Abstracts
P-glycoprotein (P-gp), a transmembrane permeability glycoprotein, is a member of ATP binding cassette (ABC) super family that functions specifically as a carrier mediated primary active efflux transporter. It is widely distributed throughout the body and has a diverse range of substrates. Several vital therapeutic agents are substrates to P-gp and their bioavailability is lowered or a resistance is induced because of the protein efflux. Hence P-gp inhibitors were explored for overcoming multidrug resistance and poor bioavailability problems of the therapeutic P-gp substrates. The sensitivity of drug moieties to P-gp and vice versa can be established by various experimental models in silico, in vitro and in vivo. Ever since the discovery of P-gp, the research plethora identified several chemical structures as P-gp inhibitors. The aim of this review was to emphasize on the discovery and development of newer, inert, non-toxic, and more efficient, specifically targeting P-gp inhibitors, like those among the natural herb extracts, pharmaceutical excipients and formulations, and other rational drug moieties. The applications of cellular and molecular biology knowledge, in silico designed structural databases, molecular modeling studies and quantitative structure-activity relationship (QSAR) analyses in the development of novel rational P-gp inhibitors have also been mentioned.
P-glycoprotein; Multidrug resistence; Cluster of differentiation 243; Sphingolipids; Competitive inhibitors
Glicoproteína-p (P-gp), uma glicoproteína de transmembrana permeável, é um membro da superfamília (ABC) de cassete de gene de ligação de ATP que funciona especificamente como um carreador mediado pelo transportador de efluxo ativo primário. É amplamente distribuído por todo o corpo e apresenta uma gama diversificada de substratos. Diversos agentes terapêuticos vitais são substratos para P-gp e sua biodisponibilidade é reduzida ou a resistência é induzida devido ao efluxo de proteínas. Portanto, os inibidores da P-gp foram explorados para a superação da resistência a múltiplas drogas e problemas de biodisponibilidade deficiente dos substratos terapêuticos da P-gp. A sensibilidade das moléculas da droga à P-gp e vice-versa, pode ser estabelecida por vários modelos experimentais in silico, in vitro e in vivo. Desde a descoberta da P-gp, diversas pesquisas identificaram várias estruturas químicas como inibidores da P-gp. O objetivo deste presente estudo foi o de enfatizar a descoberta e desenvolvimento de inibidores mais novos, inertes, atóxicos e mais eficazes, visando especificamente os da P-gp, como aqueles entre os extratos vegetais, excipientes e formulações farmacêuticas, e outras moléculas racionais de droga. As aplicações do conhecimento de biologia celular e molecular, bancos de dados estruturais in silico, estudos de modelagem molecular e análises da relação quantitativa estrutura-atividade (QSAR) no desenvolvimento de novos inibidores racionais da P-gp também foram mencionados.
Glicoproteína-p; Resistência a múltiplas drogas; Cluster de diferenciação 243; Esfingolipídeos; Inibidores competitivos
REVIEW
Overview of P-glycoprotein inhibitors: a rational outlook
Kale Mohana Raghava Srivalli; P. K. Lakshmi
Department of Pharmaceutics, G. Pulla Reddy College of Pharmacy, Osmania University, India
Correspondence Correspondence: Lakshmi, P. K. Department of Pharmaceutics, G. Pulla Reddy College of Pharmacy, Osmania University Hyderabad 500028 - Andhra Pradesh, India E-mail: drlakshmisuresh@gmail.com
ABSTRACT
P-glycoprotein (P-gp), a transmembrane permeability glycoprotein, is a member of ATP binding cassette (ABC) super family that functions specifically as a carrier mediated primary active efflux transporter. It is widely distributed throughout the body and has a diverse range of substrates. Several vital therapeutic agents are substrates to P-gp and their bioavailability is lowered or a resistance is induced because of the protein efflux. Hence P-gp inhibitors were explored for overcoming multidrug resistance and poor bioavailability problems of the therapeutic P-gp substrates. The sensitivity of drug moieties to P-gp and vice versa can be established by various experimental models in silico, in vitro and in vivo. Ever since the discovery of P-gp, the research plethora identified several chemical structures as P-gp inhibitors. The aim of this review was to emphasize on the discovery and development of newer, inert, non-toxic, and more efficient, specifically targeting P-gp inhibitors, like those among the natural herb extracts, pharmaceutical excipients and formulations, and other rational drug moieties. The applications of cellular and molecular biology knowledge, in silico designed structural databases, molecular modeling studies and quantitative structure-activity relationship (QSAR) analyses in the development of novel rational P-gp inhibitors have also been mentioned.
Uniterms: P-glycoprotein/inhibitors. Multidrug resistence. Cluster of differentiation 243. Sphingolipids. Competitive inhibitors.
RESUMO
Glicoproteína-p (P-gp), uma glicoproteína de transmembrana permeável, é um membro da superfamília (ABC) de cassete de gene de ligação de ATP que funciona especificamente como um carreador mediado pelo transportador de efluxo ativo primário. É amplamente distribuído por todo o corpo e apresenta uma gama diversificada de substratos. Diversos agentes terapêuticos vitais são substratos para P-gp e sua biodisponibilidade é reduzida ou a resistência é induzida devido ao efluxo de proteínas. Portanto, os inibidores da P-gp foram explorados para a superação da resistência a múltiplas drogas e problemas de biodisponibilidade deficiente dos substratos terapêuticos da P-gp. A sensibilidade das moléculas da droga à P-gp e vice-versa, pode ser estabelecida por vários modelos experimentais in silico, in vitro e in vivo. Desde a descoberta da P-gp, diversas pesquisas identificaram várias estruturas químicas como inibidores da P-gp. O objetivo deste presente estudo foi o de enfatizar a descoberta e desenvolvimento de inibidores mais novos, inertes, atóxicos e mais eficazes, visando especificamente os da P-gp, como aqueles entre os extratos vegetais, excipientes e formulações farmacêuticas, e outras moléculas racionais de droga. As aplicações do conhecimento de biologia celular e molecular, bancos de dados estruturais in silico, estudos de modelagem molecular e análises da relação quantitativa estrutura-atividade (QSAR) no desenvolvimento de novos inibidores racionais da P-gp também foram mencionados.
Unitermos: Glicoproteína-p. Resistência a múltiplas drogas. Cluster de diferenciação 243. Esfingolipídeos. Inibidores competitivos.
INTRODUCTION
P-glycoprotein (P-gp), the permeability glycoprotein or plasma glycoprotein is an active, efflux, membrane bound transport protein pump discovered in 1976 (Juliano, Ling, 1976). P-gp is a member of ATP binding cassette (ABC) super family, multidrug resistance (MDR)/transporter associated with antigen processing (TAP), sub-family B, and member 1, abbreviated as ABCB1. It is also called as MDR1 and PGY1. It has been recently designated as CD243 (cluster of differentiation 243). Its efflux mechanism involves the protein binding to the ATP and requires energy derived by the hydrolysis of ATP to ADP in the presence of adenosine-triphosphatase enzyme (ATPase) (Shekfeh, 2009). P-gp, which was first identified in cancer cells, is encoded by MDR1/ABCB1 gene in humans. The gene shows an exclusive over expression in cancer cells. The correlation between the up regulation of MDR1 gene mRNA transcription and the over expression of the P-gp transport system, leading to multidrug resistance (MDR) phenotype, during the drug therapy for cancer and several microbial infections has been well established. Repetitive treatment with P-gp substrates may also enhance the P-gp expression (Krishna, Mayer, 2000). A further support to the correlation between the P-gp over expression and development of MDR is given by the role of P-gp in the metabolism of endogenous sphingolipids. The sphingolipids and their metabolites were reported to confer MDR in concert with the efflux transporters (Dijkhuis et al., 2003). P-gp was found to be present on the surface of biliary canalicular hepatocytes, luminal surface of columnar epithelial cells of the lower gastrointestinal tract (GIT) including liver, pancreas, small and large intestines, jejunum and colon, apical surface of proximal convoluted tubular cells of kidney, capillary endothelial cells of blood-brain barrier (BBB), apical membrane of the placental fetal-membrane barrier function and in various other tissues like lungs, heart, adrenals, prostrate, skin, spleen and skeletal muscle (Thiebut et al., 1987). On account of its distribution, P-gp can be viewed as a unique defensive barrier network against the entry of xenobiotics into the body. This efflux carrier decreases the bioavailability of administered drugs by preventing their sufficient accumulation intracellularly. Ultimately, the efficacy of drugs is lowered. It also alters the pharmacokinetics and pharmacodynamics of its substrates by dictating their ADMET (absorption, distribution, metabolism, elimination and toxicity) characteristics (Lin, 2003). The optimal P-gp expression is always appreciated for its protective function. But, P-gp over expression leads to MDR while its low expression leads to toxic reactions.
