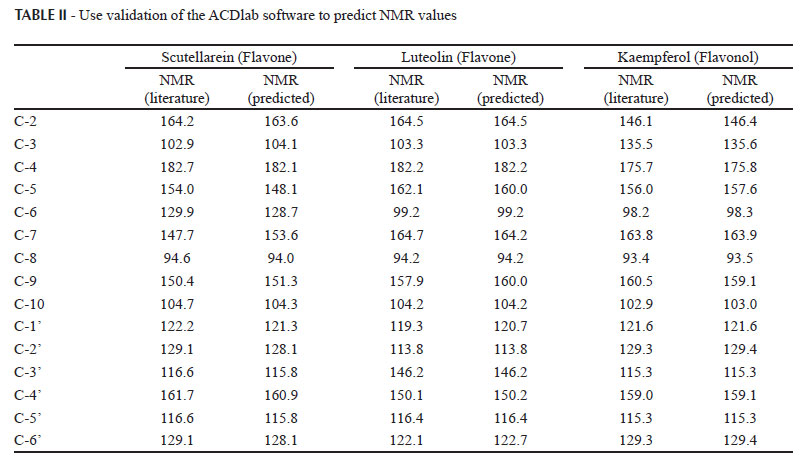
For the analysis correlating the 13C NMR with the flavonoid biological activities, the 13C NMR data of most molecules were obtained from the literature (Agrawal, 1989). For a few structures (compounds 02, 16, 17, 24, 29 and 36), the 13C NMR data were obtained using ACDlabs software (ACD/HNMR,2003). Since flavonoids are plane aromatic compounds, the additivity model can be applied satisfactorily to predict 13C NMR data. To validate the method employed in the 13C NMR data prediction, the chemical shifts of some flavonoids were predicted and compared against the literature data (Table II). The chemical shifts predicted by the software were very close to the literature reference (errors smaller than 3 ppm). Thus, for the six above-mentioned compounds, the 13C NMR data were predicted by the ACD software.
To standardize the 13C NMR analysis, since not all molecules have the glycosil and methyl groups, the 13C NMR data used in the analysis included only the chemical shifts pertaining to the flavonoid skeleton. It is important to note that the even though the glycosil and the methyl carbons, as cited earlier, interfere in the activity only by excluding a free hydroxyl group, glycosylation and methoxylation greatly influence kinetic factors such as absorption, plasma transport and clearance.
Self-organizing maps
The unsupervised training was performed using the SOM Toolbox version 2.0 for Matlab version 6.5 computing environment by MathWorks, Inc (Mathworks, 2010; Vesanto et al., 2010). The toolbox contains functions for creation, visualization and analysis of Self-Organizing Maps. The training was conducted through the Batch-training algorithm. In this algorithm, the whole dataset is presented to the network before any adjustment is made. In each training step, the dataset is partitioned according to the regions of the map weight vectors.
RESULTS AND DISCUSSION
Prediction of antioxidant activity of flavonoids through 13C NMR data
In this first approach, the molecular information yielded by the 13C NMR data was used to predict the antioxidant activity of the flavonoids, sorting them into active or inactive compounds from the ANNs.
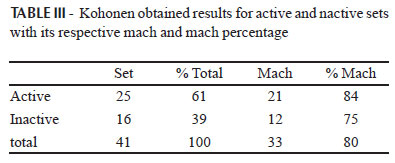
The correlation between the 13C NMR data and the biological activity was performed using SOM. To this end, the compounds were classified as active if they exhibited IC50<200 mM and inactive if they exhibited values of IC50>200 mM (Table I).
The 13C NMR chemical shifts of flavonoids were used as the input data, and the separation between active and inactive compounds was obtained. The results are shown in Table III.
