Abstracts
The objective of the present investigation was to develop bilayered tablets of lornoxicam to achieve biphasic release pattern. A bilayered tablet, consisting of an immediate and controlled release layer, was prepared by direct compression technique. The controlled release effect was achieved by using various hydrophilic natural, semi synthetic and synthetic controlled release polymers such as xanthan gum, hydroxypropyl methylcellulose (HPMC) and polyethylene oxide (PEO) to modulate the release of the drug. The in vitro drug release profiles showed the biphasic release behavior in which the immediate release (IR) layer containing the lornoxicam was released within 15 minutes, whereas the controlled release (CR) layer controlled the drug release for up to 24 h. All the bilayered tablets formulated have followed the zero order release with non-Fickian diffusion controlled release mechanism after the initial burst release. FTIR studies revealed that there was no interaction between the drug and polymers used in the study. Statistical analysis (ANOVA) showed no significant difference in the cumulative amount of drug release after 15 min, but significant difference (p < 0.05) in the amount of drug released after 24 h from optimized formulations was observed. Based on the release kinetic parameters obtained, it can be concluded that xanthan gum polymer was suitable for providing a biphasic release of lornoxicam.
Lornoxicam; Lornoxicam; Bilayered tablet; Hydrophilic polymers; Hydrophilic polymers; Drugs
O objetivo do presente trabalho foi desenvolver comprimidos bicamada de lornoxicam para atingir padrão de liberação bifásica. Preparou-se, por compressão direta, comprimido bicamada, consistindo de uma camada de liberação imediata e uma de liberação controlada. A liberação controlada foi obtida pelo uso de vários polímeros naturais hidrofílicos, semi-sintéticos e sintéticos, tais como goma xantana, hidroxipropilmetil celulose (HPMC) e óxido de polietileno (PEO) para modular a liberação do fármaco. Os perfis de liberação in vitro mostraram comportamento bifásico em que a camada de liberação imediata (IR) contendo lornoxicam foi liberada em 15 minutos, enquanto a camada de liberação controlada (CR) liberou o fármaco em mais de 24 horas, Todos os comprimidos bicamada formulados seguiram a liberação de ordem zero com mecanismo de liberação controlada por difusão não fickiana após a liberação inicial por erupção. Os estudos de FTIR revelaram que não há interação entre o fármaco e os polímeros utilizados no estudo. A análise estatística (ANOVA) não mostrou diferença significativa na quantidade acumulada de fármaco após 15 minutos de liberação, mas observou-se diferença significativa (p<0,05) na quantidade de fármaco liberado após 24 h nas formulações otimizadas. Com base nos parâmetros de cinética de liberação obtidos, pode-se concluir que a goma xantana foi adequada para se atingir liberação bifásica de lornoxicam.
Lornoxicam; Lornoxicam; Comprimido bicamada; Polímeros hidrofílicos; Polímeros hidrofílicos; Fármacos
ARTICLES
Design and evaluation of lornoxicam bilayered tablets for biphasic release
Songa Ambedkar Sunil* * Correspondence: S. A. Sunil. A.U. College of Pharmaceutical Sciences, Andhra University, Visakhapatnam-530003, India. E-mail: sunilsonga@gmail.com ; Meka Venkata Srikanth; Nali Sreenivasa Rao; Sakamuri Balaji; Kolapalli Venkata Ramana Murthy
A.U. College of Pharmaceutical Sciences, Andhra University, India
ABSTRACT
The objective of the present investigation was to develop bilayered tablets of lornoxicam to achieve biphasic release pattern. A bilayered tablet, consisting of an immediate and controlled release layer, was prepared by direct compression technique. The controlled release effect was achieved by using various hydrophilic natural, semi synthetic and synthetic controlled release polymers such as xanthan gum, hydroxypropyl methylcellulose (HPMC) and polyethylene oxide (PEO) to modulate the release of the drug. The in vitro drug release profiles showed the biphasic release behavior in which the immediate release (IR) layer containing the lornoxicam was released within 15 minutes, whereas the controlled release (CR) layer controlled the drug release for up to 24 h. All the bilayered tablets formulated have followed the zero order release with non-Fickian diffusion controlled release mechanism after the initial burst release. FTIR studies revealed that there was no interaction between the drug and polymers used in the study. Statistical analysis (ANOVA) showed no significant difference in the cumulative amount of drug release after 15 min, but significant difference (p < 0.05) in the amount of drug released after 24 h from optimized formulations was observed. Based on the release kinetic parameters obtained, it can be concluded that xanthan gum polymer was suitable for providing a biphasic release of lornoxicam.
Uniterms: Lornoxicam/bilayered tablet/development. Bilayered tablet/biphasic release. Hydrophilic polymers/use/drug release. Drugs/controlled release.
