Abstracts
Average bioequivalence of two 500 mg levofloxacin formulations available in Brazil, Tavanic(c) (Sanofi-Aventis Farmacêutica Ltda, Brazil, reference product) and Levaquin(c) (Janssen-Cilag Farmacêutica Ltda, Brazil, test product) was evaluated by means of a randomized, open-label, 2-way crossover study performed in 26 healthy Brazilian volunteers under fasting conditions. A single dose of 500 mg levofloxacin tablets was orally administered, and blood samples were collected over a period of 48 hours. Levofloxacin plasmatic concentrations were determined using a validated HPLC method. Pharmacokinetic parameters Cmax, Tmax, Kel, T1/2el, AUC0-t and AUC0-inf were calculated using noncompartmental analysis. Bioequivalence was determined by calculating 90% confidence intervals (90% CI) for the ratio of Cmax, AUC0-t and AUC0-inf values for test and reference products, using logarithmic transformed data. Tolerability was assessed by monitoring vital signs and laboratory analysis results, by subject interviews and by spontaneous report of adverse events. 90% CIs for Cmax, AUC0-t and AUC0-inf were 92.1% - 108.2%, 90.7% - 98.0%, and 94.8% - 100.0%, respectively. Observed adverse events were nausea and headache. It was concluded that Tavanic(c) and Levaquin(c) are bioequivalent, since 90% CIs are within the 80% - 125% interval proposed by regulatory agencies.
Levofloxacin/bioequivalence; Pharmacokinetics; High-performance liquid chromatography/qualitative analysis; Tavanic(c)/bioequivalence; Levaquin(c)/bioequivalence
A bioequivalência média de duas formulações de levofloxacino disponíveis no Brasil, Tavanic(c) (Sanofi-Aventis Farmacêutica Ltda, Brasil, produto referência) e Levaquin(c) (Janssen-Cilag Farmacêutica Ltda, Brasil, produto teste) foi determinada por meio da realização de ensaio aleatório, aberto, cruzado, com dois períodos e duas sequências, em 26 voluntários sadios em condições de jejum. Amostras de sangue dos voluntários foram obtidas ao longo de um período de 48 horas após administração de dose única de 500 mg de levofloxacino. As concentrações plasmáticas do fármaco foram determinadas por método cromatográfico validado. Os parâmetros farmacocinéticos Cmax, Tmax, Kel, T1/2el, AUC0-t e AUC0-inf foram calculados por análise não compartimental. A bioequivalência foi determinada pelo cálculo de intervalos de confiança 90% (IC 90%) para as razões entre os valores de Cmax, AUC0-t e AUC0-inf obtidos para os produtos teste e referência, usando dados transformados logaritmicamente. A tolerabilidade foi avaliada pelo acompanhamento dos sinais vitais e resultados de exames laboratoriais, por consultas e por relato espontâneo dos voluntários. ICs 90% para Cmax, AUC0-t e AUC0-inf foram 92.1% - 108.2%, 90.7% - 98.0%, e 94.8% - 100.0%, respectivamente. Os eventos adversos observados foram náusea e cefaleia. Concluiu-se que os produtos Tavanic(c) e Levaquin(c) são bioequivalentes, uma vez que os ICs 90% estão dentro da faixa de 80%-125% proposta pelas agências reguladoras
Levofloxacino/bioequivalência; Farmacocinética; Cromatografia líquida de alta eficiência/análise qualitativa; Tavanic(c)/bioequivalência; Levaquin(c)/bioequivalência
INTRODUCTION
Levofloxacin is the synthetic L-isomer of the racemic quinolone ofloxacin (Anderson, Perry, 2008ANDERSON, V. R.; PERRY, C. M. Levofloxacin: a review of its use as a high-dose, short-course treatment for bacterial infection. Drugs, v.68, p.535-565, 2008.; Siewert, 2006SIEWERT, S. Validation of a levofloxacin HPLC assay in plasma and dialysate for pharmacokinetic studies. J. Pharm. Biomed., v.41, p.1360-1362, 2006.). The drug is administered to treat infectious diseases, e.g. community acquired and nosocomial pneumonia, skin and skin structure infections and urinary tract infections (Siewert, 2006SIEWERT, S. Validation of a levofloxacin HPLC assay in plasma and dialysate for pharmacokinetic studies. J. Pharm. Biomed., v.41, p.1360-1362, 2006.). It has a broad spectrum antibacterial profile including Gram-positive as well as Gram-negative bacteria, and atypical organisms (Anderson, Perry, 2008ANDERSON, V. R.; PERRY, C. M. Levofloxacin: a review of its use as a high-dose, short-course treatment for bacterial infection. Drugs, v.68, p.535-565, 2008.; Croom, Goa, 2003CROOM, K. F.; GOA, K. L. Levofloxacin: a review of its use in the treatment of bacterial infection in the United States. Drugs, v.63, p.2769-2802, 2003.; Noreddin, Haynes, Zhanel, 2005NOREDDIN, A. M.; HAYNES, V. L.; ZHANEL, G. G. Pharmacokinetics and pharmacodynamics of the new quinolones. J. Pharm. Pract., v.18, p.432-443, 2005.; Takahashi, Hayakawa, Akimoto, 2003TAKAHASHI, H.; HAYAKAWA, I.; AKIMOTO, T. The history of the development and changes of quinolone antibacterial agents. Yakushigaku Zasshi, v.38, p.161-179, 2003.). Levofloxacin is well tolerated, and is associated with few of the phototoxic, cardiac or hepatic adverse events seen with some other quinolones. Adverse events occurring in ≥2% of patients receiving once-daily levofloxacin 750 mg for 5 days or once-daily levofloxacin 500 mg for 10 days are nausea, headache, diarrhea, insomnia, constipation, abdominal pain, dizziness, dyspepsia and vomiting. Rare adverse effects associated with levofloxacin include photosensitivity/phototoxicity reactions after exposure to the sun or ultraviolet (UV) light, C. difficile diarrhoea which may vary in severity from mild to fatal colitis, tendon ruptures, sensory or sensorimotor axonal poly-neuropathy, and hypersensitivity (Anderson, Perry, 2008ANDERSON, V. R.; PERRY, C. M. Levofloxacin: a review of its use as a high-dose, short-course treatment for bacterial infection. Drugs, v.68, p.535-565, 2008.; Croom, Goa, 2003CROOM, K. F.; GOA, K. L. Levofloxacin: a review of its use in the treatment of bacterial infection in the United States. Drugs, v.63, p.2769-2802, 2003.).
Oral levofloxacin undergoes complete absorption, and the time to maximum concentration (Tmax) occurs 1-2 hours after a 500 or 750 mg oral dose, in subjects with normal renal function. The absolute bioavailability is 99% or greater and the oral solution or tablet formulation have the same bioavailability as the intravenous formulation (Anderson, Perry, 2008ANDERSON, V. R.; PERRY, C. M. Levofloxacin: a review of its use as a high-dose, short-course treatment for bacterial infection. Drugs, v.68, p.535-565, 2008.). Levofloxacin serum half-life is 6-7 hours (Lee et al., 1997LEE, L. J.; HAFKIN, B.; LEE, I. D.; HOH, J.; DIX, R. Effects of food and sucralfate on a single oral dose of 500 milligrams of levofloxacin in healthy subjects. Antimicrob. Agents Chemother., v.41, p.2196-2200, 1997.; Lubasch et al., 2000LUBASCH, A.; KELLER, I.; BORNER, K.; KOEPPE, P.; LODE, H. Comparative pharmacokinetics of ciprofloxacin, gatifloxacin, grepafloxacin, levofloxacin, trovafloxacin, and moxifloxacin after single oral administration in healthy volunteers. Antimicrob. Agents Chemother., v.44, p.2600-2603, 2000.; Wagenlehner et al., 2006WAGENLEHNER, F. M.; KINZIG-SCHIPPERS, M.; TISCHMEYER, U.; WAGENLEHNER, C.; SÖRGEL, F.; DALHOFF, A.; NABER, K. G. Pharmacokinetics of ciprofloxacin XR (1000 mg) versus levofloxacin (500 mg) in plasma and urine of male and female healthy volunteers receiving a single oral dose. Int. J. Antimicrob. Ag., v.27, p.7-14, 2006.). It has a large volume of distribution allowing for penetration in various cells and tissues, including alveolar cells or macrophages and paranasal sinuses mucosa, where it can achieve concentrations that surpass plasma levels after 2-4 hours of administration (Anderson, Perry, 2008ANDERSON, V. R.; PERRY, C. M. Levofloxacin: a review of its use as a high-dose, short-course treatment for bacterial infection. Drugs, v.68, p.535-565, 2008.; Croom, Goa, 2003CROOM, K. F.; GOA, K. L. Levofloxacin: a review of its use in the treatment of bacterial infection in the United States. Drugs, v.63, p.2769-2802, 2003.). Pharmacokinetics of levofloxacin is not affected by age, gender, race, HIV (human immunodeficiency virus) status or the presence of serious community-acquired bacterial infections (Anderson, Perry, 2008ANDERSON, V. R.; PERRY, C. M. Levofloxacin: a review of its use as a high-dose, short-course treatment for bacterial infection. Drugs, v.68, p.535-565, 2008.). However, Tmax and maximum plasma concentration (Cmax) were affected by consumption of a high-fat meal, although the extent of absorption was not affected (Rodvold, Neuhauser, 2001RODVOLD, K. A.; NEUHAUSER, M. Pharmacokinetics and pharmacodynamics of fluoroquinolones. Pharmacotherapy, v.21, p.233S-252S, 2001.). Because renal clearance accounts for most of total body clearance of levofloxacin, its pharmacokinetics is affected by renal impairment but not by hepatic impairment (Rodvold, Neuhauser, 2001RODVOLD, K. A.; NEUHAUSER, M. Pharmacokinetics and pharmacodynamics of fluoroquinolones. Pharmacotherapy, v.21, p.233S-252S, 2001.). Levofloxacin and newer quinolones, when compared with older agents such as ciprofloxacin, achieve higher peak serum concentrations (Cmax), resulting in higher Cmax/MIC (minimum inhibitory concentration) ratios, which result in excellent bacterial killing (Noreddin, Haynes, Zhanel, 2005NOREDDIN, A. M.; HAYNES, V. L.; ZHANEL, G. G. Pharmacokinetics and pharmacodynamics of the new quinolones. J. Pharm. Pract., v.18, p.432-443, 2005.).