The four non-matching molecules were flavones that shared the substitution pattern C-4' hydroxyl and C-3' with no groups attached. This substitution is characteristic of inactive compounds. Therefore, active compounds that have this odd pattern were classified as inactive by the ANN. For the inactive compounds, half (2) of the non-matching molecules were flavones that have C-4' hydroxyl and C-3' methoxyl.-
Figure1 shows the contribution of each carbon to the map, based on its 13C NMR data. The distribution depicts a satisfactory separation between both groups as a consequence of a significant Kohonen performance.
Prediction of antioxidant activity of flavonoids through molecular descriptors and genetic algorithms
The molecular descriptors were computed for the compounds which were first divided into two subsets: one training set composed of 20 structures, and one external test set composed of 6 compounds, suitable for analyzing the predictive performance.
Regression analysis of the training set leads to Model I which contains RDF105m, RDF110v (Hemmer et al., 1999), RDF (Radial Distribution Function) descriptors, and O-057 (Viswanadhan et al., 1989), an atom-centered fragments descriptor, which were able to explain 88.6% of variance of antioxidant activity.
Analyzing equation 2, the value of the coefficient of internal prediction Qcv2 is highly significant (0.837), which is indicative of a robust model. The linear adjustment is shown in Figure 4 and the low errors in Table IV. The F value is also highly (41.91) significant because for 95% confidence with 3 and 16 degrees of freedom, the necessary minimum F value is 3.24.

The significant linear adjustment for training and test sets is shown in Figure 2, and the small errors in Table IV confirm that the equation was capable of predicting and differentiating the most active compound from the others. Statistically, the validation of the model is shown by analysing the significant values of the parameters r2ext (0.662) and Qext2 (0.657).
Table V shows the correlation matrix between each pair of descriptors and shows low validated values, indicating low co-linearity descriptors since they are not highly correlated.
Radial distribution function, RDFRw, can be interpreted as the probability distribution of finding an atom in a spherical volume of radius ("R") in angstroms (equation 3), weighted by the characteristic properties "w" of the atom, where rij is the interatomic distance between atoms i and j, nAT is the number of atoms in the molecule, and β is a smoothing parameter. Besides information about interatomic distances in the entire molecule, these descriptors provide further information about bond distances, ring types, planar and non-planar systems and atom types.
RDF105m and RDF110v were selected in this study (by equation 2). The former indicates that in a radius of 10.5Å, the greater the atomic masses and number of atoms in this radius, the greater the RDF value (Hemmer et al., 1999). The latter indicates that in a radius of 11.0Å, the greater the atomic van der Walls volumes as for the number of atoms in this radius, the greater the RDF value (Hemmer et al., 1999).

In Model I, RDF105m has a positive contribution while RDF110v has a negative contribution. Thus, in a radius of 10.5Å, the greater the atomic masses, the greater the biological activity value, and in a radius of 11.0Å, the greater the atomic van der Waals volumes, the lower the biological activity value.
Analyzing the molecule structures, separating them into one set possessing the greatest values of biological activity, and another exhibiting the lowest values, and then comparing these compounds by groups that could be responsible for the high or low activity, gives rise to several considerations.
Compounds that showed the highest RDF105m values (compounds 3 and 25) share a substitution pattern: O-glycosylation at C-7 and C-3' hydroxylation. These compounds also have a high biological activity value. Compound 37, which does not have this pattern, has a low RDF105m value and a low biological activity value. As the radial distribution can characterize the solvation layers around a specific atom, this descriptor can be related to the access capability of both water molecules and the radical species involved in the reaction. In this study, this ability can be associated with the presence of the glycosyl group, which shows great hydrophilicity and has hydroxyl groups that participate in the biological activity. Thus, the greater the RDF value, the greater the activity.