RESUMO
O objetivo do presente trabalho foi desenvolver comprimidos bicamada de lornoxicam para atingir padrão de liberação bifásica. Preparou-se, por compressão direta, comprimido bicamada, consistindo de uma camada de liberação imediata e uma de liberação controlada. A liberação controlada foi obtida pelo uso de vários polímeros naturais hidrofílicos, semi-sintéticos e sintéticos, tais como goma xantana, hidroxipropilmetil celulose (HPMC) e óxido de polietileno (PEO) para modular a liberação do fármaco. Os perfis de liberação in vitro mostraram comportamento bifásico em que a camada de liberação imediata (IR) contendo lornoxicam foi liberada em 15 minutos, enquanto a camada de liberação controlada (CR) liberou o fármaco em mais de 24 horas, Todos os comprimidos bicamada formulados seguiram a liberação de ordem zero com mecanismo de liberação controlada por difusão não fickiana após a liberação inicial por erupção. Os estudos de FTIR revelaram que não há interação entre o fármaco e os polímeros utilizados no estudo. A análise estatística (ANOVA) não mostrou diferença significativa na quantidade acumulada de fármaco após 15 minutos de liberação, mas observou-se diferença significativa (p<0,05) na quantidade de fármaco liberado após 24 h nas formulações otimizadas. Com base nos parâmetros de cinética de liberação obtidos, pode-se concluir que a goma xantana foi adequada para se atingir liberação bifásica de lornoxicam.
Uniterms: Lornoxicam/comprimido bicamada/desenvolvimento. Comprimido bicamada/liberação bifásica. Polímeros hidrofílicos/uso/liberação do fármaco. Fármacos/liberação controlada.
INTRODUCTION
Biphasic drug delivery systems are designed to release the drug at two different rates or at two different time periods i.e. they are either quick/slow or slow/quick. A quick/slow release system provides an initial burst release of drug followed by a constant rate (ideally) of release over a defined period of time. This type of system is used primarily when maximum relief needs to be achieved quickly, and it is followed by a controlled release phase to avoid repeated administration. Whereas release from slow/quick is vice versa and it is obtained using multi-unit dosage forms or compression coated tablets for the treatment of those diseases that follow circadian rhythm (Kenneth et al., 2003). Non-steroidal anti-inflammatory drugs (NSAIDs), antihypertensive, antihistaminic and anti-allergic agents (Maggi et al., 1999) are some classes of drugs suitable for design of bilayered (biphasic) drug delivery systems. Generally, conventional controlled release dosage forms provide delayed release of drug from the dosage form which leads to delay in attaining maximum therapeutic plasma levels immediately. To modify the release of the drug from these systems, the surface area exposed to a fluid can be restricted by the addition of barrier layers to one or both sides of the tablets (Colombo et al., 1990; Qiu et al.,1998; Chidambaram et al., 1998). There are several approaches reported to achieve biphasic drug delivery, like multi-particulate system (White et al., 1997; Morita et al., 2000), osmotic pump system (Botti et al., 2004; Li et al., 2004), pulsatile drug delivery system, compressed matrix core tablet (Eiji et al., 2000) and of these bilayered tablet system is a novel approach.
Previously, Luana Perioli et al., 2007 formulated a mucoadhesive bilayered tablet of flurbiprofen with sustained release for bucaal delivery. The first layer was responsible for retention on mucosa and the second layer for the buccal drug delivery. Remya et al., 2010 has developed a robust formulation of bilayered tablets containing ibuprofen and methocarbamol using povidone k-30 as binder. The main aim for formulation of these tablets was to attain the prolonged and repeat action of drug. Sunil Reddy et al., 2010 used the concept of bilayered technology to develop the tablets containing glimepride for immediate release using sodium starch glycolate as super disintegrant and metformin hydrochloride for sustained release by using Hydroxyl propyl methyl cellulose and sodium carboxy methyl cellulose as the matrix forming polymer, and PVPK-30 as binder. Ajit et al., 2009 designed a bilayer regioselective floating tablets to attain a biphasic release of atenolol and lovastatin to give immediate release of lovastatin and sustained release of atenolol.
Lornoxicam is NSAID of the oxicam class with analgesic, anti-inflammatory and antipyretic properties which belongs to Biopharmaceutical Class II (Low solubility and high permeability). The bioavailability of lornoxicam is 90% - 100%. Because of its relatively short plasma half-life, 3-5 h (Neil et al., 1998), it is prescribed to take lornoxicam in divided daily doses either twice or thrice daily in order to maintain the therapeutic plasma concentration. These characteristics make lornoxicam a suitable candidate for developing into bilayered tablet.
In the present study, it is aimed to design, develop and characterize a biphasic delivery dosage form as a bilayered tablet in which the immediate release by IR layer and controlled release by the CR layer using different hydrophilic polymers of synthetic (PEO), semisynthetic (HPMC) and natural (xanthan gum) origin.
MATERIAL AND METHODS
Material
Lornoxicam, a slightly soluble drug was obtained as gift sample from Macleods Pharmaceuticals Ltd., Mumbai, India. PEO WSR N 60K was purchased from Kemphasol Chemicals, Mumbai, India. HPMC was supplied by Colorcon (Orpington, UK) and Xanthan gum was purchased from M/s Girijan Co-operative Corporation,Visakhapatnam, India. Starch 1500 from Reidel Chem., Mumbai, India and polyvinyl pyrrolidone (povidone) was obtained from Lab. Chemi. Mumbai, India. All other chemicals used were of analytical grade.