The bioavailability of a drug product is defined as the rate and extent to which the active ingredient or therapeutic moiety is absorbed and becomes available at the site of drug action. Two drug products are considered to be bioequivalent if they are pharmaceutical equivalents (i.e., similar dosage forms made, perhaps, by different manufacturers) or pharmaceutical alternatives (i.e., different dosage forms) and if their rates and extents of absorption do not show a significant difference when administered at the same molar dose of the therapeutic moiety under similar experimental conditions (Chow, Liu, 2000CHOW, S. C.; LIU, J. P. Design and analysis of bioavailability and bioequivalence Studies. 2.ed. New York: Marcel Dekker, 2000. 584 p.).
Bioavailability and bioequivalence studies provide important information in the overall set of data that ensure the availability of safe and effective medicines to patients and clinicians. These studies gained increasing attention during the last decades, after it became evident that marketed products having same amounts of the same drug may exhibit marked differences between their therapeutic responses. In many instances, these differences were correlated successfully to dissimilar drug blood levels caused mainly by impaired absorption (Abdou, 1989ABDOU, H. M. Dissolution, bioavailability and bioequivalence. Easton: Mack Publishing Company, 1989. 554 p.; Chen et al., 2001CHEN, M. L.; SHAH, V.; PATNAIK, R.; ADAMS, W.; HUSSAIN, A.; CONNER, D.; MEHTA, M.; MALINOWSKI, H.; LAZOR, J.; HUANG, S. M,, HARE, D.; LESKO, L.; SPORN, D.; WILLIAMS, R. Bioavailability and bioequivalence: an FDA regulatory overview. Pharm. Res., v.18, p.1645-1650, 2001.).
According to international criteria (Agência Nacional de
Vigilância Sanitária, 2006AGÊNCIA NACIONAL DE VIGILÂNCIA SANITÁRIA. Guia para provas de
biodisponibilidade relativa/bioequivalência de medicamentos. 2006. Available at:
<http://www.in.gov.br/visualiza/index.jsp?data=24/04/2006&jornal=1&pagina=101&totalArquivos=128>.
Accessed on: 03 May 2013.
http://www.in.gov.br/visualiza/index.jsp...
; European Medicines
Agency, 2010EUROPEAN MEDICINES AGENCY. Guideline on the investigation of
bioequivalence. Committee for Medicinal Products for Human Use: 2010. 27p. Available
at
<http://www.emea.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf>.
Accessed on: 02 May 2013.
http://www.emea.europa.eu/docs/en_GB/doc...
; FDA, 2001FOOD AND DRUG ADMINISTRATION. FDA. Guidance for Industry: Statistical
approaches to establishing bioequivalence. U.S. Department of Health and Human
Services: 2001. 45p. Available at:
<http://www.fda.gov/downloads/Drugs/Guidances/ucm070244.pdf>. Accessed on: 02
May 2013.
http://www.fda.gov/downloads/Drugs/Guida...
), average
bioequivalence between two pharmaceutical products is determined by comparing its
bioavailabilities through the assessment of rate and extension of drug absorption and
through the calculation of 90% confidence intervals (90% CI) for the ratio of
Cmax, and AUC (area under the plasma concentration-time curve) values for
both products, using logarithmic transformed data. The products are considered
bioequivalent if all 90% CI fall within 80%-125%.
Especially in antimicrobial therapy, bioequivalence studies are very important, since the dosage form must provide effective plasma concentration, thereby assuring the elimination of the microorganism, which causes the infection. Should the plasma concentration fall below the level required to ensure efficacy, the infection will not be eradicated and the risk of developing resistance to the drug may increase (Porta, Chang, Storpirtis, 2005PORTA, V.; CHANG, K. H.; STORPIRTIS, S. Evaluation of the bioequivalence of capsules containing 150 mg of fluconazole. Int. J. Pharm., v.288, p.81-86, 2005.).
Several Brazilian pharmaceutical industries currently produce or import levofloxacin tablets formulations. These formulations must prove bioequivalence to the reference product to be marketed in Brazil. Thus, the aim of the present study was to compare the pharmacokinetic profile and to evaluate the bioequivalence of two 500 mg levofloxacin formulations available in Brazil, Tavanic(c) (Sanofi-Aventis Farmacêutica Ltda, Brazil, reference product) and Levaquin(c) (Janssen-Cilag Farmacêutica Ltda, Brazil, test product) for regulatory approval of Levaquin(c).
SUBJECTS AND METHODS
Ethics committee review and selection of volunteers
The study was carried out in accordance with the principles of the Declaration of
Helsinki and its amendments (World Medical
Association, 2008WORLD MEDICAL ASSOCIATION. WMA Declaration of Helsinki: ethical
principles for medical research involving human subjects. 2008. Available at:
<http://www.wma.net/en/30publications/10policies/b3/index.html>. Accessed on:
03 May 2013.
http://www.wma.net/en/30publications/10p...
) and the International Conference on Harmonization
Guideline for Good Clinical Practice (International
Conference on Harmonization, 1996INTERNATIONAL CONFERENCE ON HARMONIZATION. Guideline for good clinical
practice E6(R1). International Conference on Harmonisation of Technical Requirements
for Registration of Pharmaceuticals for Human Use: 1996. 53p. Available at:
<http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6_R1/Step4/E6_R1__Guideline.pdf>.
Accessed on: 03 May 2013.
http://www.ich.org/fileadmin/Public_Web_...
). The protocol was approved by the Ethics
Committee of the College of Medical Sciences of the University of Campinas.
Medical history, physical examination, 12-lead electrocardiography, and laboratory tests (hematology, blood biochemistry, hepatic function and urinalysis) were carried out prior to the beginning of the study and at its conclusion. Inclusion criteria were weight within 15% of ideal body weight, absence of heart, kidneys, neurological or metabolic diseases and no history of drug hypersensitivity. Exclusion criteria were abnormal values on physical examination, eletrocardiography or laboratory tests, ongoing pharmacological treatment and pregnancy. Volunteers were instructed to abstain from taking any medication one week before and during the study.
Study design and drug administration
The bioequivalence of levofloxacin tablet at a single dose of 500 mg was assessed under fasting conditions, in an open, randomized, 2x2 crossover trial, with a washout period of 6 days. Although Tmax and Cmax could be affected by consumption of a high-fat meal, effect of food was not tested, since it is not required in Brazil for registration of immediate release oral formulations.
Subjects were admitted into hospital at 9:00p.m., the day before the drug administration and were randomly assigned to group A or B. During the first period, volunteers from group A received a single 500 mg dose of reference product (Tavanic(c), Sanofi-Aventis Famacêutica Ltda, Brazil), while volunteers from group B received a single 500 mg dose of test product (Levaquin(c), Janssen-Cilag Farmacêutica Ltda, Brazil), according to a randomization schedule. In the second study period, the order was reversed.
The products were administered in the morning with 200 mL of water after a 10 hours fasting period. No food was allowed for 4 hours after ingestion of the dose. Subjects were provided with standard meals 4 hours (lunch), 7 hours (snack) and 10 hours (supper) after drug administration in each treatment. Volunteers did not ingest any alcoholic drink, coffee or xanthine containing drinks or foods during the trial.
Tolerability
Tolerability was assessed by a clinician by monitoring vital signs (blood pressure in the upper arm using sphygmomanometry, and heart rate) at 1.0, 2.0, 4.0, 6.0, 8.0, 12.0, 24.0, 36.0 e 48.0 hours after drug administration, by interviewing subjects at each sampling time and at each meal, and by registering adverse events spontaneously reported during the course of the study.