RDF105m of molecule 3 is higher than 11 and the biological activity is also higher, which makes clear the influence of a single hydroxyl group at C-3'. Examining the compounds 3 and 7 and their activities and RDF105m, reveals that the absence of a hydroxyl at C-3' could be partially compensated by a hydroxyl at C-6. For compound 8 in comparison to 3, the glycosyl group removal at C-7 decreases both activity and RDF105m value. In contrast, comparing the molecules 27 and 34 evidences that glycosyl removal at C-3 in flavonoids increases both activity and also RDF105m value. Moreover, each kind of sugar has a different effect in activity, as is seen when comparing compound 27 to compounds 30, 31 and 34, where higher activity is obtained with higher RDF105m value.
Molecules with bulky groups, especially at C-7, show the greatest probability of having a high value of RDF110v, and thus a lower value of antioxidant activity. Compound 10 has a high RDF110v and a low biological activity, whereas compound 27, which is a small molecule, has a low RDF110v value and high biological activity. It is noteworthy that solely being a small molecule does not fulfil all the requirements for high activity, since the position of the bulky and hydroxyl groups usually impacts the effectiveness of the molecule against free radicals. For instance, molecules with greater activity and low RDF110v such as compounds 24 and 28 have bulky substituents but are at C-3.
Atom-centred fragments can be understood simply as being the number of specific fragment types in a molecule. For the O-057 molecular descriptor, the increase in the number of phenol, enol or acid groups present in the structure is directly proportional to the O-057 value observed. Molecules with acid groups, such as compounds 6 and 24, have a high O-057 value and will exhibit a high antioxidant activity value. Polyhydroxylated flavonoids such as 2, 4, 5, 8, and 25 also have a high O-057 value, suggesting the positive effect of the hydroxyl group on the radical-scavenging activity.
CONCLUSIONS
Using molecular descriptors to perform a GA analysis, this study was able to show that sterical and fragment features are directly, and electronic features (van der Waals volume) indirectly, involved in the determination of the substitution patterns in the molecule that are responsible for a major effect on activity. For instance, hydroxyl at C-3', C-4' and C-6 are very important for high biological activity, while glycosylation at C-7 frequently decreases the activity.
The importance of these carbons was confirmed in the 13C NMR analysis. Through the chemical shifts of the 13C NMR, the ANN achieved a significant result, sorting the active and inactive molecules with minimal errors (20%). This supports the notion that 13C NMR data can be used to perform classification of flavonoids taking into account the biological activity, making clear that radical scavenging activity is highly correlated with the chemical environment.
A future application could be the elucidation of the role that other carbons (i.e. C-2, C-3, C-5, C-8 and C-1') play on activity. In this context, a more extensive study is needed to compare a greater number of compounds and 13C NMR data. Moreover, it would be useful to study other classes of flavonoids such as flavanones, flavanonols and anthocyanidins to determine which skeleton type is most active and which position in the structure has the greatest contribution to the activity.
REFERENCES
ADVANCED CHEMISTRY DEVELOPMENT Inc. HNMR Predictor, version 7.03. Toronto, 2003. CD-Rom.
AGRAWAL, P.K. Carbon-13 NMR of flavonoids. Amsterdam: Elsevier, 1989. 564 p.
BEGER, R., Computational modeling of biologically active molecules using NMR spectra. 2006. Drug Discov. Today, v.11, n.9-10, p.429-435, 2006.
BRINGMANN, G.; HINRICHS, J.; KRAUS, J.; WUZIK, A.; SCHULZ, T. Nondynamic kinetic resolution of configurationally stable biaryl lactones by reduction with oxazaborolidine-activated borane: AM1 studies and experimental verification. J. Org. Chem., v.65, n.8, p.2517-2527, 2000.
DEWAR, M.J.S.; ZOEBISCH, E.G.; HEALY, E.F.; STEWART, J.J.P. AM1: A new general purpose quantum mechanical molecular model. J. Am. Chem. Soc., v.107, n.13, p.3902-3909, 1985.
DEWAR, M.J.S.; HEALY, E.F.; HOLDER, A.J.; YUAN, Y.C. Comments on a comparison of AM1 with the recently developed PM3 method. J. Comput. Chem., v.11, n.4, p.541-542, 1990.