Methods
Calculation of the controlled release dose
As per the zero order release principle, the rate of delivery must be independent of the amount of drug remaining in the dosage form and constant over time. The release from the dosage form should follow zero order kinetics, as shown by the following Equation 1:

where K0 is the zero order rate constant for drug release (amount/time), ke is the first order rate constant of overall drug elimination (h-1), Cp is the desired plasma drug level (amount/volume), and Vd is the volume of drug distributed (Wai-Yip et al., 2000). If the elimination half-life of lornoxicam is 3-5 h (ke = 0.693/4.0), Cp is 0.28 mg/L (Neil et al., 1998), and Vd is 10.5 L, then K0 is 0.5093 mg/h. For a system in which the maintenance dose releases the drug by a zero order process for a specified period of time, the total dose is as follows:

where DI is the initial dose, K0 is the zero order rate Constant, tp is the time of the peak drug level and td is the total time desired for controlled release from single dose (i.e. 24 h). If the maintenance dose begins to release the drug at the time of dosing (t = 0), it will add to that which is provided by the initial dose, thus increasing the initial drug level. In this case, a correction factor is needed (K0 tp) to account for the added drug from the maintenance dose. This correction factor is the amount of drug provided during the period from t = 0 to the time of the peak drug level, tp. If DI is 3.24 mg, K0is 0.5093 mg/h, tp is 1.5 h, and td is 24 h, then as per Equation 2 the total dose would be 15.46 mg of lornoxicam. From the above calculations total dose obtained for controlled release of lornoxicam for 24 h is 15.46 mg. The total dose was rounded to 15.5 mg for the convenience.
Preparation and characterization of bilayer tablets
The dose in the formulation for IR tablet was 3.25 mg and the maintenance dose or CR dose (12.25 mg) of lornoxicam was calculated as per above given formula in Equation 2.
Formulation of the immediate release layer
The IR layer was prepared by passing the accurately weighed drug, starch 1500 and 5% PVP as a dry binder through # 40 (420 µm ASTM) as per the formulae given in Table I. The sieved blend was transferred to a poly bag and mixed for 5 minutes. The obtained blend was lubricated for 2 minutes with magnesium stearate and brilliant lake red passed thorough # 60 (250 µm ASTM) and # 100 (150 µm ASTM) respectively. Brilliant lake red was added to differentiate the IR layer from the CR layer.
Formulation of the controlled release layer
The CR layer was prepared by blending the drug and the controlled release polymer (PEO, HPMC K15M, Xanthan gum ) individually, uniformly along with diluent and PVP K30 as a dry binder in a ploy bag as per the formulae given in Table II after sifting them through # 40 (420 µm). The resulting blend was lubricated with talc and magnesium stearate passed through # 60 (250 µm).
Precompression studies of the prepared blend to be compressed into a tablet
Prior to the compression of the formulation blends into tablets, flow properties of the powders should be determined to check its suitability for compression.
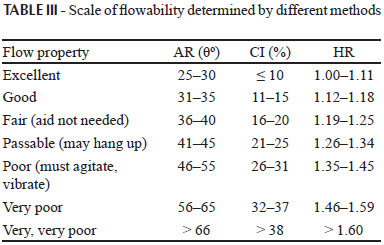
Angle of repose (AR), compressibility Index (CI) and Hausner's ratio (HR) were used to characterize flow properties of the blends. The flowability of a powder is of critical importance in the production of pharmaceutical dosage forms in order to reduce dose variations (Kulkarni et al., 2010; Fahmy et al., 2008; Amidon et al., 2009; USP32-NF27, 2009; Kulkarni et al., 2010; Bhise et al., 2008; Shah et al., 2008).
Bulk density
Bulk density of a powder is the ratio of the mass of the powder to the volume occupied by the loose powder bed. The unit volume includes the spaces between the particles and the envelope volumes of the particles themselves.
An amount of powder equivalent to 15 g was accurately weighed and placed in a 50 mL volumetric cylinder without compaction and the volume occupied was measured and the initial bulk density was calculated by the following equation. Each experiment was performed in triplicate and calculated using Equation 3.

where, ρbulk = bulk density, M = mass of the powder and Vbulk = Initial volume of powder
Tapped density
Tapped density of a powder is the ratio of the mass of the powder to the volume occupied by the powder after a fixed number of taps (100). The tapped density of a powder represents its random dense packing.
An amount of each powder of 15 g was accurately weighed and placed in a 50 mL volumetric cylinder. Then the cylinder was tapped by raising it to a height of 12-14 mm and then allowing it to fall under its own weight. The final volume was recorded and the final tapped density was calculated by the following Equation 4. Each experiment was performed in triplicate.

where, ρtap = tapped density, M = mass of the powder and Vtap = Volume of powder on tapping
The bulk and tapped densities were used to calculate the CI and the HR.