Sample collection and processing
Sample collection was achieved by means of an indwelling venous cannula of the cubital vein maintained with 1 ml of heparin diluted with normal saline solution to a final concentration of 50 mg/L. Eight milliliters of venous blood were collected to heparinized tubes according to a time schedule which included a blank before-drug sample just prior to dosing (predose) and samples at 0.5, 1.0, 1.5, 2.0, 2.5, 3.0, 3.5, 4.0, 6.0, 8.0, 12.0, 24.0, 36.0 and 48.0 hours after drug administration. One milliliter of blood was collected and discarded before each sample collection. Plasma was immediately separated by centrifugation at 1900 g for 10 min under refrigeration (4 °C). Plasma samples were divided into two aliquots, transferred to labeled tubes, and stored at -20 °C until analytic assay.
Analysis of plasma samples
For the determination of levofloxacin plasmatic levels, an accurate, precise and sensitive high performance liquid chromatography (HPLC) method with UV detection was developed and validated, based on previously described methods (Almeida et al., 2005ALMEIDA, S.; FILIPE, A.; ALMEIDA, A.; WONG, H.; CAPARROS, N.; TANGUAY, M. Comparative bioavailability of two formulations of levofloxacin and effect of sex on bioequivalence analysis. Data from a randomized, 2x2 crossover trial in healthy volunteers. Arzneimittel Forsch., v.55, p.414-419, 2005.; Baietto et al., 2009BAIETTO, L.; D'AVOLIO, A.; DE ROSA, F. G.; GARAZZINO, S.; PATANELLA, S.; SICCARDI, M.; SCIANDRA, M.; DI PERRI, G. Simultaneous quantification of linezolid, rifampicin, levofloxacin, and moxifloxacin in human plasma using high-performance liquid chromatography with UV. Ther. Drug Monit., v.31, p.104-109, 2009.; Chien et al., 1997CHIEN, S. C.; ROGGE, M. C.; GISCLON, L. G.; CURTIN, C.; WONG, F.; NATARAJAN, J.; WILLIAMS, R. R.; FOWLER, C. L.; CHEUNG, W. K.; CHOW, A. T. Pharmacokinetic profile of levofloxacin following once-daily 500-milligram oral or intravenous doses. Antimicrob. Agents Chemother., v.41, p.2256-2260, 1997., 1998CHIEN, S. C.; WONG, F. A.; FOWLER, C. L.; CALLERY-D'AMICO, S. V.; WILLIAMS, R. R.; NAYAK, R.; CHOW, A. T. Double-blind evaluation of the safety and pharmacokinetics of multiple oral once-daily 750-milligram and 1-gram doses of levofloxacin in healthy volunteers. Antimicrob. Agents Chemother. v.42, p.885-888, 1998.; Conte et al., 2006CONTE JR.; J. E.; GOLDEN, J. A.; MCLVER, M.; ZURLINDEN, E. Intrapulmonary pharmacokinetics and pharmacodynamics of high-dose levofloxacin in healthy volunteer subjects. Int. J. Antimicrob. Ag., v.28, p.114-121, 2006.; Djabarouti et al., 2004DJABAROUTI, S.; BOSELLI, E.; ALLAOUCHICHE, B.; BA, B.; NGUYEN, A. T.; GORDIEN, J. B.; BERNADOU, J. M.; SAUX, M. C.; BREILH, D. Determination of levofloxacin in plasma, bronchoalveolar lavage and bone tissues by high-performance liquid chromatography with ultraviolet detection using a fully automated extraction method. J. Chromatogr. B, v.799, p.165-172, 2004.; Ji et al., 2006JI, H. Y.; JEONG, D. W.; KIM, Y. H.; KIM, H. H.; SOHN, D. R.; LEE, H. S. Hydrophilic interaction liquid chromatography-tandem mass spectrometry for the determination of levofloxacin in human plasma. J. Pharm. Biomed., v.41, p.622-627, 2006.; Lee et al., 1997LEE, L. J.; HAFKIN, B.; LEE, I. D.; HOH, J.; DIX, R. Effects of food and sucralfate on a single oral dose of 500 milligrams of levofloxacin in healthy subjects. Antimicrob. Agents Chemother., v.41, p.2196-2200, 1997.; Liang, Kays, Sowinski, 2002LIANG, H.; KAYS, M. B.; SOWINSKI, K. M. Separation of levofloxacin, ciprofloxacin, gatifloxacin, moxifloxacin, trovafloxacin and cinoxacin by high-performance liquid chromatography: application to levofloxacin determination in human plasma. J. Chromatogr. B, v.772, p.53-63, 2002.; Lubasch et al., 2000LUBASCH, A.; KELLER, I.; BORNER, K.; KOEPPE, P.; LODE, H. Comparative pharmacokinetics of ciprofloxacin, gatifloxacin, grepafloxacin, levofloxacin, trovafloxacin, and moxifloxacin after single oral administration in healthy volunteers. Antimicrob. Agents Chemother., v.44, p.2600-2603, 2000.; Nemutlu et al., 2007NEMUTLU, E.; KIR, S.; ÖZYÜNCÜ, Ö.; BEKSAÇ, M. S. Simultaneous separation and determination of seven quinolones using HPLC: analysis of levofloxacin and moxifloxacin in plasma and amniotic fluid. Chromatographia, v.66, p.S15-S24, 2007.; Schulte et al., 2006SCHULTE, S.; ACKERMANN, T.; BERTRAM, N.; SAUERBRUCH, T.; PAAR, W. D. Determination of the newer quinolones levofloxacin and moxifloxacin in plasma by high-performance liquid chromatography with fluorescence detection. J. Chromatogr. Sci., v.44, p.205-208, 2006.; Siewert, 2006SIEWERT, S. Validation of a levofloxacin HPLC assay in plasma and dialysate for pharmacokinetic studies. J. Pharm. Biomed., v.41, p.1360-1362, 2006.; Wagenlehner et al., 2006WAGENLEHNER, F. M.; KINZIG-SCHIPPERS, M.; TISCHMEYER, U.; WAGENLEHNER, C.; SÖRGEL, F.; DALHOFF, A.; NABER, K. G. Pharmacokinetics of ciprofloxacin XR (1000 mg) versus levofloxacin (500 mg) in plasma and urine of male and female healthy volunteers receiving a single oral dose. Int. J. Antimicrob. Ag., v.27, p.7-14, 2006.; Wong, Juzwin, Flor, 1997WONG, F. A.; JUZWIN, S. J.; FLOR, S. C. Rapid stereospecific high-performance liquid chromatographic determination of levofloxacin in human plasma and urine. J. Pharm. Biomed., v.15, p.765-771, 1997.; Zhou et al., 2007ZHOU, Z. L.; YANG, M.; YU, X. Y.; PENG, H. Y.; SHAN, Z. X.; CHEN, S. Z.; LIN, Q. X.; LIU, X. Y.; CHEN, T. F.; ZHOU, S. F.; LIN, S. G. A rapid and simple high-performance liquid chromatography method for the determination of human plasma levofloxacin concentration and its application to bioequivalence studies. Biomed. Chromatogr., v.21, p.1045-1051, 2007.).
Metronidazol was used as internal standard (IS). Briefly, 250 μL of plasma and 25 μL of IS solution (80 μg/mL) were added to a 8 mL glass test tube and vortex-mixed for 30 seconds; after this procedure, a 2 mL volume of acetonitrile was added for protein precipitation. Samples were centrifuged for 10 minutes at 1900 g. The supernatant layer was filtered through a Millex GV 0.45 μm filter unit into an 8 mL conical glass tube and solvent was evaporated under a nitrogen stream while immersed in a 40 °C water bath. Each sample was reconstituted with 250 μL of mobile phase and vortex-mixed for 30 seconds. The samples were transferred to auto-sampler vials and 25 μL were injected into the HPLC system. Chromatographic separation was performed in a Gemini C18 column, 150 × 4.6 mm I.D., 5 μm particle size (Phenomenex(r), Torrance, USA), protected with an AJO-4287 C18 guard cartridge, 5 × 4.6 mm I.D., 5 μm particle size (Phenomenex(r), Torrance, USA). The mobile phase consisted of a solution of sodium dihydrogen phosphate monohydrate (25 mM) and acetonitrile in the ratio of 87:13 (v/v), without pH adjustment, filtered through a 0.22 μm filter and ultrasonically degassed for 15 min. The chromatographic column was maintained at 40 ºC during the analysis. The mobile phase flow-rate was set at 1.2 mL/min and the detection wavelength, at 280 nm.
Method validation was achieved through determination of specificity, recovery, linearity, lower limit of quantification (LLOQ), precision, accuracy and stability using standard plasma samples (0.25 μg/mL to 10.00 μg/mL) and quality control plasma samples (0.75 μg/mL, 4.00 μg/mL and 8.00 μg/mL). Preparation of calibration standard plasma samples was accomplished daily by spiking drug-free plasma with known amounts of levofloxacin. Quality control plasma samples were prepared by adding known amounts of levofloxacin to drug-free plasma, aliquotation and storage at -20 ºC. Specificity was investigated by analyzing four normal plasma samples, one hemolised plasma sample and one lipemic plasma sample for interference of endogenous compounds. The anticoagulant (heparin) interference was also verified during this stage. Recoveries of levofloxacin and IS were evaluated by comparing chromatographic peak areas from standard samples which did not undergo sample pre-treatment and from extracted plasma samples at quality control concentrations. The linearity was determined by the calibration curve ranging from 0.25 μg/mL to 10.00 μg/mL, using least-squares linear regression analysis. The LLOQ was the smallest analytical concentration which could be measured with accuracy and precision still better than 20%. Intra- and inter-assay precision and accuracy were determined by repeated analysis of quality control plasma samples on the same day and on different days. Stability of levofloxacin in plasma was evaluated after three freezing-thaw cycles and after storage at -20 ºC for 63 days. Additionally, stability of processed plasma samples during storage in the auto sampler for 24 and 48 hours at room temperature was determined.
