Resumos
A fenilcetonúria (PKU) ocorre na incapacidade para transformar fenilalanina em tirosina, trazendo efeitos tóxicos para o sistema nervoso central. Tradicionalmente, no tratamento da PKU, o aleitamento materno é substituído por fórmula láctea. Este estudo verificou os efeitos do aleitamento materno como fonte de fenilalanina no desenvolvimento de crianças com PKU. Participaram dez lactentes com PKU, que iniciaram o tratamento com a introdução de fórmula láctea antes dos 30 dias e que mantiveram o aleitamento materno por no mínimo 30 dias de vida após o início dos procedimentos. Os procedimentos basearam-se em estimar a ingestão de leite materno, com margem segura da concentração da fenilalanina, calculando o volume gástrico e oferecendo inicialmente fórmula láctea, seguida do aleitamento materno em demanda livre, em todas as mamadas. O tempo de amamentação variou de um mês e cinco dias a 14 meses. Os controles sanguíneos foram semanais. Se o nível sérico da fenilalanina estivesse >2 mg/dL e <6 mg/dL mantinha-se a prescrição; se estivesse <2 mg/dL, diminuía-se a fórmula láctea em 25%, aumentando indiretamente o aleitamento materno; se estivesse >6 mg/dL, aumentava-se a fórmula em 50%. Avaliou-se os níveis de fenilalanina, aplicou-se a Early Language Milestone Scale e Passos Básicos do Desenvolvimento. Foram considerados adequados aqueles lactentes que apresentaram índices normativos em todas as avaliações. Dos lactentes, 80% conseguiram manter limites seguros da fenilalanina e desenvolvimento nos índices normativos. Há viabilidade da continuidade do aleitamento materno no tratamento de crianças com PKU desde que os níveis de fenilalanina sejam rigorosamente controlados e que os efeitos do aleitamento materno para o desenvolvimento infantil sejam verificados.
Aleitamento materno; Fenilcetonúrias; Desenvolvimento infantil; Dietoterapia; Lactente; Metabolismo; Nutrição da criança
Phenylketonuria (PKU) is the inability to convert phenylalanine into tyrosine, causing toxic effects to the central nervous system. Traditionally, in the treatment of PKU, breastfeeding is replaced by formula milk. This study verified the effects of breastfeeding as a source of phenylalanine on the development of children with PKU. Participants were ten infants with PKU who started treatment with the introduction of formula before 30 days of life, and maintained breastfeeding for at least 30 days after the start of procedures. The procedures were based on estimating breast milk intake, with a safe margin of phenylalanine concentration, calculating stomach volume, and initially offering formula, then breastfeeding on free demand, at every feeding. Breastfeeding duration ranged from one month and five days to 14 months. Blood controls were tested weekly. If the serum level of phenylalanine was >2 mg/dL and <6 mg/dL, the prescription was kept; if it was >2 mg/dL, the formula was decreased by 25%, indirectly increasing breastfeeding; if it was <6 mg/dL the formula was increased by 50%. The phenylalanine levels were assessed, and the Early Milestone Scale and the Basic Steps of Development were applied. Those who had normative index in all evaluations were considered adequate. Eighty percent of infants were able to keep safe concentrations of phenylalanine and development within normal indices. Continued breastfeeding is viable in the treatment of children with PKU, provided that phenylalanine levels are strictly controlled and the effects of breastfeeding on child development are monitored.