Carr's compressibility index
Carr's compressibility index (CI) of the powder was found by using the following Equation 5.

CI value can reflect the ease of powder consolidation. A higher CI values indicates its poor flow.
Hausner's ratio
The powder's Hausner's ratio (HR) is expressed as HR value reflects inter particulate friction. A high HR value means poor flow calculated using Equation 6.

Angle of repose
Angle of repose (AR) has been used as an indirect method of quantifying powder flowability. AR for the blend of each formulation was determined by the fixed funnel method.
A glass funnel was secured with its tip positioned at a fixed height (h) above a graph paper placed on a horizontal surface. The powders were carefully poured through the funnel until the apex of the conical pile so formed just touched the tip of the funnel. The mean radius (r) of the base of the powder cone was determined and the AR (θ) was calculated as follows:

where, h = height of the conical pile and r = radius of the conical pile.
The procedure was done in triplicate and the average AR was calculated for each powder. AR is a characteristic property related to inter particulate friction or resistance to movement between particles. A general scale of powder flow using the CI, HR and AR are given in Table III.
Compression of the bilayer tablet
Initially the die was filled with CR blend and precompressed using 16 station, single rotary tablet compression machine (Cadmach Machinery Co.Pvt. Ltd., India) equipped with 6 mm round concave punches. Upon this precompressed CR layer, weighed quantity of IR blend was transferred manually followed by compression obtaining a bilayered tablet having hardness in the range of 6-8 kgcm-2.
Evaluation of bilayered tablets
The prepared bilayered tablets were subjected to quality control tests such as uniformity of weight, friability test, hardness, drug content and in vitro dissolution studies.
Uniformity of weight
According to Indian Pharmacopoeia (I.P.) (IP2007, 2008), 20 tablets were selected at random, weighed together and then individually. The mean and the standard deviation were determined.
Prepared tablets complies the test if not more than two of the individual weights deviate from the average weight (>250 mg) by more than the percentage (5%) and none deviate more than twice that percentage.
Friability
The friability test (IP2007, 2008) was carried out in dual chamber friabilator (Electrolab, India). Tablets equivalent to 6.5 g were weighed (W0) initially and put in a rotating drum. Then, they were subjected to 100 falls of 6 inches height (25 rpm for four min). After completion of rotations, the tablets were dedusted by using camel hair brush and weighed (W). The percent loss in weight or friability was calculated by the formula given below.
Hardness (Banker et al., 2009)
Tablets should be sufficiently hard to resist breaking during normal handling and yet soft enough to disintegrate properly after swallowing. Five tablets were randomly selected and the hardness of each tablet was measured using Monsanto hardness tester. The mean hardness was determined and expressed in kgcm-2.
Determination of drug content
From each batch of prepared tablets, 10 tablets were randomly collected and powdered (IP2007, 2008). Powder equivalent to 15.46 mg of lornoxicam was weighed accurately from each batch and were transferred separately to a 100 mL volumetric flask. Volume was made up to 100 mL with pH 7.4 phosphate buffer and subjected for vortex mixing and sonication for dissolving the drug. Appropriate dilutions were made with pH 7.4 phosphate buffer and the amount of lornoxicam was analyzed using a validated method at 378 nm using a double beam UV/visible spectrophotometer (Elico Ind Ltd, India) (Bettini et al., 2001). The drug content was calculated using Equation 8.

In vitro dissolution study
The in vitro release tests were performed using LABINDIA, Disso 2000, according to the US Pharmacopoeia 27, the compressed bilayered tablets were introduced into dissolution medium. A 900 mL of phosphate buffer pH 7.4 was used as dissolution medium, rotational speed of the paddle was set at 50 rpm at 37 ± 0.5 ºC. Aliquots (5 mL each) were withdrawn at predetermined time intervals by means of a syringe fitted with a 0.45 µm pre-filter and immediately replaced with 5 mL of fresh medium maintained at 37±0.5 ºC. The samples were analyzed for lornoxicam at 378 nm using a double beam UV/visible spectrophotometer (Elico Ind Ltd, India). All the dissolution experiments were carried out in triplicate.
Release kinetics
The analysis of drug release mechanism from a pharmaceutical dosage form is an important but complicated process and is practically evident in the case of matrix systems. As a model-dependent approach, the dissolution data was fitted to five popular release models such as zero-order, first-order, Higuchi, Hixon-Crowel and Korsmeyer Peppas equations. The order of drug release from matrix systems was described by using zero order kinetics or first orders kinetics and the mechanism of drug release by using Higuchi and Hixon-Crowel equation.
Zero order release kinetics is (Lazarus et al., 1961) defined as a linear relationship between the fractions of drug released versus time. A plot of the fraction of drug released against time will be linear if the release obeys zero order release kinetics. It is given by Equation 9 below:

where, Q is the fraction of drug released at time t and k0 is the zero order release rate constant.