Pharmacokinetic and statistical analysis
The following pharmacokinetic parameters were calculated using a non-compartmental model: Cmax, Tmax, AUC0-t (area under the plasma concentration-time curve from time zero to sample time of the last measurable levofloxacin concentration), AUC0-inf (area under the plasma concentration-time curve from time zero to infinity), Kel (elimination rate constant) and T1/2el (elimination half-life). Cmax and Tmax were obtained directly from the concentration-time curve. AUC0-t was calculated using the linear trapezoidal method. Kel was calculated by applying a log-linear regression analysis to at least the last three quantifiable concentrations of levofloxacin and T1/2el was calculated as 0.693/Kel (Dipiro et al., 1996DIPIRO, J. T.; BLOUIN, R. A.; PRUEMER, J. M.; SPRUILL, W. J. Concepts in clinical pharmacokinetics. 2.ed. Bethesda: American Society of Health-System Pharmacists, 1996. 208 p.). AUC0-inf was calculated as AUC0-t + Ct/Kel, where Ct is the last measurable levofloxacin concentration (Dipiro et al., 1996DIPIRO, J. T.; BLOUIN, R. A.; PRUEMER, J. M.; SPRUILL, W. J. Concepts in clinical pharmacokinetics. 2.ed. Bethesda: American Society of Health-System Pharmacists, 1996. 208 p.).
A sample size of 26 volunteers was chosen to evaluate bioequivalence with 80% power and to account for dropouts or withdrawals.
Analysis of variance (ANOVA) was performed on logarithmic transformed values of the pharmacokinetic parameters Cmax, AUC0-t and AUC0-inf. The ANOVA model included sequence, formulation, and period as fixed effects and subjects nested within sequence as random effect. These effects were tested at the 5% level of significance (Chow, Liu, 2000CHOW, S. C.; LIU, J. P. Design and analysis of bioavailability and bioequivalence Studies. 2.ed. New York: Marcel Dekker, 2000. 584 p.).
Bioequivalence between the products was determined by calculating 90% confidence
intervals (90% CI) for the ratio of Cmax and AUC0-t values for
the test and reference products, using logarithmic transformed data. The products
were considered bioequivalent if the 90% CI for AUC0-t and Cmax
fell within 80-125% (Agência Nacional de Vigilância
Sanitária, 2006AGÊNCIA NACIONAL DE VIGILÂNCIA SANITÁRIA. Guia para provas de
biodisponibilidade relativa/bioequivalência de medicamentos. 2006. Available at:
<http://www.in.gov.br/visualiza/index.jsp?data=24/04/2006&jornal=1&pagina=101&totalArquivos=128>.
Accessed on: 03 May 2013.
http://www.in.gov.br/visualiza/index.jsp...
; European Medicines Agency,
2010EUROPEAN MEDICINES AGENCY. Guideline on the investigation of
bioequivalence. Committee for Medicinal Products for Human Use: 2010. 27p. Available
at
<http://www.emea.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf>.
Accessed on: 02 May 2013.
http://www.emea.europa.eu/docs/en_GB/doc...
; FDA, 2001).
RESULTS
Tolerability
A total of 26 healthy volunteers, 13 females and 13 males, were included in the study after signing a consent form. The mean (range) age, weight, and height of the subjects were 33 (21-43) years; 68 (49-86) kg; and 166 (146-178) cm respectively. During the first period of the study, six volunteers from group A (reference product) and three from group B (test product) reported headache and one volunteer from group A reported nausea. During the second period, headache was reported by one volunteer from group A (test product) and two from group B (reference product) and nausea was reported by one volunteer from group B. No clinical abnormalities were found at the conclusion of the study.
Method validation and drug concentration in plasma
The HPLC method for levofloxacin quantification was specific for levofloxacin and IS, with total run time of 10 minutes, and was linear in the range of 0.25 μg/mL to 10.00 μg/mL. Intra and inter-assay accuracy ranged from 94.8% to 100.6%, and from 95.2% to 105.9%, respectively. Precision, measured as percent relative standard deviation (RSD%), was between 1.1% and 1.8%, intra-assay, and between 1.9% and 6.3%, inter-assay. Mean recovery was 83.3% for levofloxacin and 86.4% for IS. The processed samples were stable at room temperature for at least 48 hours. Levofloxacin concentrations were not altered after three freeze-thaw cycles and plasma samples were stable for at least 63 days when stored at -20 ºC.
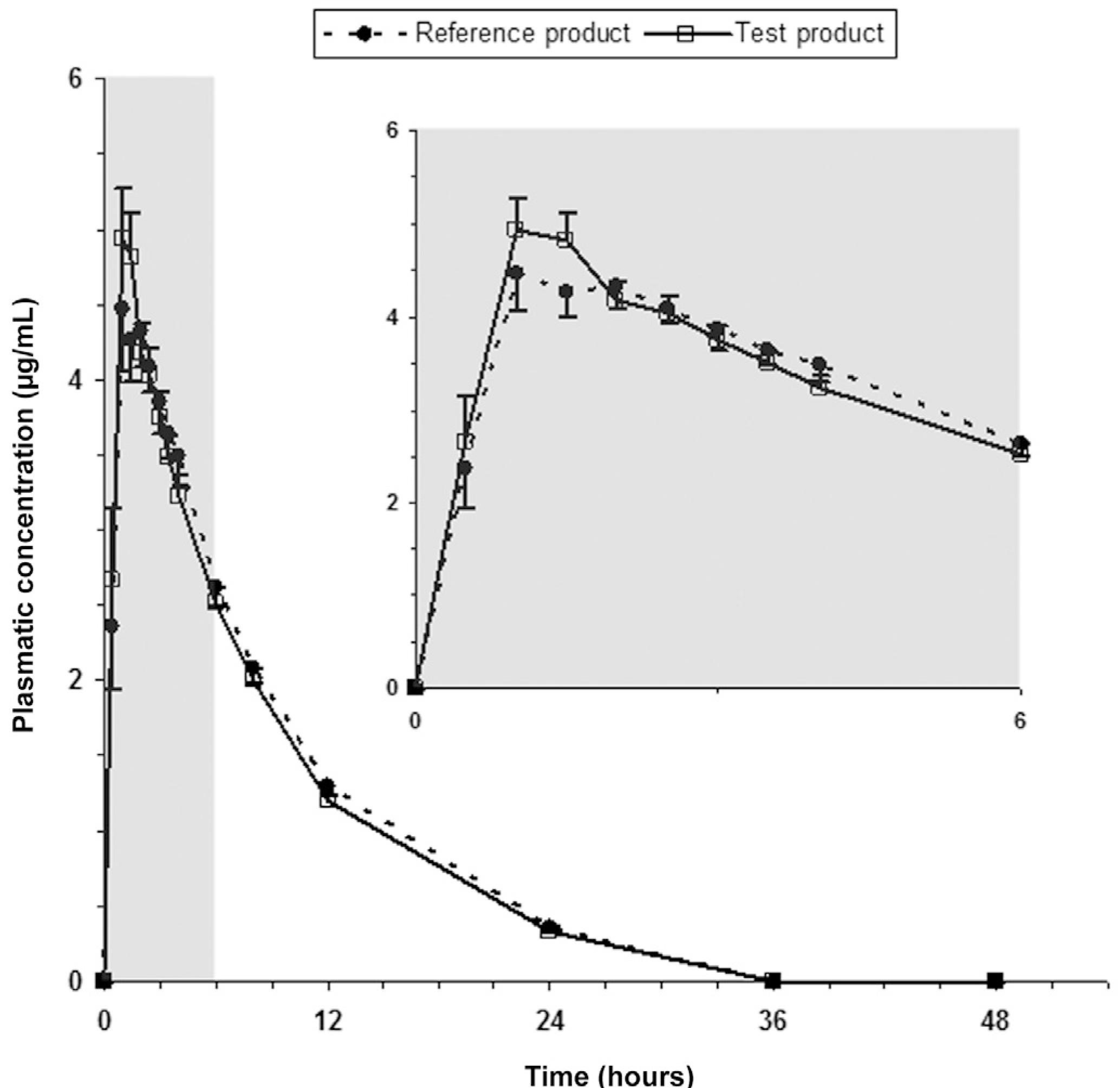
Average plasma concentrations of levofloxacin after administration of test and reference products to 26 healthy human volunteers are shown in Figure 1.
Average plasma concentrations of levofloxacin after oral administration of reference (Tavanic(r), Sanofi-Aventis Farmacêutica Ltda, Brazil) and test (Levaquin(r), Janssen-Cilag Farmacêutica Ltda, Brazil) products to 26 healthy volunteers. Bars indicate mean standard errors (lower bars for reference product and upper bars for test product).