This review presents a brief note on the mechanism and kinetics of P-gp efflux and discusses about various P-gp inhibitors identified under different categories of chemical, natural, pharmaceutical and biochemical classes. Different possible strategies that can be developed to circumvent the protein action were mentioned. Further, the interaction between the P-gp substrates/inhibitors/inducers and P-gp has been well differentiated and elucidated.
P-GLYCOPROTEIN SUBSTRATES
P-gp transporter has a diverse array of substrates which vary not only in size and structure but also in several chemical properties. Since the primary criterion subjecting a substrate to P-gp efflux is its interaction with the bilayer lipid membrane, a wide range of cationic, lipophilic and planar drugs become the protein substrates inspite of their structural dissimilarity. This explains the vast spread structural specificity or in the practical sense, the non-specific nature of P-gp (Higgins, Gottesman, 1992). However, there is one unifying structural feature that is commonly shared among all the substrates of P-gp, and that is they all possess spatially separated hydrophilic and hydrophobic moieties. The P-gp substrates reconstitute most of the clinically efficient agents. Anticancer drugs, various pharmacotherapeutic agents that act on central nervous system, cardio vascular system and antimicrobials are substrates to this efflux protein (Hunter, Hirst, 1997; Schinkel, 1999).
MECHANISM AND KINETICS OF P-GLYCOPROTEIN EFFLUX
The efflux action of the protein follows a carrier mediated primary active transport mechanism. In this process, the protein pump export needs direct ATP requirement and the energy released from the ATP hydrolysis gives the driving force for extrusion process. The efflux takes place unidirectionally (out of the cells into the extracellular space) and transfers only one molecule at a time. Thus, P-gp is a uniporter carrier protein.
Figure 1 explains the mechanism of action of the competitive and non-competitive (non-transported) inhibitors apart from the P-gp efflux kinetics. While a P-gp substrate binds to protein's transport site and gets translocated by the protein, competitive inhibitors compete with the substrate drugs for extrusion and occupy all the available protein transport sites leaving no space for the P-gp and substrate interaction.
Non-competitive inhibitors neither bind to protein's transport site nor are translocated by the protein efflux and hence are as well called as non-transported inhibitors. They non-competitively inhibit the protein efflux by binding to an allosteric modulatory site.
Since the number of protein carriers is limited, the transport system is capacity limited. The efflux kinetics is described by the equation (1), as mixed-order kinetics or Michaelis Menten kinetics or saturation or non-linear dose dependent kinetics (Jang, Wientjes, Jessie, 2001; Varma et al., 2003).

Where,
Jpgp = P-gp efflux
Vmax = maximum velocity of P-gp efflux per unit surface area
C = substrate concentration
Km = affinity constant or michaelis menten constant (dissociation constant for P-gp mediated efflux).
At low drug concentrations, where Km >> C,
The efflux follows first order kinetics. The efflux rate is proportional to the drug concentration and increases linearly with the drug concentration.
At high drug concentrations, where Km << C,
The protein carriers get saturated and the efflux occurs at constant rate. In other words, the efflux rate process approaches an asymptote and becomes independent of drug concentration. The efflux follows zero order kinetics in this case.
In cases where Km = C, the efflux rate is half its maximum velocity and assumes mixed order (exhibiting zero and first order kinetics together) kinetics.
Characterization of the interaction of certain compounds with P-Glycoprotein
It is the nature of interaction of a compound with the protein that identifies and establishes it as a P-gp inhibitor or substrate or inducer. From the Venn diagram (Figure 2) describing the interaction of different compounds with P-gp as inhibitors/substrates/inducers, it is evident that such a classification is not very straight forward. Though an effort has been made in this review to provide a vivid profile picture of those P-gp interacting compounds differentiated in the Venn diagram, the characterization of rest other modulators still remains ambiguous. Compounds like progesterone, gomisin A, valspodar, elacridar were identified as pure inhibitors since they are not P-gp substrates. They neither bind to protein's transport site nor are translocated by the protein efflux. They non-competitively inhibit the protein efflux by binding to an allosteric modulatory site (Haslam et al., 2008). Cis-flupenthixol, a typical/classical antipsychotic is another P-gp reverser identified as a non-transported inhibitor, binding to protein's allosteric binding site (Maki, Hafkemeyer, Dey, 2003). However, there are "n" numbers of P-gp inhibitors which are its substrates too. They are all competitive inhibitors as they compete with the substrate drugs for extrusion and thus favour intracellular accumulation of those substrates. Propafenone and its major metabolites 5-hydroxy propafenone and N-desalkyl propafenone are all P-gp inhibitors but propafenone and N-desalkyl propafenone are not P-gp substrates while 5-hydroxy propafenone is translocated across the cell membrane by human P-gp. Thus, 5-hydroxy propafenone can be distinguished as a competitive P-gp inhibitor while the other two compounds act non-competitively (Bachmakov et al., 2005). Quinidine and amprenavir exhibit an uncertain combination of three distinct interactions with P-gp. They act as P-gp substrates, inhibitors and also as P-gp inducers suggesting an indiscriminate characterization. Artemisinin, on the other hand, is a non-transported P-gp inducer. It is a non-substrate to P-gp and is more or less alike non-competitive inhibitor in one point that it exerts its action without being subject to P-gp efflux. The most conflicting categorization is that of piperine and apocyanin. Andrographolide, berberin, glycyrrhizin and magniferin are the other natural constituents, which are also classified under this category. The explanation for their characterization as both inducers and inhibitors is given by their "biphasic protein modulation" by which they stimulate the protein efflux at lower concentrations and inhibit the same at their higher concentrations (Najar et al., 2010). Rifampicin is classified as an inducing substrate of P-gp as it is transported by the protein and as well has an up regulating effect on P-gp expression. Digoxin, bromocriptine and cimetidine are mere P-gp substrates with neither an inhibitory nor an inductive effect on protein function (Vautier et al., 2006).
Classification of P-Glycoprotein inhibitors: the past & the present perspective
The candidates which block or bypass the P-gp efflux are called as P-gp inhibitors/P-gp modulators/chemosensitizers/reversal agents. Co-/concurrent administration of the P-gp substrate-therapeutics with these P-gp inhibitors can prevent/overcome the substrate expulsion by P-gp and render the intended therapeutic benefits of the substrate drugs. Though several P-gp inhibitors were earlier identified among the available drugs, their toxicity and drug interaction profiles drove the researchers to discover more rational inhibitors. As of now, the inhibitors were identified among various natural products, pharmaceutical inert excipients and formulations. Prodrug strategy is also being applied to escape P-gp efflux. A few novel antitumor drugs, synthetic peptides and P-gp expression suppressors are under development to circumvent the protein action. The cellular and molecular biologists are now focusing on designing inhibitors based on the information extracted about the protein's vital structural organization. Certain unique developmental strategies which on implementation could hopefully achieve rational prototype molecules have also been discussed in this review. Table I enumerates the possible rational approaches to strategically develop P-gp inhibitors (Balayssaca et al., 2005; Bansal et al., 2009a; Yuan et al., 2008; Bansal et al., 2009b).