In first order release kinetics (Wagner et al., 1969) Wagner assumed that the exposed surface area of a tablet decreased exponentially with time during dissolution process, this suggests that drug release from most of the slow release tablets could be described adequately by apparent first-order kinetics. First order kinetics is defined by Equation 10 below:

where, Q is the fraction of drug released at time t and k1 is the first order release rate constant. Thus, a plot of the logarithm of the fraction of drug remained against time will be linear if the release obeys first order release kinetics.
Higuchi equation (Higuchi et al., 1963) defines a linear dependence of the active fraction released per unit of surface (Q) on the square root of time. It is defined by Equation 11 below:

where, k2 is the release rate constant. Hence, a plot of the fraction of drug released against square root of time will be linear if the release obeys Higuchi equation. This equation describes drug release as a diffusion process based on the Fick's law, square root time dependent.
Erosion equation (Korsmeyer et al.,1983) defines the drug release based on tablet erosion alone (See Equation 12).

where, Q is the fraction of drug released at time t, K3 is the release rate constant. Thus, a plot between [1-(1-Q)1/3 ] against time will be linear if the release obeys erosion equation.
In order find out the mechanism of drug release, first 60% drug release data were fitted in Korsmeyer Peppas model in Equation13

where Mt / M8 is a fraction of drug released at time, t, K4 is the release rate constant and n is the release exponent. The n value is used to characterize different release for cylindrical shaped matrices. For the case of cylindrical tablets, 0.45 = n corresponds to a Fickian diffusion mechanism, 0.45 < n < 0.89 to non-Fickian transport, n = 0.89 to Case II (relaxational) transport, and n > 0.89 to super case II transport (Peppas et al., 1985).
Fourier Transform Infrared Spectroscopy
Infrared spectroscopy was conducted using a Shimadzu FTIR 8300 Spectrophotometer and the spectrum was recorded in the region of 4000 to 400 cm-1. The procedure consisted of dispersing a sample (drug and drug resinate mixture, 1:1 ratio) in KBr (200-400 mg) and compressing into discs by applying a pressure of 5 tons for 5 min in a hydraulic press. The pellet was placed in the light path and the spectrum was obtained. Spectra were recorded in duplicate for each of sample.
RESULTS AND DISCUSSION
Precompression studies of the prepared blend to be compressed into a tablet
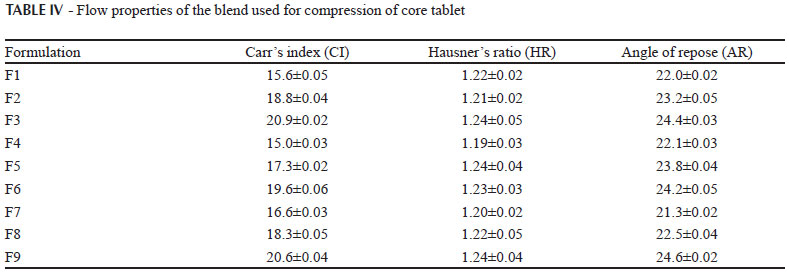
Powder flow properties are the important parameters which are crucial in processing operations such as mixing, flow of blend from hoppers to the turret for compression. One of the major problems encountered with these poor flowing powders in pharmaceutical industry is inconsistent and uniform flow from the hoppers. The angle of repose of the all blends (F1 to F9) was found to be < 25º indicating excellent properties. The Carr's index and Hausner's ratio of the all blends was found to be < 20 % and 1.20-1.25 indicating good flow properties. Blend of all the batches used for compression of core tablets exhibited good flow and compression properties as the excipients used for the formulations were of directly compressible grade. The results in Table IV had shown that the direct compression method can be used for the compression of tablets.
Evaluation of bilayered tablets
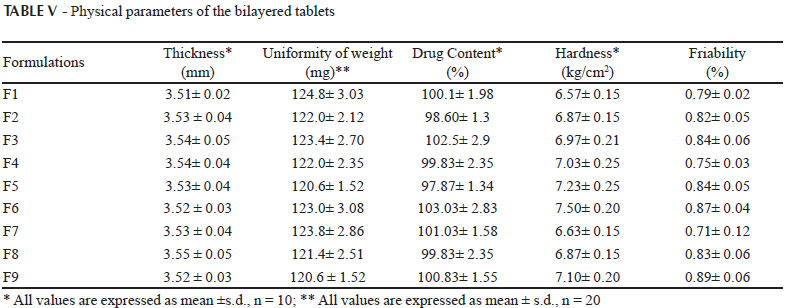
The physical attributes of the tablet were found to be satisfactory. Typical tablet defects, such as capping, chipping, and picking, were not observed. Tablet properties like weight variation, thickness, hardness, friability and drug content of each batch was represented in Table V. All batches passed the weight variation test and found to be within range (±7.5%). Friability of all batches were found less than 1% and indicates that tablet surfaces are strong enough to withstand mechanical shock or attrition during storage and transportation and until they are consumed. Hardness of the tablet was found to be 6.5-7.5 kgcm-2. Thickness and diameter were found to be fixed during the compression cycle; values were 3.5 mm, and 6 mm, respectively. Drug content of all batches was found within the limit (90-110%). Low s.d. values in drug content indicated uniformity of drug distribution in all the prepared tablets. Thus, the prepared bilayered tablets of lornoxicam were found to be of good quality fulfilling all the official compendia for tablets.