Pharmacokinetic parameters
The mean values for Cmax, Tmax, Kel, T1/2el, AUC0-t and AUC0-inf of the two products did not differ significantly, suggesting that plasma profiles generated by the test product are comparable to those produced by the reference product. Pharmacokinetic parameters values are listed in Table I.
Average (range) pharmacokinetic parameters after oral administration of reference (Tavanic(r), Sanofi-Aventis Farmacêutica Ltda, Brazil) and test (Levaquin(r), Janssen-Cilag Farmacêutica Ltda, Brazil) products to 26 healthy volunteers
ANOVA detected the occurrence of period effect for Cmax, AUC0-t
and AUC0-inf and product effect for AUC0-t (Table II). The 90% confidence intervals for the
ratio of Cmax (92.1-108.2 %), AUC0-t (90.7-98.0 %) and
AUC0-inf (94.8-100.0 %) values for test and reference products are
within the 80-125% interval proposed by United States Food and Drug Administration
(FDA) (2001)FOOD AND DRUG ADMINISTRATION. FDA. Guidance for Industry: Statistical
approaches to establishing bioequivalence. U.S. Department of Health and Human
Services: 2001. 45p. Available at:
<http://www.fda.gov/downloads/Drugs/Guidances/ucm070244.pdf>. Accessed on: 02
May 2013.
http://www.fda.gov/downloads/Drugs/Guida...
, European Medicines Agency
(EMEA) (2010)EUROPEAN MEDICINES AGENCY. Guideline on the investigation of
bioequivalence. Committee for Medicinal Products for Human Use: 2010. 27p. Available
at
<http://www.emea.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf>.
Accessed on: 02 May 2013.
http://www.emea.europa.eu/docs/en_GB/doc...
and Brazilian Agência
Nacional de Vigilância Sanitária (ANVISA) (2006) (Table II). Power of statistic test was 0.99 for Cmax, 1.00 for
AUC0-t and 1.00 for AUC0-inf.
Analysis of variance (ANOVA) for the assessment of the product, sequence and period effects, and 90% confidence intervals (90% CI) for the ratio of Cmax, AUC0-t and AUC0-inf values for the test and reference products, using logarithmic transformed data, after oral administration of reference (Tavanic(r), Sanofi-Aventis Farmacêutica Ltda, Brazil) and test (Levaquin(r), Janssen-Cilag Farmacêutica Ltda, Brazil) products to 26 healthy volunteers (a=0.05)
DISCUSSION
Bioequivalence of drug products is usually assessed through open-label, randomized crossover studies (Eradiri et al., 2007ERADIRI, O.; SISTA, S.; LAI, J. C.; NGUYEN, O. H.; SILVERSTONE, P. H. Single- and multiple-dose bioequivalence of two once-daily tramadol formulations using stereospecific analysis of tramadol and its demethylated (M1 and M5) metabolites. Curr. Med. Res. Opin., v.23, p.1593-1604, 2007.; Howard et al., 2005HOWARD, D. R.; HARIBHAKTI, R.; KITTNER, B.; AGRAWALA, P. Single-dose and steady-state bioequivalence of fexofenadine and pseudoephedrine combination tablets compared with individual formulations in healthy adults. Curr. Med. Res. Opin., v.21, p.769-776, 2005.; Schützer et al., 2004SCHÜTZER, K. M.; WALL, U.; LÖNNERSTEDT, C.; OHLSSON, L.; TENG, R.; SARICH, T. C.; ERIKSSON, U. G. Bioequivalence of ximelagatran, an oral direct thrombin inhibitor, as whole or crushed tablets or dissolved formulation. Curr. Med. Res. Opin., v.20, p.325-331, 2004.). End-points in bioequivalence trials (Cmax and AUC0-inf) are objective assessments, obtained from drug plasma concentration-time profiles. These assessments will not be affected by the knowledge of the treatment by the volunteer or the researcher. Since, in this case, it is not possible that unconscious bias will affect the observation, an open-label design can be used. In a crossover design, each subject serves as his or her own control, thus allowing a within subject comparison between formulations. It also removes the inter-subject variability from the comparison between formulations and, with a proper randomization of subjects to the sequence of formulation administrations, it provides the best unbiased estimates for the differences between formulations (Chow, Liu, 2000CHOW, S. C.; LIU, J. P. Design and analysis of bioavailability and bioequivalence Studies. 2.ed. New York: Marcel Dekker, 2000. 584 p.).
High-performance liquid chromatography (HPLC) methods with ultraviolet (UV) (Baietto et al., 2009BAIETTO, L.; D'AVOLIO, A.; DE ROSA, F. G.; GARAZZINO, S.; PATANELLA, S.; SICCARDI, M.; SCIANDRA, M.; DI PERRI, G. Simultaneous quantification of linezolid, rifampicin, levofloxacin, and moxifloxacin in human plasma using high-performance liquid chromatography with UV. Ther. Drug Monit., v.31, p.104-109, 2009.; Chien et al., 1998CHIEN, S. C.; WONG, F. A.; FOWLER, C. L.; CALLERY-D'AMICO, S. V.; WILLIAMS, R. R.; NAYAK, R.; CHOW, A. T. Double-blind evaluation of the safety and pharmacokinetics of multiple oral once-daily 750-milligram and 1-gram doses of levofloxacin in healthy volunteers. Antimicrob. Agents Chemother. v.42, p.885-888, 1998.; Djabarouti et al., 2004DJABAROUTI, S.; BOSELLI, E.; ALLAOUCHICHE, B.; BA, B.; NGUYEN, A. T.; GORDIEN, J. B.; BERNADOU, J. M.; SAUX, M. C.; BREILH, D. Determination of levofloxacin in plasma, bronchoalveolar lavage and bone tissues by high-performance liquid chromatography with ultraviolet detection using a fully automated extraction method. J. Chromatogr. B, v.799, p.165-172, 2004.; Lee et al., 1997LEE, L. J.; HAFKIN, B.; LEE, I. D.; HOH, J.; DIX, R. Effects of food and sucralfate on a single oral dose of 500 milligrams of levofloxacin in healthy subjects. Antimicrob. Agents Chemother., v.41, p.2196-2200, 1997.; Liang, Kays, Sowinski, 2002LIANG, H.; KAYS, M. B.; SOWINSKI, K. M. Separation of levofloxacin, ciprofloxacin, gatifloxacin, moxifloxacin, trovafloxacin and cinoxacin by high-performance liquid chromatography: application to levofloxacin determination in human plasma. J. Chromatogr. B, v.772, p.53-63, 2002.; Nemutlu et al., 2007NEMUTLU, E.; KIR, S.; ÖZYÜNCÜ, Ö.; BEKSAÇ, M. S. Simultaneous separation and determination of seven quinolones using HPLC: analysis of levofloxacin and moxifloxacin in plasma and amniotic fluid. Chromatographia, v.66, p.S15-S24, 2007.; Wong, Juzwin, Flor, 1997WONG, F. A.; JUZWIN, S. J.; FLOR, S. C. Rapid stereospecific high-performance liquid chromatographic determination of levofloxacin in human plasma and urine. J. Pharm. Biomed., v.15, p.765-771, 1997.), fluorescence (Almeida et al., 2005ALMEIDA, S.; FILIPE, A.; ALMEIDA, A.; WONG, H.; CAPARROS, N.; TANGUAY, M. Comparative bioavailability of two formulations of levofloxacin and effect of sex on bioequivalence analysis. Data from a randomized, 2x2 crossover trial in healthy volunteers. Arzneimittel Forsch., v.55, p.414-419, 2005.; Chien et al., 1997CHIEN, S. C.; ROGGE, M. C.; GISCLON, L. G.; CURTIN, C.; WONG, F.; NATARAJAN, J.; WILLIAMS, R. R.; FOWLER, C. L.; CHEUNG, W. K.; CHOW, A. T. Pharmacokinetic profile of levofloxacin following once-daily 500-milligram oral or intravenous doses. Antimicrob. Agents Chemother., v.41, p.2256-2260, 1997.; Lubasch et al., 2000LUBASCH, A.; KELLER, I.; BORNER, K.; KOEPPE, P.; LODE, H. Comparative pharmacokinetics of ciprofloxacin, gatifloxacin, grepafloxacin, levofloxacin, trovafloxacin, and moxifloxacin after single oral administration in healthy volunteers. Antimicrob. Agents Chemother., v.44, p.2600-2603, 2000.; Schulte et al., 2006SCHULTE, S.; ACKERMANN, T.; BERTRAM, N.; SAUERBRUCH, T.; PAAR, W. D. Determination of the newer quinolones levofloxacin and moxifloxacin in plasma by high-performance liquid chromatography with fluorescence detection. J. Chromatogr. Sci., v.44, p.205-208, 2006.; Siewert, 2006SIEWERT, S. Validation of a levofloxacin HPLC assay in plasma and dialysate for pharmacokinetic studies. J. Pharm. Biomed., v.41, p.1360-1362, 2006.; Wagenlehner et al., 2006WAGENLEHNER, F. M.; KINZIG-SCHIPPERS, M.; TISCHMEYER, U.; WAGENLEHNER, C.; SÖRGEL, F.; DALHOFF, A.; NABER, K. G. Pharmacokinetics