DRAGON software. Milano, Available at:<http://www.talete.mi.it/products/dragon_description.htm>. Accessed on: 10 jun. 2010.
FERNÁNDEZ, M.; CABALLERO, J.; HELGUERA, A.M. Quantitative structure-activity relationship to predict differential inhibition of aldose reductase by flavonoid compounds. Bioorg. Med. Chem., v.13, n.9, p.3269-3277, 2005.
FERREIRA, M.J.P.; RODRIGUES, G.V.; EMERENCIANO, V.P. MONOREG - An expert system for structural elucidation of monoterpenes. Can. J. Chem., v.79, n.12, p.1915-1925, 2001.
GASTEIGER, J.; SADOWSKI, J.; SCHUUR, J.; SELZER, P.; STEINHAUER, L.; STEINHAUER, V. Chemical Information in 3D Space. J. Chem. Inf. Model., v.36, n.5, p.1030-1037, 1996.
GASTMANS, J.P.; FURLAN, M.; LOPES, M.N.; BORGES, J.H.G.; EMERENCIANO, V.P. A inteligência artificial em química de produtos naturais. O Projeto SISTEMAT - Parte I. Quím. Nova, v.13, n.2, p.10-16, 1990a.
GASTMANS, J.P.; FURLAN, M.; LOPES, M.N.; BORGES, J.H.G.; EMERENCIANO, V.P. A inteligência artificial em química de produtos naturais. O Projeto SISTEMAT - Parte II. Quím. Nova, v.13, n.2, p.75-80, 1990b.
HALLIWELL, B.; GUTTERIDGE, J M.C. Free radicals in biology and medicine. 3.ed. Clarendon: Oxford, 2000. 936 p.
HEMMER, M.C.; STEINHAUER, V.; GASTEIGER, J. Deriving the 3D structure of organic molecules from their infrared spectra. Vibrat. Spect., v.19, n.1, p.151-164, 1999.
KOHONEN, T. Speech recognition based on topology preserving neural maps. In: ALEKSANDER, I. (Ed.) Neural computing architectures. London: Koogan 1989. p.26-40.
KOHONEN, T. Self-Organizing maps. 3.ed. New York: Springer, 2001. 495 p.
KOHONEN, T.; OJA, E. Visual feature analysis by the self-organizing maps. Neural Comput. Appl., v.7, n.3, p.273-286, 1998.
KOHONEN, T. The self-organizing map. NeuroComputing, v.21, n.1-3, p.1-6, 1998.
KUBINYI, H. Variable selection in QSAR studies. I. An evolutionary algorithm. Quant. Struct.-Act. Relat., v.13, n.3, p.285-294, 1994a.
KUBINYI, H.; Variable selection in QSAR studies. II. A highly efficient combination of systematic search and evolution. Quant. Struct.-Act. Relat.,, v.13, n.3, p.393-401, 1994b.
LAWRENCE, J. Introduction to neural networks design, theory and applications. 6.ed. London: Sylvia Luedeking 1994. 348 p.
LEARDI, R. Genetic algorithms in chemometrics and chemistry: a review. J. Chemom., v.15, n.7, p.559-569, 2001.
MASUI, H.; HONG, H. Spec2D: a structure elucidation system based on 1H NMR and H-H COSY spectra in organic chemistry. J. Chem. Inf. Model., v.46, n.2, p.775-787, 2006.
MATHWORKS INC. METLAB and Simulink for technical computing Available at: <http://www.mathworks.com>. Accessed on: 10 jul. 2010.
MOLYNEUX, P. The use of stable free radical diphenylpicrylhydrazyl. (DPPH) for estimating antioxidant activity. Songklanakarin J. Sci. Technol., v.28, n.2, p.211-219, 2004.
PASHA, F.A.; CHO, S.J.; BEG, Y.; TRIPATHI, Y.B. Quantum chemical QSAR study of flavones and their radical-scavenging activity. Med. Chem. Res., v.16, n.7-9, p.408-417,2007.