In vitro dissolution study
Bilayered tablets were developed using different retarding polymers (PEO WSR N60K, HPMC K 15 M and Xanthan gum) in different concentrations (0.5%, 1.0% and 1.5% w/w). More than 99% of the drug was released from all the tablets. The polyether chains present in PEO can form strong hydrogen bonds upon contact with water and therefore, when solid matrices brought into contact with an aqueous medium, the polymer tends to hydrate, forming a superficial gel which eventually erodes as the polymer dissolves. As the polymer dissolves the fresh surfaces are exposed to the dissolution medium, only once the gel layer is formed it starts controlling the diffusion of the polymer (Petrovic et al., 2009). The tablets F1, F2 and F3 prepared with PEO released more than 99% of the drug in 5 hrs, 8 hrs and 11 hrs respectively. F4, F5 and F6 formulations with HPMC as retarding polymer released more than 99% of the drug at the end of 5 hrs, 11 hrs and 16 hrs respectively. The results have also shown that the presence of more polymer at the surface resulted in instant formation of a gel layer upon contact with medium, which prevented initial burst of drug release. This supports Pham et al, (1994) observation that for relatively water insoluble drugs and/or lower viscosity grades of HPMC, polymer dissolution plays an important role in regulating drug release.
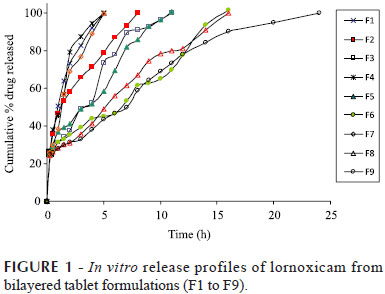
When xanthan gun was used in formulations F7, F8 and F9, maximum drug release was observed in 5 h, 16 h and 24 h respectively. Formulation F9 prepared with xanthan gum as a rate retarding polymer was selected as the optimized formulation keeping in view the minimum amount of xanthan gum required to control the release for a period of 24 h. From all the formulations it was observed that, loading dose of the lornoxicam was released from the IR layer within the first 15 min of the dissolution study. Further release of lornoxicam was studied for 24 hours. Respective dissolution profiles of all the formulations are given in Figure 1
All the polymers used in the study are hydrophilic but the difference in the percentage drug release was due to the swelling nature of the polymers. Higher the swelling nature of the polymer, higher was the ability to retard the drug release. As the concentration of the polymer was increased the % drug release decreased for all the polymers. The order of retarding capacity of the polymers was in the following order xanthan gum>HPMC K 15 M>PEO N 60K, which also states that the swelling capacity of xanthan gum was more than the other polymers.
Release kinetics
The drug release of lornoxicam from all the formulations F1 to F9 followed zero order kinetics which was indicated by higher 'r' values of zero order release model, when compared to those of first order release model. The 'r' values ranged from 0.9463 - 0.9936 and the rate order values are shown in Table VI.
The relative contributions of drug diffusion and erosion to drug release were further confirmed by subjecting the dissolution data to Higuchi model and Hixon Crowell model. Release of the drug from a matrix tablet containing hydrophilic polymers generally involves diffusion. It was found that all the formulations F1 to F9 followed diffusion mechanism as the 'r' values (0.9527 to 0.9942) were higher than for erosion release. The 'n' value for the diffusion release observed was less than 0.5 indicated that all the formulations Non-Fickian diffusion mechanism. All the formulations F1 to F9 followed Zero order release with Non-Fickian diffusion mechanism. The desired targeted release 24 hrs was achieved by formulation F9 which was prepared by using Xanthan gum in (1:1.5) ration of drug and polymer and also as the polymer is of natural origin which is of great advantageous over synthetic polymers (PEO WSR N60K) and semi synthetic polymers (HPMC K 15M), it is considered as the best formulation. Analysis of variance (ANOVA) showed no significant difference in the amount of drug released after 15 min for all the formulations, whereas significant differences (p<0.05) was observed for optimized formulations when compared to the other formulations after 24 h.
Fourier Transform Infrared Spectroscopy
Pure lornoxicam spectra showed characteristic peaks at 3090.31 cm-1 (C-H stretching for hetero aromatic functional group), 1596.79 cm-1 (N-H bending group), 1643.29 cm-1 (C=O stretching). as shown in Figure 2.
Optimized formulations F9 also exhibited the characteristic peaks of lornoxicam with no additional peaks observed in the spectra, indicating retention of chemical identity of lornoxicam. However, intensity of peaks corresponding to the drug was reduced or broadened in the optimized formulations possibly due to the mixing of other excipients as shown in Figure 3. The FTIR spectra data confirmed that xanthan gum did not alter the performance characteristics indicating their compatibility of the drug.