of ciprofloxacin XR (1000 mg) versus levofloxacin (500 mg) in plasma and urine of male and female healthy volunteers receiving a single oral dose. Int. J. Antimicrob. Ag., v.27, p.7-14, 2006.; Zhou et al., 2007ZHOU, Z. L.; YANG, M.; YU, X. Y.; PENG, H. Y.; SHAN, Z. X.; CHEN, S. Z.; LIN, Q. X.; LIU, X. Y.; CHEN, T. F.; ZHOU, S. F.; LIN, S. G. A rapid and simple high-performance liquid chromatography method for the determination of human plasma levofloxacin concentration and its application to bioequivalence studies. Biomed. Chromatogr., v.21, p.1045-1051, 2007.) or mass spectrometry (Conte et al., 2006CONTE JR.; J. E.; GOLDEN, J. A.; MCLVER, M.; ZURLINDEN, E. Intrapulmonary pharmacokinetics and pharmacodynamics of high-dose levofloxacin in healthy volunteer subjects. Int. J. Antimicrob. Ag., v.28, p.114-121, 2006.; Ji et al., 2006JI, H. Y.; JEONG, D. W.; KIM, Y. H.; KIM, H. H.; SOHN, D. R.; LEE, H. S. Hydrophilic interaction liquid chromatography-tandem mass spectrometry for the determination of levofloxacin in human plasma. J. Pharm. Biomed., v.41, p.622-627, 2006.) detection for analysis of levofloxacin in biological fluids have been published. Sample preparation procedures include protein precipitation with acetonitrile, methanol, trifluoroacetic acid or perchloric acid (Baietto et al., 2009BAIETTO, L.; D'AVOLIO, A.; DE ROSA, F. G.; GARAZZINO, S.; PATANELLA, S.; SICCARDI, M.; SCIANDRA, M.; DI PERRI, G. Simultaneous quantification of linezolid, rifampicin, levofloxacin, and moxifloxacin in human plasma using high-performance liquid chromatography with UV. Ther. Drug Monit., v.31, p.104-109, 2009.; Liang, Kays, Sowinski, 2002LIANG, H.; KAYS, M. B.; SOWINSKI, K. M. Separation of levofloxacin, ciprofloxacin, gatifloxacin, moxifloxacin, trovafloxacin and cinoxacin by high-performance liquid chromatography: application to levofloxacin determination in human plasma. J. Chromatogr. B, v.772, p.53-63, 2002.; Lubasch et al., 2000LUBASCH, A.; KELLER, I.; BORNER, K.; KOEPPE, P.; LODE, H. Comparative pharmacokinetics of ciprofloxacin, gatifloxacin, grepafloxacin, levofloxacin, trovafloxacin, and moxifloxacin after single oral administration in healthy volunteers. Antimicrob. Agents Chemother., v.44, p.2600-2603, 2000.; Schulte et al., 2006SCHULTE, S.; ACKERMANN, T.; BERTRAM, N.; SAUERBRUCH, T.; PAAR, W. D. Determination of the newer quinolones levofloxacin and moxifloxacin in plasma by high-performance liquid chromatography with fluorescence detection. J. Chromatogr. Sci., v.44, p.205-208, 2006.; Siewert, 2006SIEWERT, S. Validation of a levofloxacin HPLC assay in plasma and dialysate for pharmacokinetic studies. J. Pharm. Biomed., v.41, p.1360-1362, 2006.; Wagenlehner et al., 2006WAGENLEHNER, F. M.; KINZIG-SCHIPPERS, M.; TISCHMEYER, U.; WAGENLEHNER, C.; SÖRGEL, F.; DALHOFF, A.; NABER, K. G. Pharmacokinetics of ciprofloxacin XR (1000 mg) versus levofloxacin (500 mg) in plasma and urine of male and female healthy volunteers receiving a single oral dose. Int. J. Antimicrob. Ag., v.27, p.7-14, 2006.), liquid-liquid extraction (Chien et al., 1997CHIEN, S. C.; ROGGE, M. C.; GISCLON, L. G.; CURTIN, C.; WONG, F.; NATARAJAN, J.; WILLIAMS, R. R.; FOWLER, C. L.; CHEUNG, W. K.; CHOW, A. T. Pharmacokinetic profile of levofloxacin following once-daily 500-milligram oral or intravenous doses. Antimicrob. Agents Chemother., v.41, p.2256-2260, 1997., 1998CHIEN, S. C.; WONG, F. A.; FOWLER, C. L.; CALLERY-D'AMICO, S. V.; WILLIAMS, R. R.; NAYAK, R.; CHOW, A. T. Double-blind evaluation of the safety and pharmacokinetics of multiple oral once-daily 750-milligram and 1-gram doses of levofloxacin in healthy volunteers. Antimicrob. Agents Chemother. v.42, p.885-888, 1998.; Lee et al., 1997LEE, L. J.; HAFKIN, B.; LEE, I. D.; HOH, J.; DIX, R. Effects of food and sucralfate on a single oral dose of 500 milligrams of levofloxacin in healthy subjects. Antimicrob. Agents Chemother., v.41, p.2196-2200, 1997.; Wong, Juzwin, Flor, 1997WONG, F. A.; JUZWIN, S. J.; FLOR, S. C. Rapid stereospecific high-performance liquid chromatographic determination of levofloxacin in human plasma and urine. J. Pharm. Biomed., v.15, p.765-771, 1997.; Zhou et al., 2007ZHOU, Z. L.; YANG, M.; YU, X. Y.; PENG, H. Y.; SHAN, Z. X.; CHEN, S. Z.; LIN, Q. X.; LIU, X. Y.; CHEN, T. F.; ZHOU, S. F.; LIN, S. G. A rapid and simple high-performance liquid chromatography method for the determination of human plasma levofloxacin concentration and its application to bioequivalence studies. Biomed. Chromatogr., v.21, p.1045-1051, 2007.), solid-phase extraction (Djabarouti et al., 2004DJABAROUTI, S.; BOSELLI, E.; ALLAOUCHICHE, B.; BA, B.; NGUYEN, A. T.; GORDIEN, J. B.; BERNADOU, J. M.; SAUX, M. C.; BREILH, D. Determination of levofloxacin in plasma, bronchoalveolar lavage and bone tissues by high-performance liquid chromatography with ultraviolet detection using a fully automated extraction method. J. Chromatogr. B, v.799, p.165-172, 2004.; Nemutlu et al., 2007NEMUTLU, E.; KIR, S.; ÖZYÜNCÜ, Ö.; BEKSAÇ, M. S. Simultaneous separation and determination of seven quinolones using HPLC: analysis of levofloxacin and moxifloxacin in plasma and amniotic fluid. Chromatographia, v.66, p.S15-S24, 2007.) and ultrafiltration (Liang, Kays, Sowinski, 2002LIANG, H.; KAYS, M. B.; SOWINSKI, K. M. Separation of levofloxacin, ciprofloxacin, gatifloxacin, moxifloxacin, trovafloxacin and cinoxacin by high-performance liquid chromatography: application to levofloxacin determination in human plasma. J. Chromatogr. B, v.772, p.53-63, 2002.). Most of the methods employ isocratic mobile phase, but the use of mobile phase gradient is also described (Baietto et al., 2009BAIETTO, L.; D'AVOLIO, A.; DE ROSA, F. G.; GARAZZINO, S.; PATANELLA, S.; SICCARDI, M.; SCIANDRA, M.; DI PERRI, G. Simultaneous quantification of linezolid, rifampicin, levofloxacin, and moxifloxacin in human plasma using high-performance liquid chromatography with UV. Ther. Drug Monit., v.31, p.104-109, 2009.; Nemutlu et al., 2007NEMUTLU, E.; KIR, S.; ÖZYÜNCÜ, Ö.; BEKSAÇ, M. S. Simultaneous separation and determination of seven quinolones using HPLC: analysis of levofloxacin and moxifloxacin in plasma and amniotic fluid. Chromatographia, v.66, p.S15-S24, 2007.; Schulte et al., 2006SCHULTE, S.; ACKERMANN, T.; BERTRAM, N.; SAUERBRUCH, T.; PAAR, W. D. Determination of the newer quinolones levofloxacin and moxifloxacin in plasma by high-performance liquid chromatography with fluorescence detection. J. Chromatogr. Sci., v.44, p.205-208, 2006.; Siewert, 2006SIEWERT, S. Validation of a levofloxacin HPLC assay in plasma and dialysate for pharmacokinetic studies. J. Pharm. Biomed., v.41, p.1360-1362, 2006.). Although all of these methods are successful in the determination of levofloxacin, some of them have limitations including long chromatographic running time (Baietto et al., 2009BAIETTO, L.; D'AVOLIO, A.; DE ROSA, F. G.; GARAZZINO, S.; PATANELLA, S.; SICCARDI, M.; SCIANDRA, M.; DI PERRI, G. Simultaneous quantification of linezolid, rifampicin, levofloxacin, and moxifloxacin in human plasma using high-performance liquid chromatography with UV. Ther. Drug Monit., v.31, p.104-109, 2009.; Siewert, 2006SIEWERT, S. Validation of a levofloxacin HPLC assay in plasma and dialysate for pharmacokinetic studies. J. Pharm. Biomed., v.41, p.1360-1362, 2006.), and complicated and time-consuming sample pretreatment procedures (Chien et al., 1997CHIEN, S. C.; ROGGE, M. C.; GISCLON, L. G.; CURTIN, C.; WONG, F.; NATARAJAN, J.; WILLIAMS, R. R.; FOWLER, C. L.; CHEUNG, W. K.; CHOW, A. T. Pharmacokinetic profile of levofloxacin following once-daily 500-milligram oral or intravenous doses. Antimicrob. Agents Chemother., v.41, p.2256-2260, 1997.).