Small molecule inhibitors (SMIs)
The first, second and third generation inhibitors, developed based on screening among the available compounds, parent molecule optimizations and chemical syntheses by combinational chemistry approaches, respectively, are together called as SMIs. All these compounds, though grouped under the same heading, are structurally unrelated and do not share any properties in common, except that they are P-gp inhibitors.
First generation inhibitors
This class of inhibitors embodies those pharmacological agents which were primarily developed for other indications but later observed to be P-gp substrates cum inhibitors. Verapamil, an antihypertensive calcium channel blocker, trifluoperazine, a calmodulin antagonist, cyclosporine, an immunosuppressant, other antihypertensives such as quinidine and reserpine, yohimbine, antiestrogenic tamoxifen and toremifene, and antineoplastic vincristine, all fall under this category. Since most of these compounds were P-gp substrates themselves, they interacted with the protein, competed with the other substrates and acted as competitive inhibitors. Since all the first generation inhibitors have been identified this way, they obviously were non-selective and less potent. Their P-gp inhibitory concentrations reached high toxic levels due to which many of these inhibitors failed in clinical trials (Dantzig, Alwis, Burgess, 2003).
Second generation inhibitors
The first generation inhibitors were modified structurally viz. their chirality was altered to achieve a better or a null innate pharmacological profile so as to largely reduce the toxicity of the parent compounds. Dexverapamil, the R-isomer of verapamil without any cardiac activity, PSC 833 (valspodar), a cyclosporine A analogue lacking immunosuppressive character, MS-209 and several other first generation drug derivatives or analogues fall under this category. These resultant modulators still remained P-gp substrates themselves and showed low protein affinity. As such, their P-gp inhibitory dose was far beyond the tolerable dose levels. Due to the chiral optimization, these second generation chemosensitizers ended up as inevitable cytochrome P450 3A4 (CYP450 3A4) substrates for metabolism, which made them compete with the concurrently administered anticancer P-gp substrate drugs whose metabolism was also affected by the same system. This caused significant pharmacokinetic alterations that unpredictably affected the metabolic and clearance mechanisms of the substrate drugs which in turn brought about difficulties in adjusting the chemotherapy doses in patients. All these problems left this class of inhibitors deserted (Thomas, Coley, 2003; Darby, Callaghan, McMahon, 2011).
Third generation inhibitors
The quantitative structure-activity relationship (QSAR) application to high throughput screening techniques (HTS) and combinational chemistry methods gave 10-fold more potent compounds when compared to the first and second generations. Thus evolved third generation inhibitors were highly specific, lacked interactions with CYP450 3A4 system and required no alterations in the chemotherapy doses. XR 9576 (tariquidar, an anthranilamide derivative), a non-transported P-gp inhibitor, developed in this pace was stated to inhibit ATPase by interacting with a distinct modulatory binding site on the protein. It was believed to be the most promising but still suspended due to unfavorable toxicity reports in phase III trials in lung carcinoma cases. Other compounds discovered by this strategy include VX-710 (biricodar, a cyclopropyldibenzosuberane modulator, developed by Eli Lilly Inc.), GF 120918 (elacridar, an acridonecarboxamide derivative, developed by GlaxoSmithKline), OC 144-093, mitotane (NSC-38721), annamycin, R101933, ONT-093 and LY335979 (zosuquidar) (Ozben, 2006).
Upcoming P-glycoprotein inhibitors among the natural product extracts
Discoveries in this natural resources area are gaining increasing interest since they are safe and non-toxic. The grape fruit interactions with several drugs gave the first evidence of herbal applications in P-gp inhibition (Bailey et al., 1991). Several herbal constituents, as listed in Table II (Bansal et al., 2009b; Yuan et al., 2008) were identified as potent P-gp inhibitors. Though the studies already proved most of them effective in vitro, it takes still long time to evaluate their suitability for clinical purposes.
Apart from those tabulated inhibitors, ginsenoside Rg3, a red ginseng saponin was reported as a competitive P-gp inhibitor (Kim et al., 2003). Antineoplastic lamellarin D, a novel pro-apoptotic alkaloid agent of marine origin exhibited insensitivity to P-gp mediated drug efflux (Vanhuyse et al., 2005). Gomisin A, a dibenzocyclooctadiene compound isolated from Schisandra chinensis, showed an evidence of altering P-gp substrate interaction non-competitively and thereby reversing MDR. It is not a P-gp substrate by itself and can bind simultaneously to both P-gp and substrate. It is also known to inhibit the basal P-gp associated ATPase activity (Wan et al., 2006). CBT-1 is another novel bisbenzylisoquinoline plant alkaloid in development as a P-gp inhibitor (Robey et al., 2008). The epoxide moiety of laulimalide, a macrolide obtained from Hyatella species is a microtubule stabilizing agent which has been established as a P-gp inhibitor as well. Its antitumor activity was found to be a 100-fold more potent than that of taxol in MDR cell lines (Corley et al., 1988).
Pharmaceutical excipients
An ideal P-gp inhibitor is the one that is non-toxic with no pharmacological action of its own. Several pharmaceutical inert additives and functional excipients were investigated to study their P-gp inhibitory activity and to evaluate their role in enhancing the drug permeability across the lipid membrane. P-gp activity is modulated by the physical state of lipid bilayer where the protein actually resides. Several pharmaceutical agents of natural or synthetic origin that belong to various categories like the cosolvents, surfactants, polymer and lipid excipients were identified to have the P-gp inhibitory action (Buggins, Dickinson, Taylor, 2007). The mechanism of action differs with the type of excipient as presented in Table III (Bansal et al., 2009a). Most of these components increase the P-gp substrates absorption transport by inhibiting their secretion directed transport. They inhibit P-gp efflux by acting on the lipid membrane and exhibit fewer side effects. Surfactants and polymers solubilize and stabilize the drug molecule. They act indirectly and non-specifically by interacting with the lipid bilayer. Some surfactants can further decrease the P-gp ATPase activity. A few act simultaneously by both the mechanisms. Pluronic block copolymers and other amphiphilic diblock copolymers modulate P-gp activity by inducing the membrane permeability changes through reduction in membrane microviscosity and also by depletion of cellular ATP levels. Vesicular transport of substrates by micellar structures, modulate P-gp action mostly at concentrations greater than the critical micelle concentrations (Yuan et al., 2008).
Pharmaceutical formulations
Excipients are safe, not absorbed from the intestine or gut and have wide pharmaceutical acceptance with a fair history of being incorporated into the parenteral and external formulations as solubilizing and stabilizing agents (Buggins, Dickinson, Taylor, 2007). Besides, in the present scenario, there is increasing development of novel drug delivery systems (DDS) like microspheres, nanoparticles and liposomes, all of which have inherent P-gp evading activity (Kim, Lim, 2002). The stealth particles (stealth liposomes) are known to saturate the P-gp carrier, reverse the P-gp efflux and thus deliver concentrated drug levels across the plasma membrane (Krishna, Mayer, 2000). Polymeric conjugates and mixed micelles can bypass the P-gp efflux since they are transported into the cells via receptor mediated endocytosis in contrast to the typical free drug diffusion. The degradation products of polymers or carriers may also block P-gp by direct interaction and inhibition (Dabholkar et al., 2006; Kobayashi et al., 2007). Surfactant polymer nanoparticles were reported to overcome P-gp mediated efflux by undergoing endocytic vesicular transport (Chavanpatil et al., 2007). As such, a combination of both the approaches where P-gp inhibiting excipients as well as the novel DDS are applied together, may serve as more potent P-gp inhibitors. Systems in which the therapeutic agents and sensitizers can be incorporated into a single carrier for simultaneous delivery to the cells can be explored to further enhance the efficacy of chemotherapy (Dharmala, Yoo, Lee, 2008; Wong et al., 2004).