CONCLUSIONS
There is no controlled release tablet or bilayered tablet marketed dosage form of lornoxicam, there is only immediate release tablet available which cannot fulfill the criteria to treat chronic pain. This is possible by formulating a biphasic release system which was achieved by bilayered tablet characterized by an initial rapid release by the IR layer followed by controlled release of drug from CR layer of bilayered tablet from the optimized formulation F9 which consists of xanthan gum a natural polymer used in 1:1.5 ratio of drug and polymer ratio. This result obtained was encouraging as xanthan gum has prolonged the release for 24 hrs which is natural in origin. Drug release is dependent on both the type and amount of polymer in the tablet. This biphasic release surmount the major disadvantages of controlled release matrix tablets by providing an additional initial burst release (i.e. loading dose form the IR layer) within 15 min followed by a constant release from controlled release layer over a defined period of time. In the light of the above, it can be concluded that bilayer tablet technology can be significantly used for the treatment of chronic diseases caused by pain.
Received for publication on 02nd July 2011
Accepted for publication on 13th July 2012
- AJIT, K.; MANISH, B. Development and evaluation of regioselective bilayer floating tablets of atenolol and lovastatin for biphasic release profile. Iran. J. Pharm. Res, v.8, p.15-25, 2009.
- AMIDON, G.E.; SECREAST, P.J.; MUDIE, D. Particle, powder and compact characterization. In: QIU, Y.; CHEN, Y.; ZHANG, G.G.Z., LIU, L.; PORTER, W. (Eds.). Developing solid oral dosage forms: pharmaceutical theory and practice. New York: Academic Press, 2009. chap.8, p.163-186.
- BANKER, G.S.; ANDERSON, N.R. Tablets. In: LACHMAN, L.; LIEBERMAN, H. (Eds.). The theory and practice of industrial pharmacy New Delhi: CBS Publishers & Distributors, 2009. p.293-345.
- BETTINI, R.; CATELLANI, P.L.; SANTI, P.; MASSIMO, G.; PEPPAS, N.A.; COLOMBO, P. Translocation of drug particles in HPMC matrix gel layer: effect of drug solubility and influence on release rate. J. Control. Release, v.70, p.383-391, 2001.
- BHISE, K.; SHAIKH, S.; BORA, D. Taste mask, design and evaluation of an oral formulation using ion exchange resin as drug carrier. AAPS Pharm.Sci.Tech., v.9, p.557-562, 2008.
- BOTTI, B.; YOUAN, C. Chronopharmaceutics: gimmick or clinically relevant approach to drug delivery? J. Control. Release, v.98, p.337-353, 2004.
- CHIDAMBARAM, N.; PORTER, W.; FLOOD, K.; QUI, Y. Formulation and characterization of new layered diffusional matrices for zero-order sustained release. J. Control. Release, v.52, p.149-158, 1998.
- COLOMBO, P.; CONTE, U.; GAZZANIGA, A.; MAGGI, L.; SANGALLI, M.E.; PEPAS, N.E.; MANNA, L.A.A. Drug release modulation by physical restrictions of matrix swelling. Int. J. Pharm., v.63, p.43-48, 1990.
- FAHMY, R.H; KASSEM, M.A. Enhancement of famotidine dissolution rate through liquisolid tablets formulation: in vitro and in vivo evaluation. Eur. J. Pharm. Biopharm, v.69, p.993-1003, 2008.
- EIJI, F.; NOBUTERU, M.; KATSUJI U.; MASAO, K. Preparation of enteric coated timed-release press-coated tablets and evaluation of their function by in vitro and in vivo tests for colon targeting. Int. J. Pharm. v.204, p.7-15, 2000.
- HIGUCHI, T. Mechanism of sustained action medication. Theoretical analysis of rate release of solid drugs dispersed in solid matrices. J. Pharm. Sci., v.52, p.1145-1149, 1963.
- INDIAN PHARMACOPOEIA. 2007. Friability of uncoated tablets. Ghaziabad: The Indian Pharmacopoeia Commission, Central Indian Pharmacopoeia Laboratory, Govt. of India, Ministry of Health & Family Welfare, 2008. p.183.
- INDIAN PHARMACOPOEIA. 2007. Uniformity of content of single-dose preparations. Ghaziabad: The Indian Pharmacopoeia Commission, Central Indian Pharmacopoeia Laboratory, Govt. of India, Ministry of Health & Family Welfare, 2008. p.182.
- INDIAN PHARMACOPOEIA. 2007. Uniformity of weight of single-dose preparations. Ghaziabad: The Indian Pharmacopoeia Commission, Central Indian Pharmacopoeia Laboratory, Govt. of India, Ministry of Health & Family Welfare, 2008. p.182.
- KENNETH, C.W.; MICHAEL B.F. Press-coating of immediate release powders onto coated controlled release tablets with adhesives. J. Control. Release, v.89, p.387-395, 2003.