The method used in this study has advantages when compared with others published methods: sample pre-treatment requires a one-step preparation, adding acetonitrile for protein precipitation; the chromatographic analysis was done in isocratic conditions; and the running time was 10 minutes for each sample.
The method showed good specificity, sensitivity, linearity, precision and accuracy over the entire concentration range achieved after oral administration of tablets conataining 500 mg of levofloxacin, thereby enabling its use in bioequivalence trials of this kind of formulations.
The average plasmatic decay curves obtained for the test product (Levaquin(r)) and reference product (Tavanic(r)) have the same pattern.
Levofloxacin was absorbed rapidly, achieving mean peak plasma level between 1 and 2 hours after drug administration for both test and reference products. Mean values for pharmacokinetic parameters Cmax (reference: 5.69 μg/mL; test: 5.67 μg/mL), Tmax (reference: 1.60 h; test: 1.29 h), AUC0-t (reference: 41.52 μg·h/mL; test: 39.22 μg·h/mL), AUC0-inf (reference: 45.50 μg·h/mL; test: 44.24 μg·h/mL) and T1/2el (reference: 6.55 hours; test: 6.69 hours) were similar to that reported by other authors (Amsden, Whitaker, Johnson, 2003AMSDEN, G. W.; WHITAKER, A. M.; JOHNSON, P. W. Lack of bioequivalence of levofloxacin when coadministered with a mineral-fortified breakfast of juice and cereal. J. Clin. Pharmacol., v.43, p.990-995, 2003.; Chien et al., 1997CHIEN, S. C.; ROGGE, M. C.; GISCLON, L. G.; CURTIN, C.; WONG, F.; NATARAJAN, J.; WILLIAMS, R. R.; FOWLER, C. L.; CHEUNG, W. K.; CHOW, A. T. Pharmacokinetic profile of levofloxacin following once-daily 500-milligram oral or intravenous doses. Antimicrob. Agents Chemother., v.41, p.2256-2260, 1997.; Lee et al., 1997LEE, L. J.; HAFKIN, B.; LEE, I. D.; HOH, J.; DIX, R. Effects of food and sucralfate on a single oral dose of 500 milligrams of levofloxacin in healthy subjects. Antimicrob. Agents Chemother., v.41, p.2196-2200, 1997.; Lubasch et al., 2000LUBASCH, A.; KELLER, I.; BORNER, K.; KOEPPE, P.; LODE, H. Comparative pharmacokinetics of ciprofloxacin, gatifloxacin, grepafloxacin, levofloxacin, trovafloxacin, and moxifloxacin after single oral administration in healthy volunteers. Antimicrob. Agents Chemother., v.44, p.2600-2603, 2000.; Wagenlehner et al., 2006). ANOVA revealed the absence of sequence effect for Cmax, AUC0-t e AUC0-inf. Period effect was observed for Cmax, AUC0-t and AUC0-inf, and product effect was observed for AUC0-t. Regarding product effect, these finding does not mean that the formulations evaluated are not bioequivalent, since bioequivalence between two products is defined in terms of 90% CI. In this case, the occurrence of product effect indicates that there is a statistically significant difference between bioavailabilities of test and reference products regarding extension of drug absorption, but there is no difference regarding absorption rate, since no product effect was observed for Cmax. This difference in extension of drug absorption is not considered to be clinically important, since 90% CI is within the limits established by regulatory agencies (Pabst, Jaeger, 1990PABST, G.; JAEGER, H. Review of methods and criteria for the evaluation of bioequivalence studies. Eur. J. Clin. Pharmacol., v.38, p.5-10, 1990.; Westlake, 1979WESTLAKE, W. J. Statistical aspects of comparative bioavailability trials. Biometrics, v.35, p.273-280, 1979.). A period effect measures the differences between study periods. A well-run study with consistent treatment of subjects should produce no significant period effects. However, a significant period effect does not invalidate the study, but the cause should be investigated (Ormsby, 1994ORMSBY, E. Statistical methods in bioequivalence. In: JACKSON, A. J. (Ed.). Generics and bioequivalence. Boca Raton: CRC Press, 1994 p.1-27.). Period effect may be due to a too short washout between periods (residual or carry-over effects), or they may reflect differing procedures of sample workup or storage, because for each subject usually all samples of all periods are analyzed within the same run. They also may be provoked by acclimatization or even weather conditions, or they may reflect an effect of the diet (Pabst, Jaeger, 1990PABST, G.; JAEGER, H. Review of methods and criteria for the evaluation of bioequivalence studies. Eur. J. Clin. Pharmacol., v.38, p.5-10, 1990.). In the present study, predose samples did not exhibit any detectable levofloxacin level in all subjects, so the occurrence of residual or carry-over effects can be excluded. Also, procedures regarding volunteers' diet and rest, drug administration and sample workup and storage were the same in the two periods of the study. On the basis of these considerations. the present bioequivalence study is acceptable, despite the period effect.
The 90% CI for the ratio of Cmax (92.1%-108.2 %), AUC0-t (90.7%-98.0 %) and AUC0-inf (94.8%-100.0 %) values for the test and reference products are within the 80-125 % interval proposed by regulatory agencies, therefore test and reference products are bioequivalent according to average bioequivalence criteria, even if product effect have been detected in ANOVA.
CONCLUSIONS
Based on the pharmacokinetic and statistical results of this study, it was concluded that Tavanic(c) (Sanofi-Aventis Famacêutica Ltda, Brazil) and Levaquin(c) (Janssen-Cilag Farmacêutica Ltda, Brazil) are bioequivalent. Although a statistically significant difference between bioavailabilities of test and reference products regarding extension of drug absorption was detected trough ANOVA, this was not considered to be clinically important, since the 90% CI of the ratios for Cmax, AUC0-t and AUC0-inf were found to be within the regulatory agencies proposed acceptance limits (80-125%).
ACKNOWLEDGMENTS
This study was supported by Janssen-Cilag Farmacêutica Ltda, São Paulo, Brazil.
REFERENCES
- ABDOU, H. M. Dissolution, bioavailability and bioequivalence. Easton: Mack Publishing Company, 1989. 554 p.
- AGÊNCIA NACIONAL DE VIGILÂNCIA SANITÁRIA. Guia para provas de biodisponibilidade relativa/bioequivalência de medicamentos. 2006. Available at: <http://www.in.gov.br/visualiza/index.jsp?data=24/04/2006&jornal=1&pagina=101&totalArquivos=128>. Accessed on: 03 May 2013.
» http://www.in.gov.br/visualiza/index.jsp?data=24/04/2006&jornal=1&pagina=101&totalArquivos=128 - ALMEIDA, S.; FILIPE, A.; ALMEIDA, A.; WONG, H.; CAPARROS, N.; TANGUAY, M. Comparative bioavailability of two formulations of levofloxacin and effect of sex on bioequivalence analysis. Data from a randomized, 2x2 crossover trial in healthy volunteers. Arzneimittel Forsch., v.55, p.414-419, 2005.
- AMSDEN, G. W.; WHITAKER, A. M.; JOHNSON, P. W. Lack of bioequivalence of levofloxacin when coadministered with a mineral-fortified breakfast of juice and cereal. J. Clin. Pharmacol., v.43, p.990-995, 2003.
- ANDERSON, V. R.; PERRY, C. M. Levofloxacin: a review of its use as a high-dose, short-course treatment for bacterial infection. Drugs, v.68, p.535-565, 2008.
- BAIETTO, L.; D'AVOLIO, A.; DE ROSA, F. G.; GARAZZINO, S.; PATANELLA, S.; SICCARDI, M.; SCIANDRA, M.; DI PERRI, G. Simultaneous quantification of linezolid, rifampicin, levofloxacin, and moxifloxacin in human plasma using high-performance liquid chromatography with UV. Ther. Drug Monit., v.31, p.104-109, 2009.
- CHEN, M. L.; SHAH, V.; PATNAIK, R.; ADAMS, W.; HUSSAIN, A.; CONNER, D.; MEHTA, M.; MALINOWSKI, H.; LAZOR, J.; HUANG, S. M,, HARE, D.; LESKO, L.; SPORN, D.; WILLIAMS, R. Bioavailability and bioequivalence: an FDA regulatory overview. Pharm. Res., v.18, p.1645-1650, 2001.
- CHIEN, S. C.; ROGGE, M. C.; GISCLON, L. G.; CURTIN, C.; WONG, F.; NATARAJAN, J.; WILLIAMS, R. R.; FOWLER, C. L.; CHEUNG, W. K.; CHOW, A. T. Pharmacokinetic profile of levofloxacin following once-daily 500-milligram oral or intravenous doses. Antimicrob. Agents Chemother., v.41, p.2256-2260, 1997.