Breast feeding; Phenylketonurias; Child development; Diet therapy; Infant; Metabolism; Child nutrition
RELATO DE CASO
Acompanhamento do aleitamento materno no tratamento de crianças com fenilcetonúria
Dionísia Aparecida Cusin LamônicaI; Marisdalva Viegas StumpII; Karla Panice PedroII; Maura Contieri Rolim-LiporacciII; Ana Cláudia Gandara Casarin CaldeiraII; Fernanda da Luz Anastácio-PessanII; Mariana Germano GejãoI
IDepartamento de Fonoaudiologia, Faculdade de Odontologia de Bauru, Universidade de São Paulo - USP - Bauru (SP), Brasil
IILaboratório de Screening Neonatal, Associação de Pais e Amigos dos Excepcionais - APAE - Bauru (SP), Brasil
Endereço para correspondência Endereço para correspondência: Dionísia Aparecida Cusin Lamônica Faculdade de Odontologia de Bauru Al. Octávio Pinheiro Brisolla, 9/75, Vila Universitária, Bauru (SP), Brasil CEP: 17012-901. E-mail: dionelam@uol.com.br
RESUMO
A fenilcetonúria (PKU) ocorre na incapacidade para transformar fenilalanina em tirosina, trazendo efeitos tóxicos para o sistema nervoso central. Tradicionalmente, no tratamento da PKU, o aleitamento materno é substituído por fórmula láctea. Este estudo verificou os efeitos do aleitamento materno como fonte de fenilalanina no desenvolvimento de crianças com PKU. Participaram dez lactentes com PKU, que iniciaram o tratamento com a introdução de fórmula láctea antes dos 30 dias e que mantiveram o aleitamento materno por no mínimo 30 dias de vida após o início dos procedimentos. Os procedimentos basearam-se em estimar a ingestão de leite materno, com margem segura da concentração da fenilalanina, calculando o volume gástrico e oferecendo inicialmente fórmula láctea, seguida do aleitamento materno em demanda livre, em todas as mamadas. O tempo de amamentação variou de um mês e cinco dias a 14 meses. Os controles sanguíneos foram semanais. Se o nível sérico da fenilalanina estivesse >2 mg/dL e <6 mg/dL mantinha-se a prescrição; se estivesse <2 mg/dL, diminuía-se a fórmula láctea em 25%, aumentando indiretamente o aleitamento materno; se estivesse >6 mg/dL, aumentava-se a fórmula em 50%. Avaliou-se os níveis de fenilalanina, aplicou-se a Early Language Milestone Scale e Passos Básicos do Desenvolvimento. Foram considerados adequados aqueles lactentes que apresentaram índices normativos em todas as avaliações. Dos lactentes, 80% conseguiram manter limites seguros da fenilalanina e desenvolvimento nos índices normativos. Há viabilidade da continuidade do aleitamento materno no tratamento de crianças com PKU desde que os níveis de fenilalanina sejam rigorosamente controlados e que os efeitos do aleitamento materno para o desenvolvimento infantil sejam verificados.
Descritores: Aleitamento materno. Fenilcetonúrias. Desenvolvimento infantil. Dietoterapia. Lactente. Metabolismo. Nutrição da criança
INTRODUÇÃO
Fenilcetonúria (PKU) é uma desordem autossômica recessiva, resultante da mutação do gene localizado no cromossomo 12q22-24.1. Este defeito metabólico consiste na incapacidade para transformar fenilalanina em tirosina por ausência da enzima que catalisa esta reação, trazendo efeitos tóxicos para o sistema nervoso central (SNC). Tem caráter irreversível, provocando comprometimento cerebral difuso(1,2). As crianças não tratadas ou que não conseguem manter os níveis de fenilalanina normativos entre >2 mg/dL e <6 mg/dL(3) no primeiro ano de vida, poderão apresentar comprometimento progressivo das funções cerebrais e desenvolver deficiência intelectual, hiperatividade, déficit de atenção, crises convulsivas, atraso do desenvolvimento neuropsicomotor e comportamentos autísticos(4,5).
Tradicionalmente, no tratamento da PKU suspende-se o aleitamento materno (AM) e a fenilalanina necessária é fornecida pelo uso de fórmula láctea comercial, cuja concentração do aminoácido é conhecida. A quantidade de fórmula é determinada pelos níveis sanguíneos de fenilalanina e varia para cada indivíduo, de acordo com a tolerância à ingestão deste aminoácido(5-7).
A suspensão do AM seria necessária devido à dificuldade de quantificar a ingestão de fenilalanina durante a mamada ao seio, o que poderia ocasionar ampla variação nos níveis sanguíneos e/ou sua manutenção em patamares acima dos recomendados, incompatíveis com o tratamento adequado(8-10) para prevenir sequelas. Durante muito tempo acreditou-se não ser segura a amamentação de bebês recém-diagnosticados com PKU. Entretanto, as mães que tinham que parar abruptamente de amamentar seus filhos, após o diagnóstico, experimentavam sentimentos de tristeza e culpa, com interferência na relação com seu bebê e na aceitação do diagnóstico(9).
A fenilalanina é um aminoácido essencial também para fenilcetonúricos e a dieta deve contê-la em quantidade adequada, possibilitando o desenvolvimento nos parâmetros da normalidade(1,4,6,7,10).
Como lidar com o AM é uma questão de extrema relevância após o diagnóstico nos casos de PKU(9). Apesar da amamentação em indivíduos com PKU não ser comum, há vantagens e desvantagens que devem ser analisadas com cuidado e a tolerância individual deve ser rigorosamente monitorada.