Formulation strategies can be:
1. Encapsulating either the chemosensitizer or the drug while the other is freely delivered.
2. Co-encapsulation of the both.
Novel antineoplastic drugs
Novel 7-benzyl-4-methyl-5-[(2-substituted phenyl) ethyl] -7H-pyrrolo [2, 3-d] -pyrimidine-2-amine series showed remarkable P-gp mediated MDR reversal potential by binding to a unique site on tubulin which was distinct from other antineoplastic drug binding sites (Gangjee et al., 2007). KP772 (FFC24), a new anticancer lanthanum compound was reported to block P-gp expression especially in MDR cancerous cells (Heffeter et al., 2007). A selective tyrosine kinase (an epidermal growth factor receptor) inhibitor, gefitinab used for treating lung cancer interacted directly and inhibited P-gp function (Kitazaki et al., 2005). In vitro studies showed the selective modulation of MDR protein-ATPase activity by several 4-anilinoquinazoline-derived tyrosine kinase inhibitors (TKIs), effective at their submicromolar concentrations. Such TKIs include canertinib, EKI- 485, erlotinib, lapatinib, tyrphostin AG1478, and a phenylamino-pyrimidine derivative, imatinib. Another phenylamino-pyrimidine analogue, nilotinib (AMN107, Tasigna), was designed based on the "imatinib-ABL complex" crystal structure. Nilotinib proved to be a potent, relatively selective inhibitor of tyrosine kinase activity exhibited by the BCR-ABL gene, the platelet-derived growth factor and the mast/stem-cell growth factor receptor (Tiwari et al., 2009).
Pharmaceutical prodrugs
One of the successful rational attempts would be to reduce the substrate-protein affinity by imparting minute changes to the drug's chemical structure, wherever applicable. The limitation with this approach could be loss of the drug's pharmacological action brought about by chemical modification. In a drug discovery process, prodrugs are often designed to improve the pharmacological and pharmacokinetic properties of the drugs. Val-quinidine, a prodrug obtained by derivatization of quinidine was reported successful in circumventing P-gp transport (Jain et al., 2004). The first and third generation PAMAM dendrimer based prodrugs showed potency as membrane permeability enhancing P-gp inhibitors for P-gp substrate drugs (Thiagarajan et al., 2010). Pegylated substrate-prodrugs and substrate encapsulated liposomal pegylation can escape the P-gp efflux (Immordino et al., 2003). Formulations using PEG-derivatized phospholipids altered the pharmacokinetics dramatically by imparting long elimination half-lives and small volumes of distribution (Dadashzadeh, Vali, Rezaie, 2008). Substrates can be structurally modified to form conjugates of substrate and monoclonal antibody (MAb) for delivery as MAb-prodrug system (Guillemard, Saragovi, 2004). Paclitaxel 2'-ethyl carbonate, a substrate-prodrug synthesized using radical co-polymerization and substrate-copolymer conjugates of HPMA are the examples of conjugate DDS which proved to be efficient P-gp bypassing systems (Tanino et al., 2007; Stastny et al., 1999).
Inhibitors of de-Novo origin
Generation of chemical atomic structures and prediction of the binding ability of such virtually designed ligands and proteins by applying docking programs and molecular modeling methods is called de-Novo design. Homology modeling based on disulphide cross linking established an atomic detailed model for the human ABC transporter, P-gp. However, the secondary and tertiary structures of P-gp still remain incompletely elucidated due to the protein's crystallization inability for carrying out X-ray crystallographic analysis. Atomic structural resolution models are being generated for P-gp based on the protein homology strategy by utilizing the cellular and molecular biology knowledge of other ABC transporters like LmrA, a bacterial P-gp homologue (from Lactococcus lactis) and MsbA, an E. coli derived MDR-ABC transporter homologue (Pleban et al., 2005; Eckford, Sharom, 2005; Chang, Roth, 2001). The earliest research study suggested the presence of two functional substrate binding sites on the protein's transmembrane segments. They were named as H and R sites based on their respective selectivity for Hoechst 33342 and Rhodamine 123. They are considered as the protein's "active transport sites" since the two sites functioned with positive cooperation to bring about P-gp efflux transport. Drug binding to any one of these sites stimulated the transport by the other. However, the presence of at least a four more drug binding sites was later reported. Distinguishing these newly identified sites as transport sites or modulatory sites still remains debatable (Haslam et al., 2008; Sharom, Lugo, Eckford, 2005; Martin et al., 2000). Any inhibitor that can bind to the transport site can serve as a competitive inhibitor for P-gp substrates. Polyvalence, the presence of multiple drug binding sites within a large and flexible "common protein drug pocket", paves a path to develop a series of inhibitors. Reports stated that homodimers of stipiamide separated with spacers of defined-length reversed with greater efficacy, the P-gp drug efflux (Sauna et al., 2004). Therefore, P-gp efflux blocking can be accomplished either by developing inhibitor compounds that can compete with the substrates for drug binding active transport sites or by those inhibitors which can bind to distinct modulatory sites affecting the allosteric regulation. The latter is categorized as non-transported inhibitors as they are not substrates for P-gp efflux and the former can act as competitive P-gp inhibitors (105). The thiomer excipients were reported to covalently modify the cysteine residues on TMDs and thereby inhibit P-gp. Inhibitor compounds which can specifically bring about targeted modification of P-gps NBDs also represent a challenging contribution since all the ABC transporters possess highly conserved NBDs (Werle, Hoffer, 2006). If NBDs are blocked, ATP hydrolysis are affected leading to P-gp functional paralysis. Besides, several signaling residues have been identified on the TM segments like G346, I306, L339 and N508. They are involved in the allosteric communication pathways that are set during drug occupancy. Any chemosensitizing candidate that can cause mutation of these residues can alter the ATP binding and hydrolysis steps assuring promising inhibition of the protein efflux (Storm et al., 2007).
Peptide inhibitors
Some hydrophobic, linear and cyclic transmembrane peptides were designed based on the protein's primary structure of the TMDs, presuming their involvement in disrupting the proper assembly of the transporter. There were reports that these peptides could effectively resensitize the resistant cancer cells in vitro, to doxorubicin, independent of chirality and without demonstrating any eminent cell toxicity (Sharom et al., 1998). Research on reversin 121 [N (α) -Boc-l-Asp (OBn) -l-Lys (Z) -OtBu], a second generation P-gp inhibitor led to the conception of fully non-competitive and potent peptidomimetic inhibitors. The replacement of the compound aspartyl residue by trans-4-hydroxy-l-Proline (4 (R) Hyp) gave two new molecules whose IC50 values were 2- and 7-folds lower than that of the parent compound. They were respectively named as reversin 11 and reversin 15. The only difference between these molecules lies in the presence of a reduced carbonyl group of the peptidyl bond in reversin 15. These compounds were reported as specifically P-gp targeting non-transported inhibitors by binding to an allosteric modulatory site other than H and R sites on the protein (Arnaud et al., 2010).