- CHIEN, S. C.; WONG, F. A.; FOWLER, C. L.; CALLERY-D'AMICO, S. V.; WILLIAMS, R. R.; NAYAK, R.; CHOW, A. T. Double-blind evaluation of the safety and pharmacokinetics of multiple oral once-daily 750-milligram and 1-gram doses of levofloxacin in healthy volunteers. Antimicrob. Agents Chemother. v.42, p.885-888, 1998.
- CHOW, S. C.; LIU, J. P. Design and analysis of bioavailability and bioequivalence Studies. 2.ed. New York: Marcel Dekker, 2000. 584 p.
- CONTE JR.; J. E.; GOLDEN, J. A.; MCLVER, M.; ZURLINDEN, E. Intrapulmonary pharmacokinetics and pharmacodynamics of high-dose levofloxacin in healthy volunteer subjects. Int. J. Antimicrob. Ag., v.28, p.114-121, 2006.
- CROOM, K. F.; GOA, K. L. Levofloxacin: a review of its use in the treatment of bacterial infection in the United States. Drugs, v.63, p.2769-2802, 2003.
- DIPIRO, J. T.; BLOUIN, R. A.; PRUEMER, J. M.; SPRUILL, W. J. Concepts in clinical pharmacokinetics. 2.ed. Bethesda: American Society of Health-System Pharmacists, 1996. 208 p.
- DJABAROUTI, S.; BOSELLI, E.; ALLAOUCHICHE, B.; BA, B.; NGUYEN, A. T.; GORDIEN, J. B.; BERNADOU, J. M.; SAUX, M. C.; BREILH, D. Determination of levofloxacin in plasma, bronchoalveolar lavage and bone tissues by high-performance liquid chromatography with ultraviolet detection using a fully automated extraction method. J. Chromatogr. B, v.799, p.165-172, 2004.
- ERADIRI, O.; SISTA, S.; LAI, J. C.; NGUYEN, O. H.; SILVERSTONE, P. H. Single- and multiple-dose bioequivalence of two once-daily tramadol formulations using stereospecific analysis of tramadol and its demethylated (M1 and M5) metabolites. Curr. Med. Res. Opin., v.23, p.1593-1604, 2007.
- EUROPEAN MEDICINES AGENCY. Guideline on the investigation of bioequivalence. Committee for Medicinal Products for Human Use: 2010. 27p. Available at <http://www.emea.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf>. Accessed on: 02 May 2013.
» http://www.emea.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf - HOWARD, D. R.; HARIBHAKTI, R.; KITTNER, B.; AGRAWALA, P. Single-dose and steady-state bioequivalence of fexofenadine and pseudoephedrine combination tablets compared with individual formulations in healthy adults. Curr. Med. Res. Opin., v.21, p.769-776, 2005.
- INTERNATIONAL CONFERENCE ON HARMONIZATION. Guideline for good clinical practice E6(R1). International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use: 1996. 53p. Available at: <http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6_R1/Step4/E6_R1__Guideline.pdf>. Accessed on: 03 May 2013.
» http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6_R1/Step4/E6_R1__Guideline.pdf - JI, H. Y.; JEONG, D. W.; KIM, Y. H.; KIM, H. H.; SOHN, D. R.; LEE, H. S. Hydrophilic interaction liquid chromatography-tandem mass spectrometry for the determination of levofloxacin in human plasma. J. Pharm. Biomed., v.41, p.622-627, 2006.
- LEE, L. J.; HAFKIN, B.; LEE, I. D.; HOH, J.; DIX, R. Effects of food and sucralfate on a single oral dose of 500 milligrams of levofloxacin in healthy subjects. Antimicrob. Agents Chemother., v.41, p.2196-2200, 1997.
- LIANG, H.; KAYS, M. B.; SOWINSKI, K. M. Separation of levofloxacin, ciprofloxacin, gatifloxacin, moxifloxacin, trovafloxacin and cinoxacin by high-performance liquid chromatography: application to levofloxacin determination in human plasma. J. Chromatogr. B, v.772, p.53-63, 2002.
- LUBASCH, A.; KELLER, I.; BORNER, K.; KOEPPE, P.; LODE, H. Comparative pharmacokinetics of ciprofloxacin, gatifloxacin, grepafloxacin, levofloxacin, trovafloxacin, and moxifloxacin after single oral administration in healthy volunteers. Antimicrob. Agents Chemother., v.44, p.2600-2603, 2000.
- NEMUTLU, E.; KIR, S.; ÖZYÜNCÜ, Ö.; BEKSAÇ, M. S. Simultaneous separation and determination of seven quinolones using HPLC: analysis of levofloxacin and moxifloxacin in plasma and amniotic fluid. Chromatographia, v.66, p.S15-S24, 2007.
- NOREDDIN, A. M.; HAYNES, V. L.; ZHANEL, G. G. Pharmacokinetics and pharmacodynamics of the new quinolones. J. Pharm. Pract., v.18, p.432-443, 2005.
- ORMSBY, E. Statistical methods in bioequivalence. In: JACKSON, A. J. (Ed.). Generics and bioequivalence. Boca Raton: CRC Press, 1994 p.1-27.
- PABST, G.; JAEGER, H. Review of methods and criteria for the evaluation of bioequivalence studies. Eur. J. Clin. Pharmacol., v.38, p.5-10, 1990.
- PORTA, V.; CHANG, K. H.; STORPIRTIS, S. Evaluation of the bioequivalence of capsules containing 150 mg of fluconazole. Int. J. Pharm., v.288, p.81-86, 2005.
- RODVOLD, K. A.; NEUHAUSER, M. Pharmacokinetics and pharmacodynamics of fluoroquinolones. Pharmacotherapy, v.21, p.233S-252S, 2001.
- SCHULTE, S.; ACKERMANN, T.; BERTRAM, N.; SAUERBRUCH, T.; PAAR, W. D. Determination of the newer quinolones levofloxacin and moxifloxacin in plasma by high-performance liquid chromatography with fluorescence detection. J. Chromatogr. Sci., v.44, p.205-208, 2006.
- SCHÜTZER, K. M.; WALL, U.; LÖNNERSTEDT, C.; OHLSSON, L.; TENG, R.; SARICH, T. C.; ERIKSSON, U. G. Bioequivalence of ximelagatran, an oral direct thrombin inhibitor, as whole or crushed tablets or dissolved formulation. Curr. Med. Res. Opin., v.20, p.325-331, 2004.
- SIEWERT, S. Validation of a levofloxacin HPLC assay in plasma and dialysate for pharmacokinetic studies. J. Pharm. Biomed., v.41, p.1360-1362, 2006.
- TAKAHASHI, H.; HAYAKAWA, I.; AKIMOTO, T. The history of the development and changes of quinolone antibacterial agents. Yakushigaku Zasshi, v.38, p.161-179, 2003.
- FOOD AND DRUG ADMINISTRATION. FDA. Guidance for Industry: Statistical approaches to establishing bioequivalence. U.S. Department of Health and Human Services: 2001. 45p. Available at: <http://www.fda.gov/downloads/Drugs/Guidances/ucm070244.pdf>. Accessed on: 02 May 2013.
» http://www.fda.gov/downloads/Drugs/Guidances/ucm070244.pdf - WAGENLEHNER, F. M.; KINZIG-SCHIPPERS, M.; TISCHMEYER, U.; WAGENLEHNER, C.; SÖRGEL, F.; DALHOFF, A.; NABER, K. G. Pharmacokinetics of ciprofloxacin XR (1000 mg) versus levofloxacin (500 mg) in plasma and urine of male and female healthy volunteers receiving a single oral dose. Int. J. Antimicrob. Ag., v.27, p.7-14, 2006.
- WESTLAKE, W. J. Statistical aspects of comparative bioavailability trials. Biometrics, v.35, p.273-280, 1979.
- WONG, F. A.; JUZWIN, S. J.; FLOR, S. C. Rapid stereospecific high-performance liquid chromatographic determination of levofloxacin in human plasma and urine. J. Pharm. Biomed., v.15, p.765-771, 1997.
- WORLD MEDICAL ASSOCIATION. WMA Declaration of Helsinki: ethical principles for medical research involving human subjects. 2008. Available at: <http://www.wma.net/en/30publications/10policies/b3/index.html>. Accessed on: 03 May 2013.
» http://www.wma.net/en/30publications/10policies/b3/index.html - ZHOU, Z. L.; YANG, M.; YU, X. Y.; PENG, H. Y.; SHAN, Z. X.; CHEN, S. Z.; LIN, Q. X.; LIU, X. Y.; CHEN, T. F.; ZHOU, S. F.; LIN, S. G. A rapid and simple high-performance liquid chromatography method for the determination of human plasma levofloxacin concentration and its application to bioequivalence studies. Biomed. Chromatogr., v.21, p.1045-1051, 2007.
Publication Dates
-
Publication in this collection
Jan-Mar 2015
History
-
Received
02 Nov 2013 -
Accepted
16 Oct 2014