Considerando os estudos acumulados sobre os benefícios do AM(11-13) para a criança, do ponto de vista biológico e psicossocial, este estudo teve o objetivo de verificar os efeitos do AM como fonte de fenilalanina para o desenvolvimento infantil e refletir sobre a importância do AM para esta população.
APRESENTAÇÃO DOS CASOS CLÍNICOS
O projeto foi aprovado pelo Comitê de Ética em Pesquisa da Faculdade de Odontologia de Bauru da Universidade de São Paulo, protocolo número 088/2008. Foram cumpridos todos os critérios da Resolução 196/96. Os responsáveis legais pelos participantes assinaram o Termo de Consentimento Livre e Esclarecido, permitindo que os dados constantes no prontuário fossem utilizados para este estudo.
Participaram dez lactentes com PKU triados pelo "Teste do Pezinho", e diagnosticados por meio de dosagens sanguíneas, coletadas por punção venosa.
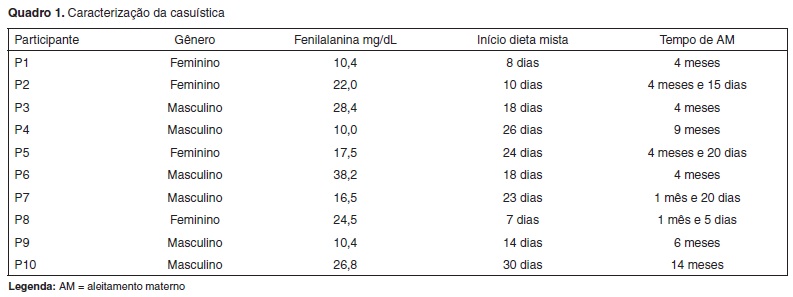
Para compor a casuística foram adotados os seguintes critérios de inclusão: crianças com PKU, sem doenças crônicas, que iniciaram o tratamento com restrição da ingestão do aminoácido fenilalanina antes dos 30 dias de vida e que mantiveram o AM por no mínimo 30 dias. A caracterização da casuística, quanto ao gênero, nível de fenilalanina no diagnóstico comprobatório, início do tratamento com dieta composta por AM e fórmula (mista) e tempo de AM encontram-se no Quadro 1.
As mães receberam orientações semanais quanto à importância do AM e da necessidade do controle dos níveis plasmáticos da fenilalanina para prevenir efeitos deletérios para o SNC e os reflexos deste no desenvolvimento infantil.
O procedimento do AM baseou-se em estimar a ingestão de miligramas de leite materno, de acordo com faixa etária, com margem segura da concentração da fenilalanina, calculando o volume gástrico e oferecendo, inicialmente a fórmula láctea, e em seguida o peito, em demanda livre em todas as mamadas.
Os controles sanguíneos de fenilalanina foram semanais. Os valores de referência da fenilalanina no plasma, no primeiro ano de vida, correspondem a >2 mg/dL e <6 mg/dL(3). A dosagem da fenilalanina para controle dos níveis sanguíneos ao longo do tratamento com AM foi realizada por meio do procedimento de ultramicrofluorimetria após eluição.
Se o nível sérico da fenilalanina estivesse entre >2 mg/dL e <6 mg/dL mantinha-se a prescrição; se estivesse <2 mg/dL, diminuía-se a fórmula láctea em 25%, aumentando indiretamente o AM; se estivesse >6 mg/dL, aumentava-se a fórmula láctea em 50%.
Nos retornos semanais foram avaliados os níveis de fenilalanina, e aplicados a Early Language Milestone Scale(14) (ELM) e Passos Básicos do Desenvolvimento(15) (PBD). A escala ELM avalia as funções auditiva receptiva, auditiva expressiva e visual. O PBD avalia aspectos do desenvolvimento motor, cognitivo, linguagem e pessoal-social. Foram considerados adequados quanto ao desenvolvimento àqueles lactentes que apresentaram índices normativos em todas as avaliações.
A viabilidade da continuidade do AM no tratamento de crianças com PKU foi verificada por meio dos valores mínimos, máximos, mediana e desvio-padrão (DP) dos resultados dos níveis sanguíneos de fenilalanina e dos resultados do acompanhamento do desenvolvimento pelas escalas ELM e PBD (Tabela 1).