RASTIJA, V.; MEDIĆ-ŠARIĆ, M. QSAR Study of antioxidant activity of wine polyphenols. Eur. J. Med. Chem., v.44, n.1, p.400-408, 2009.
ROY, A. B.; BASAK, S. C.; HARRISS, D. K.; MAGNUSON, V. R. Neighborhood Complexities and Symmetry of Chemical Graphs and Their Biological Applications. In; AVULA, X.J.R.; KALMAN, R.E.; LIPAIS, A.I.; RODIN, E.Y. (Eds.). Mathematical modelling in science and technology. Oxford: Pergamon Press, 1984. p.745-750.
SCOTTI, L.; SCOTTI, M.T.; CARDOSO, C.; PAULETTI, P.; CASTRO-GAMBOA, I.; BOLZANI, V.S.; VELASCO, M.V.R.; MENEZES, C.S.M.; FERREIRA, E.I. Modelagem molecular aplicada ao desenvolvimento de moléculas com atividade antioxidante visando o uso cosmético. Braz. J. Pharm. Sci., v.43, n.2, p.153-167, 2007.
SCOTTI, L.; SCOTTI, M.T.; PASQUALOTO, K.F.M.; BOLZANI, V.S. Molecular physicochemical parameters predicting antioxidant activity of Brazilian natural products. Braz. J. Pharmacog., v.19, n.4, p.908-913, 2009.
TODESCHINI, R.; CONSONNI, V. Handbook of molecular descriptors. Weinheim: Wiley-VCH, 2000. 667 p.
VANDERHOEVEN, S.J.; TROKE, J.; TRANTER, G.E.; WILSON, I.D.; NICHOLSON, J.K.; LINDON, J.C. Nuclear magnetic resonance (NMR) and quantitative structure-activity relationship (QSAR) studies on the transacylation reactivity of model 1β-O-acyl glucuronides. II: QSAR modelling of the reaction using both computational and experimental NMR parameters. Xenobiotica, v.34, n.10, p.889-900, 2004.
VESANTO, J.; HIMBERG, J.; ALHONIEMI, E.; PARHANKANGAS, J. SOM Toolbox for Matlab 5. Available at: <http://www.cis.hut.fi/projects/somtoolbox>. Accessed on: 10 jul. 2010.
VISWANADHAN, V.N.; GHOSE, A.K.; REVANKAR, G.R.; ROBINS, R.K. Atomic physicochemical parameters for three dimensional structure directed quantitative structure-activity relationships. 4. Additional parameters for hydrophobic and dispersive interactions and their application for an automated superposition of certain naturally occurring nucleoside antibiotics. J. Chem. Inf. Model., v.29, n.3, p.163-172, 1989.
YOKOZAWA, T.; CHEN, C.P.; DONG, E.; TANAKA, T.; NONAKA, G.-I.; NISHIOKA, I., Study on the inhibitory effect of tannins and flavonoids against the 1,1-Diphenyl-2-picrylhydrazyl radical - A newly found effect of hydrolysable-type tannin-containing crude drug and gallotannin Biochem. Pharmacol., v.56, n.2, p.213-222, 1998.
Received for publication on 23rd October 2010
Accepted for publication on 20th January 2011
- ADVANCED CHEMISTRY DEVELOPMENT Inc. HNMR Predictor, version 7.03. Toronto, 2003. CD-Rom.
- AGRAWAL, P.K. Carbon-13 NMR of flavonoids Amsterdam: Elsevier, 1989. 564 p.
- BEGER, R., Computational modeling of biologically active molecules using NMR spectra. 2006. Drug Discov. Today, v.11, n.9-10, p.429-435, 2006.
- BRINGMANN, G.; HINRICHS, J.; KRAUS, J.; WUZIK, A.; SCHULZ, T. Nondynamic kinetic resolution of configurationally stable biaryl lactones by reduction with oxazaborolidine-activated borane: AM1 studies and experimental verification. J. Org. Chem., v.65, n.8, p.2517-2527, 2000.