- KORSMEYER, R.; GURNY, R.; PEPPAS, N. Mechanisms of solute release from porous hydrophilic polymers. Int. J. Pharm., v.15, p.25-35, 1983.
- KULKARNI, A.S.; ALOORKAR, N.H.; MADHAV, S.M.; JAYASHREE, B.G. Liquisolid systems: a review. Int. J. Pharm. Sci. Nanotech., v.3, p.795-802, 2010.
- KULKARNI, A.S.; GAJA, J.B. Formulation and evaluation of liquisolid compacts of diclofenac sodium. PDA J. Pharm. Sci. Tech., v.64, p.222-232, 2010.
- LAZARUS, J.; COOPER, J. Absorption, testing, and clinical evaluation of oral prolonged-action drugs. J. Pharm. Sci., v.50, p.715-732, 1961.
- LI, Y.H.; ZHU, J.B. Modulation of combined-release behaviors from a novel "tablets-in-capsule system. J. Control. Release, v.95, p.381-389, 2004.
- LUANA, P.; VALERIA, A.; STEFANO, G.; MAURIZIO, R.; PAOLO, B.; CARLO, R. Mucoadhesive bilayered tablets for buccal sustained release of flurbiprofen. AAPS PharmSciTech, v.8, p.E20-E27, 2007.
- MAGGI, L.; MACHISTE, E.O.; TORRE, M.L.; CONTE. U. Formulation of biphasic release tablets containing slightly soluble drugs. Eur. J. Pharm. Biopharm., v.48, p.37-42, 1999.
- MORITA, R.; HONDA, R.; TAKAHASHI, Y. Development of oral controlled release preparations, a PVA swelling controlled release system (SCRS) - I. Design of SCRS and its release controlling factor. J. Control. Release, v.63, p.297-304, 2000.
- NEIL, M.S.; NEAL, M.D. Clinical pharmacokinetics of lornoxicam a short half-life oxicam. Clin. Pharmacokinet, v.34, p.421-428, 1998.
- PEPPAS, N.A. Analysis of Fickian and non-Fickian drug release from polymers. Pharm. Acta. Helv., v.60, p.110-111, 1985.
- PERIOLI, L.; AMBROGI, V.; GIOVAGNOLI, S.; RICCI, M.; BLASI, P.; ROSSI, C. Mucoadhesive bilayered tablets for buccal sustained release of flurbiprofen. AAPS Pharm.Sci.Tech., v.8, p.E1-E8, 2007.
- PETROVIC, J.; IBRIC, S.; BETZ, G.; PAROJCIC, J.; DURIC, Z. Application of dynamic neural networks in the modeling of drug release from polyethylene oxide matrix tablets. Eur. J. Pharm. Sci, v.38, p.172-180, 2009.
- PHARM, A.T.; LEE, P.I. Probing the mechanism of drug release from hydroxypropyl methylcellulose matrices. Pharm. Res v.11, p.1379-1384, 1994.
- QIU, Y.; CHIDAMBARAM, N.; FLOOD, K. Design and evaluation of layered diffusional matrices for zero-order sustained release. J. Control. Release, v.51, p.23-130, 1998.
- REMYA, P.N.; DAMODHARAN, N.; SULAKSHAN KUMAR, C.V. Formulation and evaluation of bilayered tablets of ibuprofen and methocarbamol. Int. J. Pharm. Tech. Res., v.2, p.1250-1255, 2010.
- SHAH, R.B.; TAWAKKUL, M.A.; KHAN, M.A. Comparative evaluation of flow for pharmaceutical powders and granules. AAPS Pharm.Sci.Tech., v.9, p.250-258, 2008.
- SUNIL, R.; PAVAN KUMAR, P.; RAJANARAYANA, K.; MADHUSUDAN RAO, Y. Formulation and release characteristic of a bilayer matrix tablet containing glimepride immediate release component and metformin hydrochloride as sustained release component. Int. J. Pharm. Sci. Nanotech., v.3, p.851-859., 2010.
- UNITED STATES PHARMACOPOEIA 32. NATIONAL FORMULARY 27. Powder flow. Rockville: United States Pharmacopeial Convention Inc., 2012. v.1, p.801-804.
- WAGNER, J.G. Interpretation of percent dissolved-time plots derived from In vitro testing of conventional tablets and capsules. J. Pharm. Sci., v.58, p.1253-1257, 1969.
- WAI-YIP, L.T.; ROBINSON, R.J. Controlled drug delivery systems. In: GENNARO, A.R., (Ed.). Remington: the science and practice of pharmacy. 20.ed. Philadelphia: University of Sciences in Philadelphia, 2000. p.903-929.
- WHITE, W.B.; MEHROTRA, D.V.; BLACK, H.R.; FAKOUHI, T.D. Effects of controlled-onset extended-release verapamil on nocturnal blood pressure (dippers versus nondippers). Am. J. Cardiol., v.80, p.469-474, 1997.
Publication Dates
-
Publication in this collection
14 Feb 2013 -
Date of issue
Dec 2012
History
-
Received
02 July 2011 -
Accepted
13 July 2012