P-gp expression suppressers
Since P-gp expression increases by upregulation of MDR1 gene RNA levels, compounds like trypthanthrin and trifluoperazine, that reverse the MDR1 expression or down regulate the MDR1 gene RNA levels are anticipated to find scope as novel P-gp inhibitors (Yu et al., 2007; Shin et al., 2006). Chemicals such as copper N-(2-hydroxy acetophenone) glycinate (CuNG) and oxalyl bis (N-phenyl)hydroxomic acid (OBPHA) were reported to resensitize the MDR cells to chemotherapy by down regulating the P-gp over expression (Majumder et al., 2006). The reversal of P-gp mediated MDR, achieved by a form of gene silencer, short hairpin shaped interfering double stranded RNA (siRNA) in the post-transcriptional phase exhibited selectivity and plasmid/vector targeted delivery limitations (Katoh, Ueno, Takakura, 2008). The expression of MDR1 product, P-gp, requires activation of the MDR1 promoter which is favored by phosphorylated RNA helicase A (RHA). Since the phosphorylation is catalyzed by DNA-dependent protein kinase, selection of drugs that can inhibit this subunit can be one of the alternative approaches to abolish MDR developed by P-gp (Zhong, Safa, 2007). Dopamine and cAMP regulated phosphoprotein (DARPP-32) induced downregulation of P-gp expression is another hopeful reversal strategy (Hong et al., 2008). Elevated intracellular reactive oxygen species (ROS) involved in HIF-1α expression regulation and/or its stability stands out as one more feasible strategy to circumvent P-gp efflux. Multicellular tumor spheroids were identified which could induce endogenous oxidative stress. Such stress conditions elevate ROS levels which in turn stimulate the oxidative defensive systems of the body and the defensive systems block the P-gp efflux (Khaitan, Dwaraknath, 2009).
Future prospects
Rationalization of the course of evolution and emergence of P-glycoprotein inhibitors
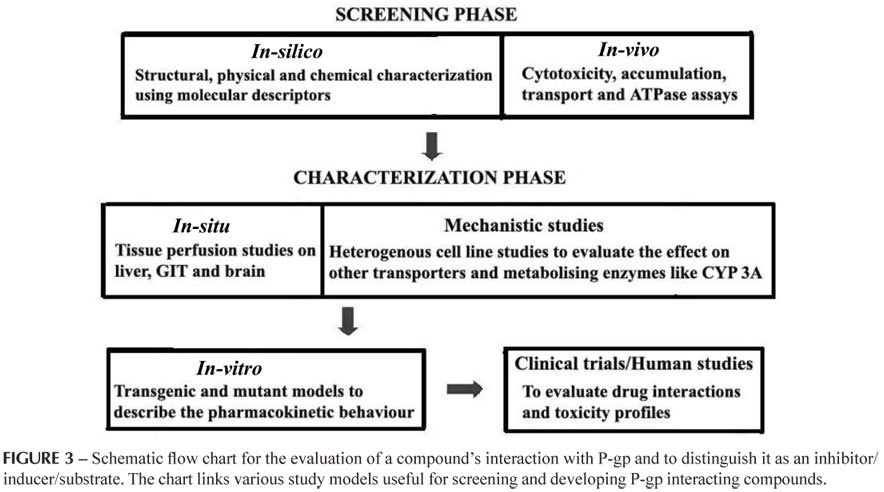
A rational drug design strategy can be used for characterizing compounds as P-gp inhibitors/substrates/inducers as it provides an approach to better understand the enigma of P-gp in drug development and delivery. It includes the in silico, in vitro screening techniques and de-Novo design to study P-gp interaction with different compounds so as to establish the protein's structural basis for substrate and modulator recognition. The two stages of rational drug design strategy for the evolutionary design and evaluation of P-gp inhibitors can be classified as discovery or screening and developmental or characterization phases as described in the Figure 3.
The screening phase involves in silico and in vitro studies where the compounds or chemical entities are screened for their P-gp affinity. The in silico analysis involves the use of chemical libraries and structural databases for screening. The in vitro tests used to identify P-gp interacting compounds include cytotoxicity assays, accumulation/efflux assays (based on fluorescent or photo affinity studies), transport assays and ATPase assays.
Thus identified chemical structures can be established as P-gp inhibitors/substrates/inducers, based on the tests carried out in the characterization phase which includes in-situ, mechanistic, in vivo and human studies. These studies provide a deeper insight into the affinity and specificity of the P-gp inhibitors/substrates/inducers. By using this sequential strategy, the compounds can be completely characterized starting from their extent of sensitivity to P-gp or their P-gp sensitizing efficiency to their drug-interaction and toxicity profiles (Bansal et al., 2009b). This kind of a schematic approach can rationalize the discovery and development of newer, inert/safer, non-toxic, and more efficient, specifically targeting P-gp inhibitors.
CONCLUSION
P-gp efflux drastically affects the bioavailability of its substrates by decreasing their effective plasma therapeutic concentration levels. Co-/concurrent administration of the P-gp substrate-therapeutics with the P-gp inhibitors can prevent/overcome the substrate expulsion by P-gp and render the intended therapeutic benefits of the substrate drugs. Hence, the research in this area remains an ever-challenging mission to the scientists. However, as a concluding remark, it should be noted that a caution must be exercised. The drug therapy/treatment for diseases using those pharmacological agents co-/concurrently administered with P-gp reversal agents should be carefully monitored after assessing all the possible risks associated with their usage.
Several efficient P-gp transport blockers were discovered earlier with the aim of enhancing the bioavailability of vital therapeutic P-gp substrates, most of them demonstrated shortcomings like unwanted side effects and toxicities which impeded their clinical utility. Hence there has always been a solid thrust on the pharmaceutical industry to develop new chemical entities to avoid or at least overcome these restraining phenomena. The recognition of inhibitors among the natural product extracts, inert pharmaceutical excipients and formulations to serve the cause is gaining utmost importance at present and as such, several inhibitor candidates identified under these categories have been compiled in this review. The safe, non-toxic nature of herbs and the inert, non-gut absorbent characters of excipients make them stand out unique forever. Though the upcoming researches identify and develop uncountable candidates from these classes, only the clinical trial reports can establish them as perfect rational P-gp inhibitors. A deeper insight into the rational drug design becomes indispensable at this stage of research to come out with more promising inert and non-toxic P-gp inhibitors. Unearthing the promiscuity of the P-gp structure, expression and substrate interaction is likely to provide a better understanding and open new vistas to develop novel inhibitory strategies. The development of novel reversal agents requires that the interaction between P-gp and the model compounds is well characterized. An attempt has been made in the current review to provide a P-gp interaction profile of a few compounds. The review discussed on several pharmaceutical prodrugs, synthetic peptides, certain novel antitumor drugs, de-Novo synthesized inhibitors, and P-gp expression suppressors which were reported to successfully escape the P-gp efflux. The development of further specific P-gp targeting inhibitors is foreseen in the nearest future to tackle and overcome the P-gp induced MDR and bioavailability problems.
Received for publication on 31st October 2011
Accepted for publication on 02nd May 2012
- ANUCHAPREEDA, S.; LEECHANACHAI, P.; SMITH, M.M.; AMBUDKAR, S.V.; LIMTRAKUL, P.N. Modulation of P-glycoprotein expression and function by curcumin in multidrug-resistant human KB cells. Biochem. Pharmacol, v.64, p.573-582, 2002.
- AOKI, S.; CAO, L.W.; MATSUI, K.; RACHMAT, R.; AKIYAMA, S.; KOBAYASHI, M. Kendarimide A, a novel peptide reversing P-glycoprotein-mediated multidrug resistance in tumor cells, from a marine sponge of Haliclona sp. Tetrahedron, v.60, p.7053-7059, 2004.
- ARNAUD, O.; KOUBEISSI, A.; ETTOUATI, L.; TERREUX R.; ALAMÉ, G.; GRENOT, C.; DUMONTET, C.; DI PIETRO, A.; PARIS, J.; FALSON, P. Potent and fully noncompetitive peptidomimetic inhibitor of multidrug resistance P-glycoprotein. J. Med. Chem., v.53, p.6720-6729, 2010.
- BACHMAKOV, I.; REKERSBRINK, S.; HOFMANN, U.; EICHELBAUM, M.; FROMM, M. F. Characterisation of (R/S)-propafenone and its metabolites as substrates and inhibitors of P-glycoprotein. Naunyn-Schmiedeberg's Arch. Pharm., v.371, p.195-201, 2005.