DISCUSSÃO
A manutenção do AM na PKU, apesar de ser um grande desafio, é de extrema relevância, pois há vantagens na amamentação que não podem ser descartadas(7-9,11-13). São inúmeros os benefícios que a prática do AM oferece do ponto de vista biológico e psicossocial. Dentre os benefícios está o fortalecimento do vínculo afetivo entre mãe e bebê, a estimulação funcional necessária para a maturação e o desenvolvimento neuromuscular das funções orais e da respiração nasal os quais, juntos, auxiliarão no crescimento e no desenvolvimento harmônico do sistema estomatognático e estético da face(12,13).
Ao ser amamentada a criança recebe estímulos térmicos, olfativos, visuais, auditivos e motores que favorecem seu desenvolvimento global, sendo de extrema relevância para o desenvolvimento da linguagem e para o fortalecimento das relações afetivas(11).
Dentre as principais orientações recebidas pelas mães em relação ao AM está a importância do leite materno na proteção quanto à doenças(13). Entretanto, no caso de crianças com PKU, estes conceitos devem ser redimensionados, uma vez que o AM para estas crianças deve ser controlado, pois a ingestão da fenilalanina, com a alteração de seu metabolismo, pode gerar variações dos níveis sanguíneos em patamares incompatíveis com o tratamento adequado(8-10) para prevenir as sequelas desta doença.
Estudos(1,5,6) revelaram que o leite materno apresenta níveis de fenilalanina baixos em comparação com outras fórmulas para lactentes, entretanto há a necessidade de verificação do metabolismo do bebê com PKU, pois se os níveis de fenilalanina forem elevados, há riscos para o desenvolvimento infantil.
Por outro lado, o AM na PKU(7-9) necessita ser bem orientado, e é necessário o compromisso materno de cumprir as exigências dos controles dos níveis de fenilalanina, por meio de exames sanguíneos frequentes. Este é um grande desafio e um dos fatores interferentes no tempo de amamentação. As razões para a interrupção do AM, tais como retorno ao trabalho, estresse, redução na produção do leite materno, dentre outras, não são diferentes das demais razões para que qualquer mãe interrompa o AM(9), com o adicional, para as famílias de crianças com PKU, da necessidade do controle rigoroso nos níveis de fenilalanina, por meio de exames de sangue frequentes. Neste estudo, o tempo de amamentação variou de 1 mês e cinco dias a 14 meses (Quadro 1) e não foi considerado o tempo de amamentação anterior ao início dos procedimentos. Um estudo(7) apresentou que a duração do AM em fenilcetonúricos foi de apenas 2,5 meses e em outro(4), a duração variou de 35 dias a um ano. Somente o primeiro estudo(7) relatou que o tempo de AM computado foi após o início dos procedimentos.
Embora o número de participantes deste estudo seja pequeno, os resultados mostraram que aplicando o protocolo de amamentação utilizado, as concentrações de fenilalanina poderiam ser mantidas dentro de níveis de segurança. Outros estudos apresentaram resultados semelhantes(4,7,9). Cabe ressaltar, que a análise das proporções dos níveis sanguíneos de fenilalanina foi apenas descritiva, pois o número de exames foi diferente para cada criança. Um estudo utilizou critérios semelhantes(7).
Na Tabela 1, observa-se que P3 e P6 apresentaram dificuldade no controle dos níveis plasmáticos em algumas das amostras, com mediana e desvio-padrão acima de 6 mg/dL. Nestes casos, o procedimento foi interrompido e foi realizado o controle com o uso exclusivo de fórmula láctea e exames sanguíneos. Os demais participantes conseguiram manter, em sua maioria, limites seguros da fenilalanina.
Um estudo(8) apresentou que 78% dos fenilcetonúricos tratados com AM e fórmula láctea conseguiram manter controle metabólico em níveis seguros, durante os seis meses de duração do estudo. O controle metabólico também sofre influências das mutações genéticas nos mecanismos envolvendo o nível de fenilalanina no sangue(6-10), ou seja, na PKU, combinações nas mutações do gene podem significar diferenças na tolerância da fenilalanina e no controle precoce metabólico, que afeta o tratamento. Daí a importância dos exames sanguíneos frequentes e do estudo genético para a compreensão desses casos, no gerenciamento do tratamento desses indivíduos. Deste modo, há crianças que mesmo com diagnóstico e tratamento iniciados precocemente, com rigoroso controle de dieta, não conseguem manter níveis seguros de fenilalanina no plasma e, desta forma, podem apresentar alterações do desenvolvimento. Cabe ressaltar, entretanto, que as consequências neurológicas dos efeitos bioquímicos podem não só estar ligadas aos altos níveis de fenilalanina no plasma, mas também a outros fatores, como os efeitos dos aminoácidos livres no cérebro, a influência da dopamina e dos neurotransmissores e o efeito da síntese proteica e mielínica no cérebro(2,3).