- DEWAR, M.J.S.; ZOEBISCH, E.G.; HEALY, E.F.; STEWART, J.J.P. AM1: A new general purpose quantum mechanical molecular model. J. Am. Chem. Soc., v.107, n.13, p.3902-3909, 1985.
- DEWAR, M.J.S.; HEALY, E.F.; HOLDER, A.J.; YUAN, Y.C. Comments on a comparison of AM1 with the recently developed PM3 method. J. Comput. Chem., v.11, n.4, p.541-542, 1990.
- DRAGON software. Milano, Available at:<http://www.talete.mi.it/products/dragon_description.htm>. Accessed on: 10 jun. 2010.
- FERNÁNDEZ, M.; CABALLERO, J.; HELGUERA, A.M. Quantitative structure-activity relationship to predict differential inhibition of aldose reductase by flavonoid compounds. Bioorg. Med. Chem., v.13, n.9, p.3269-3277, 2005.
- FERREIRA, M.J.P.; RODRIGUES, G.V.; EMERENCIANO, V.P. MONOREG - An expert system for structural elucidation of monoterpenes. Can. J. Chem., v.79, n.12, p.1915-1925, 2001.
- GASTEIGER, J.; SADOWSKI, J.; SCHUUR, J.; SELZER, P.; STEINHAUER, L.; STEINHAUER, V. Chemical Information in 3D Space. J. Chem. Inf. Model., v.36, n.5, p.1030-1037, 1996.
- GASTMANS, J.P.; FURLAN, M.; LOPES, M.N.; BORGES, J.H.G.; EMERENCIANO, V.P. A inteligência artificial em química de produtos naturais. O Projeto SISTEMAT - Parte I. Quím. Nova, v.13, n.2, p.10-16, 1990a.
- GASTMANS, J.P.; FURLAN, M.; LOPES, M.N.; BORGES, J.H.G.; EMERENCIANO, V.P. A inteligência artificial em química de produtos naturais. O Projeto SISTEMAT - Parte II. Quím. Nova, v.13, n.2, p.75-80, 1990b.
- HALLIWELL, B.; GUTTERIDGE, J M.C. Free radicals in biology and medicine. 3.ed. Clarendon: Oxford, 2000. 936 p.
- HEMMER, M.C.; STEINHAUER, V.; GASTEIGER, J. Deriving the 3D structure of organic molecules from their infrared spectra. Vibrat. Spect, v.19, n.1, p.151-164, 1999.
- KOHONEN, T. Speech recognition based on topology preserving neural maps. In: ALEKSANDER, I. (Ed.) Neural computing architectures London: Koogan 1989. p.26-40.
- KOHONEN, T. Self-Organizing maps 3.ed. New York: Springer, 2001. 495 p.
- KOHONEN, T.; OJA, E. Visual feature analysis by the self-organizing maps. Neural Comput. Appl., v.7, n.3, p.273-286, 1998.
- KOHONEN, T. The self-organizing map. NeuroComputing, v.21, n.1-3, p.1-6, 1998.
- KUBINYI, H. Variable selection in QSAR studies. I. An evolutionary algorithm. Quant. Struct.-Act. Relat., v.13, n.3, p.285-294, 1994a.
- KUBINYI, H.; Variable selection in QSAR studies. II. A highly efficient combination of systematic search and evolution. Quant. Struct.-Act. Relat.,, v.13, n.3, p.393-401, 1994b.
- LAWRENCE, J. Introduction to neural networks design, theory and applications. 6.ed. London: Sylvia Luedeking 1994. 348 p.
- LEARDI, R. Genetic algorithms in chemometrics and chemistry: a review. J. Chemom., v.15, n.7, p.559-569, 2001.