- BAILEY, D.; SPENCE, J.; MUNOZ, C.; ARNOLD, J. Interaction of citrus juices with felodipine and nifedipine. Lancet, v.337, p.268-269, 1991.
- BALAYSSACA, D.; AUTHIER, N.; CAYRE, A.; COUDORE, F. Does inhibition of P-glycoprotein lead to drugdrug interactions? Toxicol. Lett., v.156, p.319-329, 2005.
- BANSAL, T.; AKHTAR, N.; JAGGI, M.; KHAR, R.K.; TALEGAONKAR, S. Novel formulation approaches for optimising delivery of anticancer drugs based on P-glycoprotein modulation. Drug Discov. Today, v.14, p.1067-1074, 2009a.
- BANSAL, T.; JAGGI, M.; KHAR, R.K.; TALEGAONKAR, S. Emerging Significance of Flavonoids as P-Glycoprotein Inhibitors in Cancer Chemotherapy. J. Pharm. Pharm. Sci., v.12, p.46-78, 2009b.
- BATRAKOVA, E.V.; KABANOV, A.V. Pluronic block copolymers: evolution of drug delivery concept from inert nanocarriers to biological response modifiers. J. Control. Rel., v.130, p.98-106, 2008.
- BHARDWAJ, R.K.; GLAESER, H.; BECQUEMONT, L.; KLOTZ, U.; GUPTA, S.K.; FROMM, M.F. Piperine, a major constituent of black pepper, inhibits human P-glycoprotein and CYP3A4, J. Pharmacol. Exp. Ther., v.302, p.645-650, 2002.
- BUGGINS, T.R.; DICKINSON, P.A.; TAYLOR, G. The effects of pharmaceutical excipients on drug disposition. Adv. Drug Deliv. Rev., v.59, p.1482-1503, 2007.
- CHANG, G.; ROTH, C.B. Structure of MsbA from E. coli: a homolog of the multidrug resistance ATP binding cassette (ABC) transporters. Science, v.293, p.1793-1800, 2001.
- CHAVANPATIL, M.D.; KHDAIR, A.; GERARD, B.; BACHMEIER, C.; MILLER, D.W.; SHEKHAR, M.P.; PANYAM, J. Surfactant-polymer nanoparticles overcome Pglycoprotein-mediated drug efflux. Mol. Pharm., v.4, p.730-738, 2007.
- CHOI, J.S.; JO, B.W.; KIM, Y.C. Enhanced paclitaxel bioavailability after oral administration of paclitaxel or prodrug to rats pretreated with quercetin. Eur. J. Pharm. Biopharm., v.57, p.313-318, 2004.
- CHOI, J.S.; SHIN, S.C. Enhanced paclitaxel bioavailability after oral coadministration of paclitaxel prodrug with naringin to rats. Int. J. Pharm., v.292, p.149-156, 2005.
- CORLEY, D.G.; HERB, R.; MOORE, R.E.; SCHEUER, P.J. Laulimalide, new potent cytotoxic macrolides from a marine sponge and a nudibranch predator. J. Org. Chem., v.53, p.3644-3646, 1988.
- CORNAIRE, G.; WOODLEY, J.; HERMANN, P.; CLOAREC, A.; ARELLANO, C.; HOUIN, G. Impact of excipients on the absorption of P-glycoprotein substrates in vitro and in vivo. Int. J. Pharm., v.278, p.119-131, 2004.
- DABHOLKAR, R.D.; SAWANT, R.M.; MONGAYT, D.A.; DEVARAJAN, P.V.; TORCHILIN, V.P. Polyethylene glycol-phosphatidylethanolamine conjugate (PEG-PE)-based mixed micelles: some properties, loading with paclitaxel, and modulation of P-glycoprotein-mediated efflux. Int. J. Pharm., v.315, p.148-157, 2006.
- DADASHZADEH, S.; VALI, A.M.; REZAIE, M. The effect of PEG coating on in vitro cytotoxicity and in vivo disposition of topotecan loaded liposomes in rats. Int. J. Pharm., v.353, p.251-259, 2008.
- DANTZIG, A.H.; DE ALWIS, D.P.; BURGESS, M. Considerations in the design and development of transport inhibitors as adjuncts to drug therapy. Adv. Drug Deliv. Rev., v.55, p.133-150, 2003.
- DARBY, R.A.; CALLAGHAN, R.; MCMAHON, R.M. P-Glycoprotein inhibition; the past, the present and the future. Curr. Drug Metab., v.12, p.722-731, 2011.
- DHARMALA, K.; YOO, J.W.; LEE, C.H. Development of chitosan-SLN microparticles for chemotherapy: in vitro approach through efflux-transporter modulation. J. Control Rel., v.131, p.190-197, 2008.
- DIJKHUIS, A.; DOUWES, J.; KAMPS, W.; SIETSMA, H.; KOK, J.W. Differential expression of sphingolipids in P-glycoprotein or multidrug resistance-related protein 1 expressing human neuroblastoma cell lines. FEBS Lett., v.548, p.28-32, 2003.
- ECKFORD, P.D.W.; SHAROM, F.J. The reconstituted P-glycoprotein multidrug transporter is a flippase for glucosylceramide and other simple glycosphingolipids. Biochem. J., v.389, p.517-526, 2005.
- FOSTER, B.C.; FOSTER, M.S.; VANDENHOEK, S.; KRANTIS, A.; BUDZINSKI, J.W.; ARNASON, J.T.; GALLICANO, K.D.; CHOUDRI, S. An in vitro evaluation of human cytochrome P450 3A4 and P-glycoprotein inhibition by garlic. J. Pharm. Pharm. Sci., v.4, p.176-184, 2001.
- GANGJEE, A.; YU, J.; COPPER, J.E.; SMITH, C.D. Discovery of novel antitumor antimitotic agents that also reverse tumor resistance. J. Med. Chem., v.50, p.3290-3301, 2007.
- GUILLEMARD, V.; SARAGOVI, H.U. Prodrug chemotherapeutics bypass P-glycoprotein resistance and kill tumors in vivo with high efficacy and target dependent selectivity. Oncogene, v.23, p.3613-3621, 2004.
- HASLAM, I.S.; JONES, K.; COLEMAN, T.; SIMMONS, N.L. Induction of P-glycoprotein expression and function in human intestinal epithelial cells (T84). Biochem. Pharmacol., v.76, p.850-861, 2008.
- HEFFETER, P.; JAKUPEC, M.A.; KORNER, W.; CHIBA, P.; PIRKER, C.; DORNETSHUBER, R.; ELBLING, L.; SUTTERLUTY, H.; MICKSCHE, M.; KEPPLER, B.K.; BERGER, W. Multidrug-resistant cancer cells are preferential targets of the new antineoplastic lanthanum compound KP772 (FFC24). Biochem. Pharmacol., v.73, p.1873-1186, 2007.
- HIGGINS, C.F.; GOTTESMAN, M.M. Is the multidrug transporter a flippase? Trends Biochem. Sci., v.17, p.18-21, 1992.
- HONG, L.; ZHAO, Y.; WANG, J.; HAN, Y.; GUO, W.; JIN, H.; ZHAI, H.; BAI, F.; ZHANG, X.; QIAO, T.; CHEN, Z.; FAN, D. Reversal of multidrug resistance of adriamycin-resistant gastric adenocarcinoma cells through the up-regulation of DARPP-32. Dig. Dis. Sci., v.53, p.101-107, 2008.
- HUNTER, J.; HIRST, B.H. Influence of secretory systems on drug delivery/targeting. intestinal secretion of drugs. The role of P-glycoprotein and related drug efflux systems in limiting oral drug absorption. Adv. Drug Deliv. Rev., v.25, p.129-157, 1997.
- IKEGAWA, T.; USHIGOME, F.; KOYABU, N.; MORIMOTO, S.; SHOYAMA, Y.; NAITO, M.; TSURUO, T.; OHTANI, H.; SAWADA, Y. Inhibition of P-glycoprotein by orange juice components, polymethoxyflavones in adriamycin-resistant human myelogenous leukemia (K562/ADM) cells. Cancer Lett, v.160, p.21-28, 2000.