Quanto ao acompanhamento do desenvolvimento, P3 e P6 apresentaram-se fora dos padrões de normalidade nas escalas de desenvolvimento. As habilidades avaliadas nos procedimentos experimentais foram relativas ao desempenho nas atividades motoras, linguísticas e cognitivas. Achados semelhantes foram encontrados em um estudo(6) no qual 20% da casuística também apresentou alterações.
O distúrbio metabólico da feninalanina interfere na síntese protéica cerebral, na formação da mielina, nos neurotransmissores, prejudicando, particularmente, as vias dopaminérgicas das regiões do córtex pré-frontal e podem trazer disfunções para o hemisfério esquerdo(5), particularmente na substância branca, com reflexos nas funções neuropsicológicas, linguagem e aprendizagem(4,5).
Diversos estudos apresentaram que em fenilcetonúricos há a necessidade de controle rigoroso da dieta e dos níveis de fenilalanina, que devem ser bem estabelecidos na infância(1,6-10).
O tratamento da PKU é complexo, de longa duração e requer mudanças nas ações por parte do paciente e de sua família, para a prevenção das alterações do desenvolvimento, visando sua integração social, familiar e escolar. O sucesso do tratamento, como em qualquer doença crônica, depende da disponibilidade para seguir as recomendações prescritas e devem fazer parte de um programa contínuo, por toda a vida do indivíduo fenilcetonúrico.
COMENTÁRIOS FINAIS
Há viabilidade da continuidade do AM no tratamento de crianças com PKU desde que os níveis de fenilalanina sejam rigorosamente controlados e que os efeitos do AM para o desenvolvimento infantil sejam verificados. Neste estudo, 80% dos lactentes conseguiram manter limites seguros da fenilalanina e desenvolvimento nos índices normativos. Devem ser levados em conta os demais benefícios que a família e a criança com PKU ganham com o AM, quanto aos aspectos físicos, psicológicos e sociais.
REFERÊNCIAS
1. Alderete MS, Mendez RM, Mónzon MF. Avances terapéuticos en fenilcetonúria. Rev Posgrado Catedra Med. 2006;(154):21-3.
2. Sprosen Fj, Enns G. Future treatment strategies in phenylketonuria. Molecular Genetics and Metabolism. 2010;99(Suppl 1):S90-5.
3. Ahring K, Bélanger-Quintana A, Dokoupil K, Gokmen Ozel H, Lammardo AM, MacDonald A, et al. Dietary management practices in phenylketonuria across European centres. Clin Nutr. 2009;28(3):231-6. 4. Moyle JJ, Fox AM, Arthur M, Bynevelt M, Burnett JR. Meta-Analysis of neuropsychological symptoms of adolescents and adults with PKU. Neuropsychol Rev. 2007;17(2):91-101.
5. de Groot MJ, Hoeksma M, Blau N, Reiijngoud DJ, van Sprosen FJ. Pathogenesis of cognitive dysfunction in phenylketonuria: Review of Hypoteses. Mol Genet Metab. 2010;99(1):86-9.
6. Giovannini M, Verduci E, Salvatici E, Fiori L, Riva E. Phenylketonuria: Dietary and therapeutic challenges. J Inherit Metab Dis. 2007;30(2):145-52.
7. Kanufre VC, Starling AP, Leão E, Aguiar MJ, Santos JS, Soares RD, et al., Breastfeeding in the treatment of children with phenylketonuria. J Pediatr (Rio J). 2007;83(5):447-52.
8. Cornejo V, Manríquez V, Colombo M, Mabe P, Jímenez M, De La Parra A et al. Fenilquetonuria de diagnóstico neonatal y lactancia materna. Rev Med Chile. 2003;131(11):1280-7.
9. van Rijn M, Bekhof J, DijkstraT, Smit PG, Moddermam P, van Spronsen FJ. A different approach to breast-feeding of the infant with phenylketonuria. Eur J Pediatrics. 2003;162(5):323-6.
10. Feillet F, Agostoni C. Nutritional issues in treating phenylketonuria. J Inherit Metab Dis. 2010;33(6):659-64. 11. Marques R, Lopez FA, Braga JA. O crescimento de crianças alimentadas com leite materno exclusivo nos primeiros seis meses de vida. J Pediatr (Rio J). 2004;80(2):99-105.