- MASUI, H.; HONG, H. Spec2D: a structure elucidation system based on 1H NMR and H-H COSY spectra in organic chemistry. J. Chem. Inf. Model., v.46, n.2, p.775-787, 2006.
- MATHWORKS INC. METLAB and Simulink for technical computing Available at: <http://www.mathworks.com>. Accessed on: 10 jul. 2010.
- MOLYNEUX, P. The use of stable free radical diphenylpicrylhydrazyl. (DPPH) for estimating antioxidant activity. Songklanakarin J. Sci. Technol., v.28, n.2, p.211-219, 2004.
- PASHA, F.A.; CHO, S.J.; BEG, Y.; TRIPATHI, Y.B. Quantum chemical QSAR study of flavones and their radical-scavenging activity. Med. Chem. Res., v.16, n.7-9, p.408-417,2007.
- ROY, A. B.; BASAK, S. C.; HARRISS, D. K.; MAGNUSON, V. R. Neighborhood Complexities and Symmetry of Chemical Graphs and Their Biological Applications. In; AVULA, X.J.R.; KALMAN, R.E.; LIPAIS, A.I.; RODIN, E.Y. (Eds.). Mathematical modelling in science and technology Oxford: Pergamon Press, 1984. p.745-750.
- SCOTTI, L.; SCOTTI, M.T.; CARDOSO, C.; PAULETTI, P.; CASTRO-GAMBOA, I.; BOLZANI, V.S.; VELASCO, M.V.R.; MENEZES, C.S.M.; FERREIRA, E.I. Modelagem molecular aplicada ao desenvolvimento de moléculas com atividade antioxidante visando o uso cosmético. Braz. J. Pharm. Sci., v.43, n.2, p.153-167, 2007.
- SCOTTI, L.; SCOTTI, M.T.; PASQUALOTO, K.F.M.; BOLZANI, V.S. Molecular physicochemical parameters predicting antioxidant activity of Brazilian natural products. Braz. J. Pharmacog., v.19, n.4, p.908-913, 2009.
- TODESCHINI, R.; CONSONNI, V. Handbook of molecular descriptors. Weinheim: Wiley-VCH, 2000. 667 p.
- VANDERHOEVEN, S.J.; TROKE, J.; TRANTER, G.E.; WILSON, I.D.; NICHOLSON, J.K.; LINDON, J.C. Nuclear magnetic resonance (NMR) and quantitative structure-activity relationship (QSAR) studies on the transacylation reactivity of model 1β-O-acyl glucuronides. II: QSAR modelling of the reaction using both computational and experimental NMR parameters. Xenobiotica, v.34, n.10, p.889-900, 2004.
- VESANTO, J.; HIMBERG, J.; ALHONIEMI, E.; PARHANKANGAS, J. SOM Toolbox for Matlab 5. Available at: <http://www.cis.hut.fi/projects/somtoolbox>. Accessed on: 10 jul. 2010.
- VISWANADHAN, V.N.; GHOSE, A.K.; REVANKAR, G.R.; ROBINS, R.K. Atomic physicochemical parameters for three dimensional structure directed quantitative structure-activity relationships. 4. Additional parameters for hydrophobic and dispersive interactions and their application for an automated superposition of certain naturally occurring nucleoside antibiotics. J. Chem. Inf. Model., v.29, n.3, p.163-172, 1989.
- YOKOZAWA, T.; CHEN, C.P.; DONG, E.; TANAKA, T.; NONAKA, G.-I.; NISHIOKA, I., Study on the inhibitory effect of tannins and flavonoids against the 1,1-Diphenyl-2-picrylhydrazyl radical - A newly found effect of hydrolysable-type tannin-containing crude drug and gallotannin Biochem. Pharmacol., v.56, n.2, p.213-222, 1998.
Publication Dates
-
Publication in this collection
05 Aug 2011 -
Date of issue
June 2011
History
-
Accepted
20 Jan 2011 -
Received
23 Oct 2010