- IMMORDINO, M.L.; BRUSA, P.; ARPICCO, S.; STELLA, B.; DOSIO, F.; CATTEL, L. Preparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing docetaxel. J. Control Rel., v.91, p.417-429, 2003.
- JAIN, R.; MAJUMDAR, S.; NASHED, Y.; PAL, D.; MITRA, A.K. Circumventing P-glycoprotein-mediated cellular efflux of quinidine by prodrug derivatization. Mol. Pharm., v.1, p.290-299, 2004.
- JAIN, S.; LAPHOOKHIEO, S.; SHI, Z.; FU, L.W.; AKIYAMA, S.; CHEN, Z.S.; YOUSSEF, D.T.; VAN SOEST, R.W.; EL SAYED, K.A. Reversal of P-glycoprotein-mediated multidrug resistance by sipholane triterpenoids. J. Nat. Prod., v.70, p.928-931, 2007.
- JANG, S.H.; WIENTJES, M.G.; JESSIE, L. Kinetics of P-glycoprotein-mediated efflux of paclitaxel. J. Pharmacol. Exp. Ther., v.298, p.1236-1242, 2001.
- JIAO, W.; DONG, W.; LI, Z.; DENG, M.; LU, R. Lathyrane diterpenes from Euphorbia lathyris as modulators of multidrug resistance and their crystal structures. Bioorg. Med. Chem., v.17, p.4786-4792, 2009.
- JODOIN, J.; DEMEULE, M.; BELIVEAU, R. Inhibition of the multidrug resistance Pglycoprotein activity by green tea polyphenols. Biochim. Biophys. Acta., v.1542, p.149-159, 2002.
- JULIANO, R.L.; LING, V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim. Biophys. Acta, v.455, p.152-162, 1976.
- KATOH, S.Y.; UENO, M.; TAKAKURA, N. Involvement of MDR1 function in proliferation of tumour cells. J. Biochem., v.143, p.517-524, 2008.
- KHAITAN, D.; DWARAKNATH, B.S. Endogenous and induced oxidative stress in multi-cellular tumour spheroids: implications for improving tumour therapy. Indian J. Biochem. Biophys., v.46, p.16-24, 2009.
- KIM, C.K.; LIM, S.J. Recent progress in drug delivery systems for anticancer agents. Arch. Pharm. Res., v.25, p.229-239, 2002.
- KIM, S.; KWON, H.; CHI, D.; SHIM, J.; PARK, J.; LEE, Y.H.; PYO, S.; RHEE, D. Reversal of P-glycoprotein-mediated multidrug resistance by ginsenoside Rg3 Biochem. Pharmacol., v.65, p.75-82, 2003.
- KITAZAKI, T.; OKA, M.; NAKAMURA, Y.; TSURUTANI, J.; DOI, S.; YASUNAGA, M.; TAKEMURA, M.; YABUUCHI, H.; SODA, H.; KOHNO, S. Gefitinib, an EGFR tyrosine kinase inhibitor, directly inhibits the function of P-glycoprotein in multidrug resistant cancer cells. Lung Cancer, v.49, p.337-343, 2005.
- KOBAYASHI, T.; ISHIDA, Y.; OKADA, S.; ISE, S.; HARASHIMA, H.; KIWADA, H. Effect of transferrin receptor-targeted liposomal doxorubicin in P-glycoprotein-mediated drug resistant tumor cells. Int. J. Pharm., v.329, p.94-102, 2007.
- KRISHNA, R.; MAYER, L.D. Multidrug resistance (MDR) in cancer mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. Eur. J. Pharm. Sci., v.11, p.265-283, 2000.
- LIMTRAKUL, P.; KHANTAMAT, O.; PINTHA, K. Inhibition of P-glycoprotein activity and reversal of cancer multidrug resistance by Momordica charantia extract. Cancer Chemother. Pharmacol., v.54, p.525-530, 2004.
- LIMTRAKUL, P.; SIWANON, S.; YODKEEREE, S.; DUANGRAT, C. Effect of Stemona curtisii root extract on P-glycoprotein and MRP-1 function in multidrug-resistant cancer cells. Phytomedicine, v.14, p.381-389, 2007.
- LIN, J.H. Drug-drug interaction mediated by inhibition and induction of P-glycoprotein. Adv. Drug Deliv. Rev., v.55, p.53-81, 2003.
- LO, Y.L. Relationships between the hydrophiliclipophilic balance values of pharmaceutical excipients and their multidrug resistance modulating effect in Caco-2 cells and rat intestines. J. Control. Rel., v.90, p.37-48, 2003.
- MAJUMDER, S.; DUTTA, P; MUKHERJEE, P.; DATTA, E.R.; EFFERTH, T.; BHATTACHARYA, S.; CHOUDHURI, S.K. Reversal of drug resistance in P-glycoprotein-expressing T-cell acute lymphoblastic CEM leukemia cells by copper N-(2-hydroxy acetophenone) glycinate and oxalyl bis (N-phenyl) hydroxamic acid. Cancer Lett., v.244, p.16-23, 2006.
- MAKI, N.; HAFKEMEYER, P.; DEY, S. Allosteric modulation of human P-glycoprotein. Inhibition of transport by preventing substrate translocation and dissociation. J. Biol. Chem., v.278, p.18132-18139, 2003.
- MARTIN, C.; BERRIDGE, G.; HIGGINS, C.F.; MISTRY, P.; CHARLTON, P.; CALLAGHAN, R. Communication between multiple drug binding sites on P-glycoprotein. Mol. Pharmacol., v.58, p.624-632, 2000.
- MATHIJSSEN, R.H.; VERWEIJ, J.; DE BRUIJN, P.; LOOS, W.J.; SPARREBOOM, A. Effects of St. John's wort on irinotecan metabolism. J. Natl. Cancer Inst., v.94, p.1247-1249, 2002.
- MUNOZ-MARTINEZ, F.; MENDOZA, C.R.; BAZZOCCHI, I.L.; CASTANYS, S.; JIMENEZ, I.A.; GAMARRO, F. Reversion of human Pgp-dependent multidrug resistance by new sesquiterpenes from Zinowiewia costaricensis. J. Med. Chem., v.48, p.4266-4275, 2005.
- NAJAR, I.A.; SACHIN, B.S.; SHARMA, S.C.; SATTI, N.K.; SURI, K.A.; JOHRI, R.K. Modulation of P-glycoprotein ATPase Activity by Some Phytoconstituents. Phytother. Res., v.24, p.454-458, 2010.
- OZBEN, T. Mechanisms and strategies to overcome multiple drug resistance in cancer. FEBS Lett., v.580, p.2903-2909, 2006.
- PLEBAN, K.; KAISER, D.; KOPP, S.; PEER, M.; CHIBA, P.; ECKER, G.F. Targeting drug-efflux pumps - a pharmacoinformatic approach. Acta Biochim. Pol., v.52, p.737-740, 2005.
- ROBEY, R.W.; SHUKLA, S.; FINLEY, E.M.; OLDHAM, R.K.; BARNETT, D.; AMBUDKAR, S.V.; FOJO, T.; BATES, S.E. Inhibition of P-glycoprotein (ABCB1) - and multidrug resistance-associated protein 1 (ABCC1)-mediated transport by the orally administered inhibitor, CBT-1. Biochem. Pharmacol., v.75, p.1302-1312, 2008.
- SACHS-BARRABLE, K.; THAMBOO, A.; LEE, S.D.; WASAN, K.M. Lipid excipients Peceol and Gelucire 44/14 decrease P-glycoprotein mediated efflux of rhodamine 123 partially due to modifying P-glycoprotein protein expression within Caco-2 cells. J. Pharm. Pharm. Sci., v.10, p.319-331, 2007.