12. Antunes Ldos S, Antunes LA, Corvino MP, Maia LC. Amamentação natural como fonte de prevenção em saúde. Cien Saude Coletiva. 2008;13(1):103-9. 13. Demitto MO, Silva TC, Páschoa AR, Mathias TA, Bercini LO. Orientações sobre amamentação na assistência pré-natal: uma revisão. Rev Rene. 2010;Número especial(11):223-9.
14. Coplan J. Early Language Milestone Scale. 2nd ed. Texas: Pro-Ed; 1993.
15. Perez-Ramos AM. Passos Básicos do Desenvolvimento Infantil. Projeto Multinacional de Educação. MEC/OEA. Brasília 1975.
Conflito de interesses: Não
Recebido em: 22/12/2011
Aceito em: 4/5/2012
Trabalho realizado no Laboratório de Screening Neonatal, Associação de Pais e Amigos dos Excepcionais de Bauru - APAE - Bauru (SP), Brasil, em parceria com o Departamento de Fonoaudiologia, Faculdade de Odontologia de Bauru - FOB/USP - Bauru (SP), Brasil.
- 1. Alderete MS, Mendez RM, Mónzon MF. Avances terapéuticos en fenilcetonúria. Rev Posgrado Catedra Med. 2006;(154):21-3.
- 2. Sprosen Fj, Enns G. Future treatment strategies in phenylketonuria. Molecular Genetics and Metabolism. 2010;99(Suppl 1):S90-5.
- 3. Ahring K, Bélanger-Quintana A, Dokoupil K, Gokmen Ozel H, Lammardo AM, MacDonald A, et al. Dietary management practices in phenylketonuria across European centres. Clin Nutr. 2009;28(3):231-6.
- 4. Moyle JJ, Fox AM, Arthur M, Bynevelt M, Burnett JR. Meta-Analysis of neuropsychological symptoms of adolescents and adults with PKU. Neuropsychol Rev. 2007;17(2):91-101.
- 5. de Groot MJ, Hoeksma M, Blau N, Reiijngoud DJ, van Sprosen FJ. Pathogenesis of cognitive dysfunction in phenylketonuria: Review of Hypoteses. Mol Genet Metab. 2010;99(1):86-9.
- 6. Giovannini M, Verduci E, Salvatici E, Fiori L, Riva E. Phenylketonuria: Dietary and therapeutic challenges. J Inherit Metab Dis. 2007;30(2):145-52.
- 7. Kanufre VC, Starling AP, Leão E, Aguiar MJ, Santos JS, Soares RD, et al., Breastfeeding in the treatment of children with phenylketonuria. J Pediatr (Rio J). 2007;83(5):447-52.
- 8. Cornejo V, Manríquez V, Colombo M, Mabe P, Jímenez M, De La Parra A et al. Fenilquetonuria de diagnóstico neonatal y lactancia materna. Rev Med Chile. 2003;131(11):1280-7.
- 9. van Rijn M, Bekhof J, DijkstraT, Smit PG, Moddermam P, van Spronsen FJ. A different approach to breast-feeding of the infant with phenylketonuria. Eur J Pediatrics. 2003;162(5):323-6.
- 10. Feillet F, Agostoni C. Nutritional issues in treating phenylketonuria. J Inherit Metab Dis. 2010;33(6):659-64.
- 11. Marques R, Lopez FA, Braga JA. O crescimento de crianças alimentadas com leite materno exclusivo nos primeiros seis meses de vida. J Pediatr (Rio J). 2004;80(2):99-105.
- 12. Antunes Ldos S, Antunes LA, Corvino MP, Maia LC. Amamentação natural como fonte de prevenção em saúde. Cien Saude Coletiva. 2008;13(1):103-9.
- 13. Demitto MO, Silva TC, Páschoa AR, Mathias TA, Bercini LO. Orientações sobre amamentação na assistência pré-natal: uma revisão. Rev Rene. 2010;Número especial(11):223-9.
- 14. Coplan J. Early Language Milestone Scale. 2nd ed. Texas: Pro-Ed; 1993.
- 15. Perez-Ramos AM. Passos Básicos do Desenvolvimento Infantil. Projeto Multinacional de Educação. MEC/OEA. Brasília 1975.
Datas de Publicação
-
Publicação nesta coleção
07 Jan 2013 -
Data do Fascículo
2012
Histórico
-
Recebido
22 Dez 2011 -
Aceito
04 Maio 2012