- SAUNA, Z.E.; ANDRUS, M.B.; TURNER, T.M.; AMBUDKAR, S.V. Biochemical basis of polyvalency as a strategy for enhancing the efficacy of P-glycoprotein (ABCB1) modulators: stipiamide homodimers separated with defined-length spacers reverse drug efflux with greater efficacy. Biochemistry, v.43, p.2262-2271, 2004.
- SCHINKEL, A.H. P-Glycoprotein, a gatekeeper in the blood-brain barrier. Adv. Drug Deliv. Rev., v.36, p.179-194, 1999.
- SHAROM, F.J.; LUGO, M.R.; ECKFORD, P.D. New insights into the drug binding, transport and lipid flippase activities of the pglycoprotein multidrug transporter. J. Bioenerg. Biomembr., v.37, p.481-487, 2005.
- SHAROM, F.J.; PEIHUA, L.U.; RONGHUA, L.I.U.; XIAOHONG, Y.U. Linear and cyclic peptides as substrates and modulators of P-glycoprotein: peptide binding and effects on drug transport and accumulation. Biochem. J., v.333, p.621-630, 1998.
- SHEKFEH, S. Binding sites of P-glycoprotein. Knol, 2009 (Version 5). Available at: http://knol.google.com/k/suhaib-shekfeh/binding-sites-of-p-glycoprotein/2v4mv6iwmzf2t/3 Accessed on 15 nov 2010.
- SHIN, S.Y.; CHOI, B.H.; KIM, J.R.; KIM, J.H.; LEE, Y.H. Suppression of P-glycoprotein expression by antipsychotics trifluoperazine in adriamycin-resistant L1210 mouse leukemia cells. Eur. J. Pharm. Sci., v.28, p.300-306, 2006.
- STASTNY, M.; STROHALM, J.; PLOCOVA, D.; ULBRICH, K.; RÍHOVA, B. A possibility to overcome P-glycoprotein (PGP)-mediated multidrug resistance by antibody-targeted drugs conjugated to N-(2- hydroxypropyl)methacrylamide (HPMA) copolymer carrier. Eur. J. Cancer, v.35, p.459-466, 1999.
- STORM, J.; O'MARA, M.L.; CROWLEY, E.H.; PEALL, J.; TIELEMAN, D.P.; KERR, I.D.; CALLAGHAN, R. Residue G346 in transmembrane segment six is involved in inter-domain communication in P-glycoprotein. Biochemistry, v.46, p.9899-9910, 2007.
- TAKANAGA, H.; OHNISHI, A.; MATSUO, H.; SAWADA, Y. Inhibition of vinblastine efflux mediated by P-glycoprotein by grapefruit juice components in caco-2 cells. Biol. Pharm. Bull., v.21, p.1062-1066, 1998.
- TANINO, T.; NAWA, A.; KONDO, E.; KIKKAWA, F.; DAIKOKU, T.; TSURUMI, T.; LUO, C.; NISHIYAMA, Y.; TAKAYANAGI, Y.; NISHIMORI, K.; ICHIDA, S.; WADA, T.; MIKI, Y.; IWAKI, M. Paclitaxel-2'-ethylcarbonate prodrug can circumvent P-glycoprotein- mediated cellular efflux to increase drug cytotoxicity. Pharm. Res., v.24, p.555-565, 2007.
- THIAGARAJAN, G.; RAY, A.; MALUGIN, A.; GHANDEHARI, H. PAMAM-Camptothecin conjugate inhibits proliferation and induces nuclear fragmentation in colorectal carcinoma cells. Pharm. Res., v.27, p.2307-2316, 2010.
- THIEBUT, F.; TSURUO, T.; HAMADA, H.; GOTTESMAN, M.M.; PASTAN, I. Cellular localisation of the multidrug resistance gene product P-glycoprotein in normal human tissues. Proc. Natl. Acad. Sci. USA, v.84, p.7735-7738, 1987.
- THOMAS, H.; COLEY, H.M. Overcoming multidrug resistance in cancer: an update on the clinical strategy of inhibiting P-glycoprotein. Cancer Control, v.10, p.159-165, 2003.
- TIWARI, A.K.; SODANI, K.; WANG, S.; KUANG, Y.; ASHBY, C.R.; CHEN, X.; CHEN, Z. Nilotinib (AMN107, Tasigna1) reverses multidrug resistance by inhibiting the activity of the ABCB1/Pgp and ABCG2/BCRP/MXR transporters. Biochem. Pharmacol., v.78, p.153-161, 2009.
- VANHUYSE, M.; KLUZA, J.; TARDY, C.; OTERO, G.; CUEVAS, C.; BAILLY, C.; LANSIAUX, A. Lamellarin D: a novel pro-apoptotic agent from marine origin insensitive to P glycoprotein-mediated drug efflux. Cancer Lett., v.221, p.165-175, 2005.
- VARMA, M.V.S.; ASHOKRAJ, Y.; DEY, C.S.; PANCHAGNULA, R. P-glycoprotein inhibitors and their screening: a perspective from bioavailability enhancement. Pharmacol. Res., v.48, p.347-359, 2003.
- VAUTIER, S.; LACOMBLEZ, L.; CHACUN, H.; PICARD, V.; GIMENEZ, F.; FARINOTTI, R.; FERNANDEZ, C. Interactions between the dopamine agonist, bromocriptine and the efflux protein, P-glycoprotein at the blood-brain barrier in the mouse. Eur. J. Pharm. Sci., v.27, p.167-174, 2006.
- WAN, C.; ZHU, G.; SHEN, X.; CHATTOPADHYAY, A.; DEY, S.; FONG, W. Gomisin A alters substrate interaction and reverses P-glycoprotein-mediated multidrug resistance in HepG2-DR cells. Biochem. Pharmacol., v.72, p.824-837, 2006.
- WERLE, M.; HOFFER, M. Glutathione and thiolated chitosan inhibit multidrug resistance P-glycoprotein activity in excised small intestine. J. Control Rel., v.111, p.41-46, 2006.
- WONG, H.L.; BENDAYAN, R.; RAUTH, A.M.; WU, X.Y. Development of solid lipid nanoparticles containing ionically complexed chemotherapeutic drugs and chemosensitizers. J. Pharm. Sci., v.93, p.1993-2008, 2004.
- YOSHIDA, N.; KOIZUMI, M.; ADACHI, I.; KAWAKAMI, J. Inhibition of P-glycoprotein-mediated transport by terpenoids contained in herbal medicines and natural products. Food Chem. Toxicol., v.44, p.2033-2039, 2006.
- YU, S.T.; CHEN, T.M.; TSENG, S.Y.; CHEN, Y.H. Tryptanthrin inhibits MDR1 and reverses doxorubicin resistance in breast cancer cells. Biochem. Biophys. Res. Commun., v.358, p.79-84, 2007.
- YUAN, H.; LI, X.; WU, J.; LI, J.; QU, X.; XU, W.; TANG, W. Strategies to overcome or circumvent p-glycoprotein mediated multidrug resistance. Curr. Med. Chem., v.35, p.470-476, 2008.
- ZHANG, S.; MORRIS, M.E. Effects of the flavonoids biochanin A, morin, phloretin, and silymarin on P-glycoprotein-mediated transport. J. Pharmacol. Exp. Ther., v.304, p.1258-1267, 2003.
- ZHONG, X.; SAFA, A.R. Phosphorylation of RNA helicase A by DNA-dependent protein kinase is indispensable for expression of the MDR1 gene product P-glycoprotein in multidrug-resistant human leukemia cells. Biochemistry, v.46, p.5766-5775, 2007.
Publication Dates
-
Publication in this collection
18 Oct 2012 -
Date of issue
Sept 2012
History
-
Received
31 Oct 2011 -
Accepted
02 May 2012